

Abstract
Bilateral breast cancers (BBC) are currently treated as independent tumors arising in the same patient. Herein, we investigated whether BBC indeed evolve independently at the genomic level. We examined paired targeted next generation sequencing genotypes from 155 paraffin tumors corresponding to 76 BBC patients (75 women and one man; 52 concurrent and 24 metachronous), for coding mutations (amino acid changing, minor allele frequency <0.1%) and single nucleotide polymorphism (SNP) zygosity. Germline genotypes were available for 29 patients. Mutations were present in 80 tumors (54/76 patients; 71%), were mostly tumor-private (90%), more frequent in TP53 (19%), PIK3CA (14%), CDH1, GATA3, MLL3. TP53 mutations were more frequent in metachronous tumors (P<0.001); hormone receptor negative (P<0.001); with higher Ki-67 (P=0.002); and, in younger patients (P=0.01). Hypermutated tumors, all TP53 mutated, were diagnosed as the first incidence in 5 patients; their metachronous counterparts were mutation poor without TP53 involvement. Paired tumors shared common mutations at intratumoral frequency >20% in 10/54 comparable BBC (18.5%), 8/10 concurrent. SNP zygosity status was less preserved in metachronous, compared to concurrent disease. Pathogenic germline mutations were present in 10/29 patients, 9 in BRCA1 and one in TP53 (p.Phe341Val, first report in the germline). BBC demonstrated extensive inter- and intra-patient heterogeneity in the present thus far largest series of corresponding paired genotypes. The majority evolve independently and unpredictably, supporting current clinical practice. A considerable minority though, retains clonal origin and may be regarded as a distinct group for therapeutic interventions among concurrent BBC.
Keywords: Bilateral, breast cancer, targeted next generation sequencing, coding mutations, concurrent, metachronous, contralateral, clonality, hypermutation
Introduction
Breast cancer is the most common type of cancer among women, with 231,840 new patients diagnosed every year in the United States [1]. Due to novel diagnostic and therapeutic approaches breast cancer prognosis and survival have significantly improved. More than 90% of breast cancer patients are alive at 5 years and 78% at 15 years after diagnosis. An important issue in these women is the risk of developing a second primary cancer.
Bilateral breast cancer has an incidence of 4-20% in women diagnosed with breast cancer [2,3]. Bilateral tumors are diagnosed simultaneously or within three months after the initial diagnosis (concurrent) in 0.3-12% of patients, while in 5-10% of patients the contralateral tumor is diagnosed more than three months after the first one (metachronous) [4,5].
It is of great clinical importance to identify whether contralateral tumors represent a second primary or a metastatic spreading of the initial breast tumor. Such a distinction alters the physician’s treatment plan and ultimately the prognosis of the disease. To date, metachronous contralateral tumors are treated as second primary tumors; concurrent tumors are evaluated in parallel and treatment decisions are made upon the characteristics of the more aggressive tumor.
Different studies attempting to define the origin of the contralateral tumor based on clinical and histopathologic criteria [2,4,6] did not reach definitive conclusions. Further studies explored the molecular characteristics of bilateral breast cancers. Few of these studies indicated that the contralateral tumor might represent a metastatic lesion [7,8], while the majority demonstrated a high degree of discordance and advocated for a separate tumor origin [9-12]. Due to limited sample size, however, the results of these studies need to be reviewed with caution.
This is the first study to investigate large-scale genomic characteristics of bilateral breast cancer. Using targeted next generation sequencing (NGS), we explored the mutational profile of bilateral breast cancers, both concurrent and metachronous in 76 patients. We demonstrated that the majority of the tumor pairs do not share the same genetic alterations, supporting the independent nature of the contralateral tumor.
Methods
Study outline, patients and tumors
Clinical data and formalin-fixed paraffin-embedded (FFPE) tumor tissue samples from 76 patients with operable breast cancer were retrospectively retrieved from the Clinical Data Bank and Tumor Repository of the Hellenic Cooperative Oncology Group (HeCOG). Patients (75 women, 1 man) had been treated in Oncology departments of HeCOG-affiliated hospitals from 2000 to 2015. Bilateral tumors had been characterized at local pathology laboratories for histological type, tumor size, grade, nodal status, multifocality, and ER/PgR/HER2 expression with immunohistochemistry (IHC). Central pathology review of FFPE hematoxylin and eosin (H&E) sections was undertaken at the Laboratory of Molecular Oncology (MOL, Hellenic Foundation for Cancer Research/ HeCOG/Aristotle University of Thessaloniki [AUTH]) by an experienced breast pathologist (M.B.) who recorded the presence of infiltrative carcinoma, in situ carcinoma, pre-cancerous lesions and normal tissue. Marked tissue areas with known tumor cell content (TCC%) were manually macrodissected and processed for DNA extraction and targeted next generation sequencing (NGS) genotyping with a previously validated custom tissue panel [13,14]. The same panel was used for genotyping 30 blood samples from the above patients. The study was approved by the Bioethics Committees of Aristotle University of Thessaloniki (2./4.2.2015). Written informed consent had been obtained from all patients allowing the use of their biologic material for research purposes. The study outline with respect to the performed tissue panel investigations is shown in the REMARK diagram in Figure 1.
Figure 1.
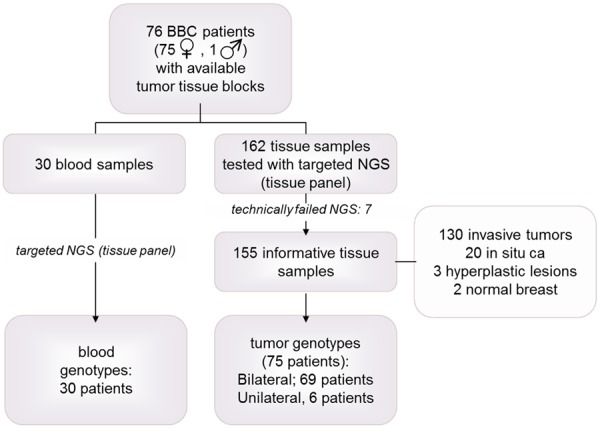
Study outline (REMARK).
Tumors were also centrally assessed for stromal tumor infiltrating lymphocytes (TILs) density based on Salgado et al [15] as previously described [16] on whole H&E sections; and, for clinical subtypes with ER/PgR/HER2/Ki67 IHC and HER2 FISH [17] on in-house low-density tissue microarrays (TMA) that contained two 1.5 mm cores per tumor. Ki67 cut-off at 20% was applied [18] to distinguish Luminal A and Luminal B tumors.
In addition, germline status was available for 29 of the above patients from NCSR “Demokritos”. These data were obtained in the frame of a separate study that was locally approved (240/EHΔ/11.3) and was in agreement with the 1975 Helsinki statement, revised in 1983. Peripheral blood DNA had been tested as previously described [19] for the five Greek founder and one recurrent BRCA1 mutations; if found wild-type, samples were further tested by Sanger sequencing for BRCA1 and BRCA2 mutations or by massively parallel sequencing with the Trusight Cancer panel on Illumina MiSeq (Illumina, San Diego, USA). The median read depth was ~200×, with 50-fold being the minimum cut-off for variant calling.
Detailed patient clinicopathologic characteristics, including germline data for cancer predisposition genes can be found at (https://figshare.com/s/39adae1d323bde0884d5, Table S1, in file: BBC supplementary data).
Targeted NGS genotyping
The majority of tumor samples (78%) had TCC ≥50%, but samples with as low as 15% TCC were also processed based on our previous experience [14]. DNA was extracted from macrodissected tissue fragments with the QIAamp® DNA Mini kit (Hilden, Germany) according to the manufacturer’s instructions; quantity was measured with the Qubit fluorometer (Life Technologies, Paisley, UK). Criteria for processing FFPE samples for NGS genotyping were ≥2 ng/ul DNA amplifiable at Ct≤32 for two control qPCR assays. Peripheral blood DNA had been isolated based on the salt-extraction procedure [20].
The tissue panel [14] targeted coding regions and single nucleotide polymorphisms (SNPs) in 61 genes most frequently implicated in breast cancer [21,22] and was applied for library construction and NGS on an Ion Proton System (Ion Torrent/Invitrogen, Paisley, UK). Samples were accepted for further evaluation if all amplicons had been read >100 times. Variants obtained from Ion Reporter v.4 were filtered out if not annotated; if indels with GC-stretches (reading artifacts with semiconductor sequencing); and, if p (system quality metric including false discovery rate) >0.0001. Variants were accepted for analysis when position and variant coverage were higher than 100 and 40, respectively. In order to avoid false negative calls, and hence, false heterogeneity interpretation, we evaluated amplicon read efficiency in matched sample pairs prior to assessing common (shared) and private variants. As above, variants were compared among samples if corresponding amplicons had been read >100 times in each sample under comparison.
Bioinformatics-statistics
We examined the presence and pattern of coding mutations and the preservation of SNP zygosity in paired samples from the same patient. Coding mutations corresponded to amino acid changing variants with minor allele frequency (MAF) <0.1% or not reported in dbSNP from NCBI and in 5000 Exomes. Initially, we assessed mutation pathogenicity according to available information in COSMIC and by using ANNOVAR [2]. However, because this and any available algorithm prioritize genomic variants in the germline for their disease relevance and may, therefore, not adequately predict pathogenicity and functional implications of the same mutations in tumors [3], we did not further pursue with characterization of these features. We assessed SNP zygosity in matched sample pairs (bilateral tumors, tumor-blood) as an indicator for genomic stability. For this, we evaluated matched samples with ≥5 SNPs (range: 5-25 SNPs per case). Differences of ≤|20| in variant allele frequencies (VAF) at SNP positions were considered as corresponding to stable, the rest to altered zygosity status. Cases were considered as (a) stable, if >90% of SNPs retained their zygosity status upon paired comparisons, and (b) of intermediate stability, for zygosity preservation in 50-90% of SNPs. Only adequately covered positions were considered for matching mutations and SNP comparisons among samples from the same patient.
In order to avoid statistical bias, we divided concurrent breast cancer tumors based on location (right or left) as previously done. Our two groups were well balanced between different histopathologic parameters, as expected. Metachronous tumors were registered based on their presentation; side 1 included all 1st incidence tumors, while side 2 the respective metachronous ones.
Frequencies and percentages were used to present categorical variables, while various measures (mean, SD, median, min and max) were used for continuous variables. Classic statistics for associations between variables included chi-square and Mann-Whitney or Kruskal-Wallis tests, where appropriate. Correlations were calculated using the Spearman’s rank correlation coefficient (Rho). Wilcoxon singed-rank test was used for comparing equal distributions of paired samples. All tests were 2-sided with the significance level set at α=0.05. Contingency tables were created with JMP v.10; descriptive statistics for parameter associations and correlation of continuous variables were performed by using the SPSS v.15 and the SAS software for statistical analysis (SAS for Windows, version 9.3, SAS Institute Inc., Cary, NC, USA).
Eligible samples
We excluded one pair of bilateral tumors (2 samples) due to inadequate DNA template, 1 sample with no tumor tissue and 4 samples identified as technical outliers based on the criteria mentioned above. Our final cohort consisted of 185 samples (155 FFPE and 30 peripheral blood) corresponding to 75 patients (Figure 1); matched bilateral tumor samples from 69 patients were eligible for analysis. The obtained values for mean depth of the eligible samples were median: 1638; mean ± SD: 3021 ± 2760; min-max: 350-21500. Despite outlier differences, mean depth did not significantly differ between tissue and blood samples (Mann-Whitney P=0.6328).
Results
Clinicopathological characteristics of BBC
Concurrent disease was noticed in 52 patients (Table 1). The time interval for manifestation of metachronous disease in 24 patients ranged between 0.5-19 yrs (mean ± SD: 6.3±4.8 yrs; median: 5.6 yrs). Menopausal status of the 75 female patients and age of all patients at first diagnosis are shown in Table 1. Twenty-eight patients (36.8%) were <50 yrs. The single male patient was 78y.o.
Table 1.
Patients, disease and tumor characteristics
| All cases: | 76 | |||||
| Age | ||||||
| N cases | 76 | |||||
| Mean ± SD | 57.2 ± 15.4 | |||||
| Median | 57.2 | |||||
| Min-max | 30-87.5 | |||||
| N (%) | ||||||
| Disease presentation | ||||||
| N cases | 76 | |||||
| Concurrent | 52 (68.4) | |||||
| Metachronous | 24 (31.6) | |||||
| Menopausal status | ||||||
| N cases | 71 | |||||
| Pre | 25 (40) | |||||
| Post | 46 (60) | |||||
| SIDE 1 | SIDE 2 | TOTAL | ||||
| Positive nodes | ||||||
| N cases | 60 | 51 | 111 | |||
| Mean ± SD | 3.1 ± 5.8 | 1.1 ± 1.9 | 2.5 ± 3.0 | |||
| Median | 0 | 0.5 | 0 | |||
| Min-max | 0-23 | 0-9 | 0-23 | |||
| Tumor size | ||||||
| N cases | 71 | 70 | 141 | |||
| Mean ± SD | 2.3 ± 1.9 | 2.5 ± 3.0 | 2.4 ± 2.5 | |||
| Median | 1.8 | 1.8 | 1.8 | |||
| Min-max | 0.1-11 | 0.2-21 | 0.1-21 | |||
| Ki67 | ||||||
| N cases | 68 | 68 | 136 | |||
| Mean ± SD | 26.6 ± 25.4 | 29 ± 24.5 | 27.9 ± 24.8 | |||
| Median | 18 | 23 | 20 | |||
| Min-max | 1-90 | 1-95 | 1-95 | |||
| Tumor infiltrating lymphocytes (TILs) | ||||||
| N cases | 68 | 63 | 131 | |||
| Mean ± SD | 9.5 ± 15.5 | 11.1 ± 17.2 | 10.3 ± 16.3 | |||
| Median | 3 | 4 | 3 | |||
| Min-max | 1-75 | 1-80 | 1-80 | |||
| SIDE 1 | SIDE 2 | TOTAL | ||||
| N | % | N | % | N | % | |
| Main lesion, carcinoma | ||||||
| In situ | 7 | 9.2 | 7 | 9.3 | 14 | 9.2 |
| Invasive | 69 | 90.8 | 68 | 90.7 | 137 | 90.8 |
| Concordance* invasive ca: 64/75 (85.3%) | ||||||
| Histology | ||||||
| Ductal, NST | 51 | 67.1 | 48 | 64 | 99 | 65.1 |
| Other | 25 | 32.9 | 27 | 36 | 52 | 34.9 |
| Concordance histology: 44/75 (61.3%) | ||||||
| Grade | ||||||
| I | 11 | 15.7 | 8 | 11.9 | 19 | 13.9 |
| II | 27 | 39.7 | 27 | 39.7 | 54 | 39.4 |
| III | 32 | 45.7 | 32 | 47.8 | 64 | 46.7 |
| Concordance grade: 37/63 (58.7%) | ||||||
| In situ lesion | ||||||
| Present | 47 | 61.2 | 43 | 57.3 | 90 | 59.6 |
| Absent | 29 | 38.2 | 32 | 42.7 | 61 | 40.4 |
| Concordance in situ presence: 39/76 (51.3%) | ||||||
| Clinical subtype | ||||||
| HER2-Enriched | 5 | 6.9 | 3 | 4.2 | 8 | 5.5 |
| Luminal A | 34 | 45.9 | 31 | 43.1 | 65 | 44.5 |
| Luminal B | 21 | 28 | 21 | 29.2 | 42 | 28.8 |
| Luminal-HER2 | 5 | 6.8 | 8 | 11.1 | 13 | 8.9 |
| TNBC | 9 | 12.2 | 9 | 12.5 | 18 | 12.3 |
| Concordance, clinical subtype: 37/72 (51.4%) | ||||||
| TILs | ||||||
| Low (≤5%) | 50 | 73.6 | 41 | 65.1 | 91 | 69.5 |
| High (5-50%) | 16 | 23.5 | 19 | 30.1 | 35 | 26.7 |
| LPBC (>50%) | 2 | 2.9 | 3 | 4.8 | 5 | 3.8 |
| Concordance, TILs: 43/59 (72.9%) | ||||||
| Multifocality | ||||||
| No | 54 | 72 | 49 | 67.1 | 103 | 69.6 |
| Yes | 21 | 28 | 24 | 32.9 | 45 | 30.4 |
| Concordance, multifocality: 48/72 (66.7%) | ||||||
| Size, categorical | ||||||
| <2 cm | 38 | 52.1 | 42 | 59.3 | 80 | 55.5 |
| ≥2 cm | 35 | 47.9 | 29 | 39.7 | 64 | 44.5 |
| Concordance, size: 36/69 (52.2%) | ||||||
| Nodal status | ||||||
| 0-3 LN | 49 | 80.3 | 46 | 90.2 | 95 | 84.8 |
| ≥4 LN | 12 | 19.7 | 5 | 0.8 | 17 | 15.2 |
| Concordance, nodal status: 35/44 (79.5%) | ||||||
concordance between Side 1 and Side 2.
In total, 76 bilateral breast tissue surgical specimens were analyzed (152 unilateral specimens), involving 65 cases with bilateral invasive carcinomas, 3 cases with bilateral CIS and 8 with unilateral invasive and contralateral carcinoma in situ (CIS). CIS were observed in 90 specimens (59.6%), 89 of them of ductal origin (DCIS). This incidence is significantly higher compared to the approximately 26% reported for DCIS diagnoses [23]. Incidence and bilateral heterogeneity of clinicopathological parameters are described in Table 1. Histological parameters and their concordance status were not associated with patient age, disease presentation, tumor size or nodal status.
Comparisons of clinicopathological parameters, including disease presentation characteristics, are shown in Table 2. Compared to concurrent BBC, metachronous disease was significantly associated with younger patient age and premenopausal status, in line with previous reports [4,24]. Metachronous BBC were rich in TNBC and HER2-enriched clinical subtypes (36%); the rate of ER/PgR-positive tumors was similar in the 1st (61%) and 2nd (61%) incidence but concordance of bilateral ER/PgR phenotypes was approximately 70%. The majority of concurrent BBC (96%) included at least one ER/PgR-positive tumor and this phenotype was preserved bilaterally in 92% of the cases. The overall incidence and bilateral concordance of the ER/PgR phenotype was significantly lower in metachronous compared to concurrent BBC (Table 2).
Table 2.
Comparison of clinicopathological and genomic characteristics in concurrent and metachronous BBC
| Concurrent | Metachronous | p-value | |||
|---|---|---|---|---|---|
| All patients: 75 | 51 | 24 | |||
| Age | <0.001 | ||||
| N (75) | 51 | 24 | |||
| Mean ± SD | 62.4 ± 14.2 | 45.9 ± 12.4 | |||
| Median | 64.7 | 40.8 | |||
| Min-max | 30-87.5 | 33.2-74.4 | |||
| N mut per case (N=75) | 0.19 | ||||
| Mean ± SD | 1.65 ± 1.64 | 7.3 ± 14.2 | |||
| Median | 3 | 1.5 | |||
| Min-max | 0-6 | 0-56 | |||
| N genes per case (N=75) | 0.23 | ||||
| Mean ± SD | 1.4 ± 1.4 | 4.9 ± 8.9 | |||
| Median | 1 | 1 | |||
| Min-max | 0-6 | 0-33 | |||
| N | % | N | % | ||
| Menopausal status (N=72) | <0.001 | ||||
| Premenopausal | 9 | 18.4 | 17 | 73.9 | |
| Postmenopausal | 39 | 79.6 | 6 | 26.1 | |
| N/A | 1 | 8 | 0 | 0 | |
| Concordant histology (N=75) | 0.25 | ||||
| Yes | 29 | 56.9 | 17 | 70.8 | |
| No | 22 | 43.1 | 7 | 29.2 | |
| Concordant grade (N=75) | 0.84 | ||||
| Yes | 24 | 47 | 13 | 54.2 | |
| No | 19 | 37.3 | 8 | 33.3 | |
| N/A | 8 | 15.7 | 3 | 12.5 | |
| Concordance for in situ (N=75) | 0.12 | ||||
| Yes | 29 | 56.9 | 9 | 37.5 | |
| No | 22 | 43.1 | 15 | 62.5 | |
| Concordant ER/PR status (N=73) | 0.013 | ||||
| Yes | 46 | 92 | 16 | 69.6 | |
| No | 4 | 8 | 7 | 30.4 | |
| Clinical subtype (N=147) | <0.001 | ||||
| ER+(LUMA, LUMB, LUM-HER2 | 92 | 92 | 30 | 63.8 | |
| ER-(HER2-enriched, TNBC) | 8 | 8 | 17 | 36.2 | |
| Both ER/PgR+ vs. both TNBC (N=73) | <0.001 | ||||
| Both ER/PgR+ | 44 | 88 | 11 | 47.8 | |
| Both TNBC | 1 | 2 | 5 | 21.8 | |
| Other | 5 | 10 | 7 | 30.4 | |
| Concordant size (N=63) | 0.78 | ||||
| Yes | 24 | 51.1 | 12 | 54.5 | |
| No | 23 | 48.9 | 10 | 45.5 | |
| Multifocality (N=75) | 0.31 | ||||
| Yes | 13 | 72.5 | 9 | 37.5 | |
| No | 38 | 27.5 | 14 | 58.3 | |
| N/A | 0 | 0 | 1 | 4.2 | |
| TP53 mutations (N=75) | <0.001 | ||||
| Mutated | 12 | 23.5 | 16 | 66.7 | |
| WT | 39 | 76.5 | 8 | 33.3 | |
| PIK3CA mutations (N=75) | 0.62 | ||||
| Mutated | 20 | 39.2 | 8 | 33.3 | |
| WT | 31 | 60.8 | 16 | 66.7 | |
| GATA3 mutations (N=75) | 0.88 | ||||
| Mutated | 7 | 13.7 | 3 | 12.5 | |
| WT | 44 | 86.7 | 21 | 87.5 | |
| MLL3 mutations (N=75) | 0.16 | ||||
| Mutated | 2 | 3.9 | 3 | 12.5 | |
| WT | 49 | 96.1 | 21 | 87.5 | |
| Hypermutated samples (N=75) | <0.001 | ||||
| Hyper | 0 | 0 | 5 | 20.8 | |
| No | 51 | 100 | 19 | 79.2 | |
| Preserved SNPs>90%* (N=64) | 0.1 | ||||
| Yes | 37 | 82.2 | 12 | 63.2 | |
| No | 8 | 17.8 | 7 | 36.8 | |
| Preserved zygocity in>90% of SNPs* (N=64) | 0.042 | ||||
| Yes | 29 | 64.4 | 7 | 36.8 | |
| No | 16 | 35.6 | 12 | 63.2 | |
| T1 vs. T2 stability (N=64) | 0.058 | ||||
| Stable | 26 | 57.8 | 5 | 26.3 | |
| Intermediate | 14 | 31.1 | 9 | 47.4 | |
| Unstable | 5 | 11.1 | 5 | 26.3 | |
N in parentheses: number of patients compared in each case;
T1 vs. T2 comparisons.
Stromal TILs density was assessable in 133 tumors including 60 matched bilateral; it was low (median: 4%) and it was not associated with ER/PgR status, other than previously reported [16,25], probably due to over-representation of ER/PgR-positive tumors in this cohort. TILs were mostly homogeneous bilaterally (Fisher’s exact P=0.018 for comparisons with the median 4% as a cut-off) and did not differ between concurrent and metachronous tumors.
Mutation analysis
In the informative 185 samples of our study, NGS revealed 2148 variants eligible for analysis (Figure 2A); among these, 258 (34.2%) were coding mutations, the rest being synonymous changes in coding regions or SNPs (Figure 2B). A detailed list of the identified coding mutations can be found at (https://figshare.com/s/39adae1d323bde0884d5, Table S2, in file: BBC supplementary data). Coding mutations were mostly missense (89%), 4% were nonsense and 7% were frameshift indels. Most frequently mutated genes were TP53 (19%) and PIK3CA (14%), followed by CDH1 (7%), GATA3 (6%) and MLL3 (5%). Recurrent coding mutations present in >2 tumors were observed only in PIK3CA hotspots (6x p.Glu542Lys; 6x p.Glu542Lys; 12x p.His1047Arg). Out of 197 unique mutations, 50.1% were characterized as probably damaging with ANNOVAR; 15.2% as possibly damaging; and 34% as benign (Table S3, in file: BBC supplementary data, https://figshare.com/s/39adae1d323bde0884d5).
Figure 2.
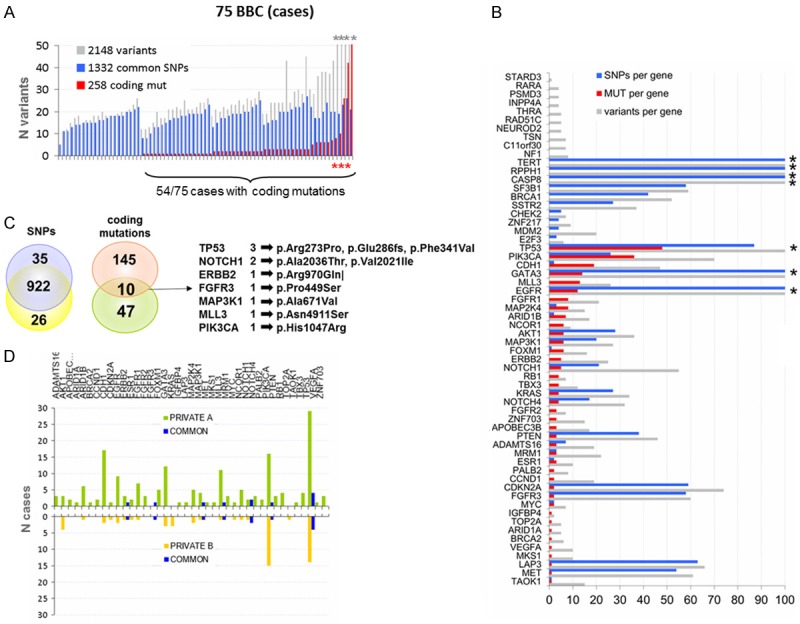
Distribution of NGS variants in BBC. A. Distribution of variants of any type in the study cases. All variants, SNPs and coding mutations are shown per case. For similar sequencing efficiency metrics (total reads, on target reads, uniformity of reads), 5-214 variants were identified per case (mean ± SD: 23 ± 30; median: 22), up to 54 in the same gene (mean ± SD: 15 ± 7; median: 12). Asterisks: truncated Y-axis for cases with >50 variants. B. Distribution of variants per gene. Grey bars: total number of variants of any type. Coding mutations were identified in 41 out of 61 genes in the MPS panel (red bars). The total number of mutations per gene varied from 1 to 49 (median: 3 mutations per gene). In additional 10 genes, only SNPs were present (blue bars). The number of SNPs per sample ranged from 10-20. Asterisks: >100 variants for these genes. C. Common and private SNPs and coding mutations. SNPs were preserved at 94% in both sides (922/983 comparable SNPs); by contrast, only 4.9% common coding mutations were observed (10/202 comparable mutations). The remaining 192 comparable mutations were present unilaterally, as shown. D. Distribution of gene coding mutations on either side; common, private A and private B mutations represented by blue, green and orange bars, respectively.
Coding mutations were found in 80/130 invasive tumors (62%), in 8/20 (40%) DCIS, in all 3 hyperplastic and in the 2 normal informative samples. Mutations were identified in 72% of the cases (Figure 2A).
The 69 cases with matched bilateral tumor samples shared 50% of all variants. Among these, SNP variants were up to 94% common bilaterally, whereas mutations were mostly private in either side (Figure 2C). Out of 202 mutations that were comparable in both sides only 10 were bilaterally common (Figure 2C and 2D; Table 3). The rate of BBC with common mutations was 18.5% among the 54 cases with mutant tumors, and 13.3% among patients with informative tumors in both sides.
Table 3.
Description of shared coding mutations identified in matched bilateral tumors and in peripheral blood samples (tissue panel)
| Sample id | Gene | Mutation location | Coding | Protein | Function | MUT allele freq blood | MUT allele freq T1 | MUT allele freq T2 | COSMIC for the present mutation |
|---|---|---|---|---|---|---|---|---|---|
| BIL-006 | FGFR3 | T1, T2 | c.1345C>T | p.Pro449Ser | Missense | 0.52 | 0.5 | Not registered | |
| BIL-013*,$ | TP53 | T1, T2 | c.818G>C | p.Arg273Pro | Missense | 0.35 | 0.76** | COSM165077 | |
| BIL-017 | PIK3CA | T1, T2 | c.3140A>G | p.His1047Arg | Missense | 0.3 | 0.22 | COSM94986 | |
| BIL-028 | ERBB2 | T1, T2 | c.2909G>A | p.Arg970Gln | Missense | 0.63 | 0.55 | Not registered | |
| BIL-033 | NOTCH1 | T1, T2 | c.6106G>A | p.Ala2036Thr | Missense | 0.23 | 0.28 | COSMIC: p.A2036V | |
| BIL-035 | MAP3K1 | T1, T2 | c.2012C>T | p.Ala671Val | Missense | 0.41 | 0.48 | COSM3674469 | |
| BIL-042*,$ | TP53 | BL, T1, T2 | c.1021T>G | p.Phe341Val^ | Missense | 0.51 | 0.58 | 0.84** | Not registered, p.R342*; p.R342P |
| BIL-062 | TP53 | T1, T2 | c.6061G>A | p.Val2021Ile | Missense | 0.43 | 0.4 | Not registered | |
| BIL-064 | MLL3 | T1, T2 | c.14732A>G | p.Asn4911Ser | Missense | 0.51 | 0.64 | Not registered | |
| BIL-067$ | TP53 | T1, T2 | c.856_857delGA | p.Glu286fs | Frameshift/Deletion | 0.26 | 0.31 | Not registered | |
| BIL-020 | FOXM1 | BL | c.490C>T | p.Arg164Trp | Missense | 0.47 | COSM1476487 | ||
| BIL-020 | APOBEC3B | BL | c.568A>G | p.Arg190Gly | Missense | 0.52 | Not registered | ||
| BIL-048 | EGFR | BL^^ | c.3244A>T | p.Ile1082Leu | Missense | 0.49 | Not registered |
shared TP53 mutation in blood (BL) and in both tumors (T1, T2).
metachronous cases, one with common blood/tumor TP53 mutation.
cases with germline mutations found upon testing for cancer predisposing genes.
increased VAF indicating loss of the wild-type allele in the metachronous tumor.
non-informative amplicons and positions in the tumors of this case.
Comparison of bilateral mutation profiles
Metachronous tumors had more than double mutations as compared to concurrent ones (174 vs. 84, respectively) but this difference did not reach statistical significance (chi-square, P=0.155). The majority of mutations concerned 5 tumors unilaterally (Figure 3A), which also carried multiple mutations in ≥2 genes (Figure 3A and 3C), a feature that was absent in all other tumors. Based on the characteristics, we called these 5 tumors hypermutated. Sequencing performance of these samples did not differ from samples with lower mutation numbers or without mutations (Figure 3A). All 5 hypermutated tumors were the 1st incidence of metachronous disease (Figure 3A and 3C). TP53 had a significantly higher mutation rate in metachronous than in concurrent tumors (Figure 3B and 3C; Table 2). The 39.2% incidence of PIK3CA mutations (57% if examined among tumors with mutations) in the mostly ER/PgR positive concurrent tumors (Figure 3B) was in line with previous observations for this phenotype [26,27]. There was no difference in mutation prevalence of the remaining recurrently mutated genes between the two subgroups.
Figure 3.

Mutation patterns in concurrent and metachronous BBC. (A) Distribution of mutations and mutated genes in tumors of both sides. SNP numbers are shown for comparison of sequencing performance of the samples; these did not differ for samples in side 1 vs. side 2 (Mann-Whitney P=0.5656) and showed a good correlation upon bivariate comparisons (Spearman’s rho 0.641; P<0.0001). The matched tumor was not available in one of the hypermutated cases (arrow). (B and C) mutation maps in concurrent (B) and metachronous (C) cases. The number of mutations per gene is indicated on the color scale. Indicated are germline mutations; hypermutated tumors; tumors with shared mutations. Most frequently mutated gene in concurrent samples is PIK3CA, followed by TP53 and GATA3. Metachronous tumors carry more frequently mutations in TP53, followed by PIK3CA and CDH1.
Mutation rate in different genes was associated with various clinicopathological parameters (Table 2). TP53 mutations in tumors were associated with younger age (Kruskal-Wallis, P=0.013), higher Ki67 labeling (chi-square, P=0.002) and ER/PgR-negative clinical subtypes (chi-square, P<0.001). TP53 mutations were present in all 5 hypermutated tumors but in none of the lobular or mucinous types.
Stromal TILs, as a continuous variable, did not correlate with any clinicopathological or mutation parameter. However, all hypermutated tumors had <4% TILs. This feature was present in the metachronous tumors of the same patients as well. High stromal TILs (median cut-off) were often present in tumors with TP53 mutations (23/40 tumors in both sides; 57%), while tumors without TP53 mutations more frequently had low TILs (65/82; 79%); this pattern was observed in both sides but was statistically significant unilaterally (chi-square, P=0.008). The opposite pattern applied for PIK3CA mutations, which were infrequent in tumors with high TILs (9/40 tumors in both sides; 22%); significance was again obtained unilaterally only (P=0.041) probably due to small group numbers.
Variant allele frequencies (VAF) and SNP zygosity status
VAF distribution significantly differed between SNPs and coding mutations (P<0.0001), while mutation VAFs were significantly lower in side 1 compared to side 2 (P=0.0001) (Figure 4A). Excluding the hypermutated tumors in side 1 resulted in similar mutation VAFs for both sides with mean values around 30% (Figure 4B). Distribution of SNP VAF’s peaked around 50% and towards 100% (Figure 4A), following the expected germline zygosity pattern. SNP VAFs fairly correlated between tumors and between germline and each tumor (Figure 4C). In cases with hypermutated tumors, SNP VAFs between metachronous tumors showed a weaker but still fair correlation (Spearman’s rho=0.700).
Figure 4.
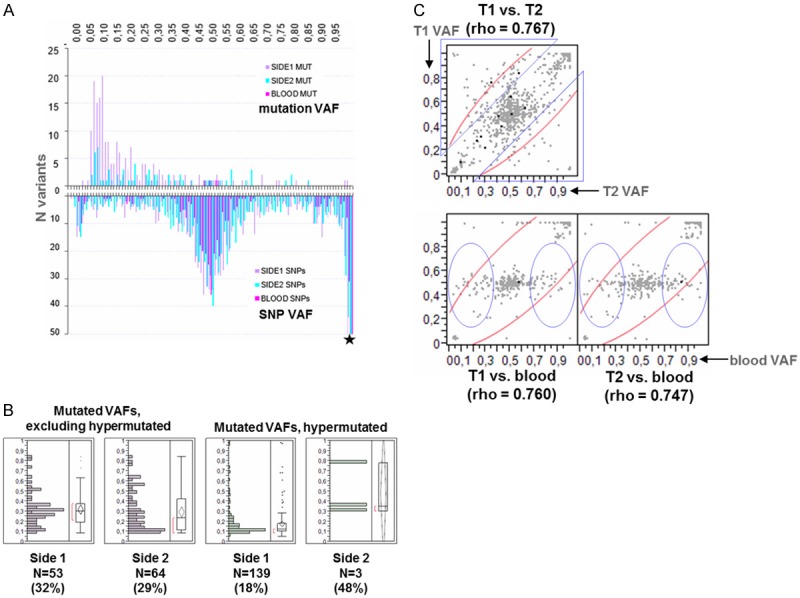
Bilateral comparisons of Variant Allele Frequencies (VAFs). A. VAF distribution for coding mutations and SNPs in both sides and in the germline. Germline variants as identified with the tissue panel. VAF distribution for coding mutations was skewed towards 0, with mean (median) values of 21% (14%) in side 1 and 30% (21%) in side 2. Most mutation VAFs were <25%, particularly in Side 1. B. Coding mutation VAFs are significantly lower in hypermutated as compared to non-hypermutated tumors (Mann-Whitney P<0.001). Numbers in parentheses: mean values for VAFs. N: number of mutations. Note that the contralateral tumors paired to hypermutated ones exhibited only one mutation in 3 out of 4 comparable cases, although they were diagnosed years later after the hypermutated tumors. C. Bilateral correlations of SNP VAFs in matched samples. Tumors in side 1 (T1) and in side 2 (T2) were compared with each other and with germline. SNPs were identified with the tissue panel. Triangles and circles: altered zygosity for these SNPs. Spearman’s rho values yielded P’s <0.0001 in all comparisons.
SNPs were compared for preserved incidence, zygosity and overall stability in 64 out of 76 cases. SNPs were shared bilaterally at a high rate in both concurrent (82.2%) and metachronous (63%) tumors. SNP zygosity status was less preserved in metachronous as compared to concurrent cases, but SNP stability reached only a trend of statistical significance (Table 2). In 25/30 cases with available peripheral blood samples, concurrent and metachronous disease did not differ in terms of germline SNP preservation. SNP zygosity was more frequently preserved (>90%) in ER/PgR-positive bilateral tumors, as opposed to bilateral TNBC or cases with different subtypes (chi-square, P=0.005). Stromal TILs did not associate with SNP zygosity and stability.
Mutations in non-cancer samples and in peripheral blood DNA
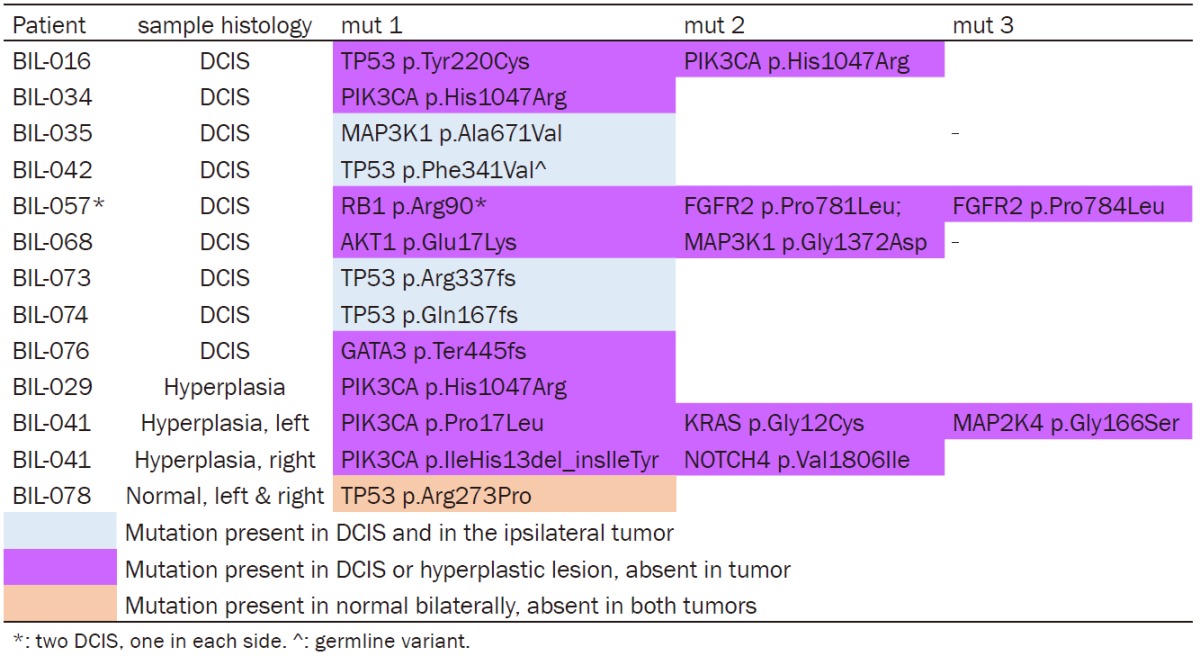
We identified 13 coding mutations in 10 DCIS (Table 4). Three out of the four TP53 and one MAP3K1 mutation were preserved in the ipsilateral tumors, while the rest were DCIS-private. VAFs of the shared mutations in DCIS and matched tumors were >25%. Coding mutations were observed in the examined 3 hyperplastic and 2 normal samples (Table 4) with VAFs <20%. All hyperplastic lesions carried mutations in PIK3CA, 2 of them in other genes as well. All identified mutations in the hyperplastic lesions were private. The 2 normal bilateral samples were obtained from our male patient. Interestingly, they were both positive for the TP53 p.Arg273Pro mutation, which was absent from the matched tumors. The TP53 p.Arg273Pro missense mutation has been previously characterized as gain of function mutation providing cancer cells with growth and survival advantage [28]. The genotypes of all samples for these cases are presented in Figure 5.
Table 4.
Amino acid changing mutations in ductal carcinoma in situ (DCIS), hyperplastic lesions and normal breast tissue
|
Figure 5.
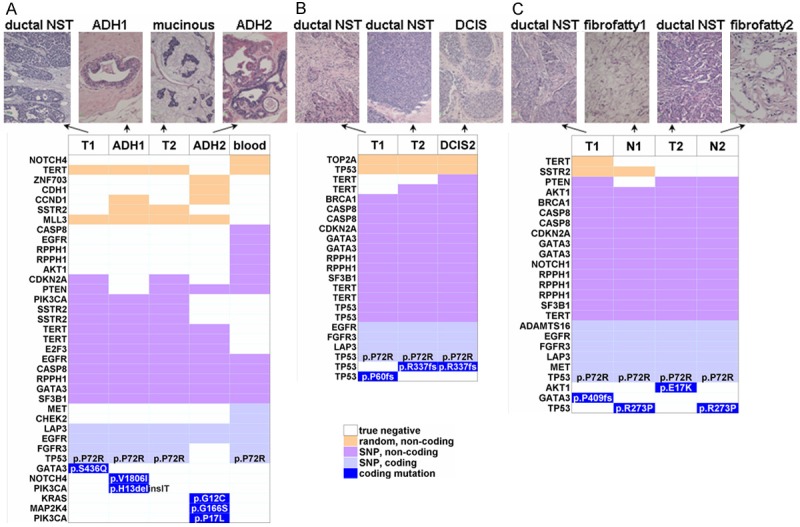
Example bilateral breast cancer (BBC) genotypes with corresponding histology. Three different cases are shown: BIL-041 (A), BIL-073 (B), BIL-078 (C), with case IDs as in Table S1. (A and C) Concurrent; (B) Metachronous. Note the different bilateral tumor genotypes in all cases; different mutations in hyperplastic lesions (ADH) in (A); shared mutations in the in situ carcinoma (DCIS) and in the unilateral tumor in (B), which were also morphologically consistent; shared TP53 mutation in the bilateral normal samples in the male BBC case in (C). SNPs were consistently shared among samples in (B) and (C) but less so in the case in (A). NST: non-specific type.
We also tested 30 peripheral blood DNA samples with the tissue panel that, of note, was not designed for interrogating the status of cancer predisposing genes [14]. This step was undertaken for assessing the somatic nature of tissue mutations. Out of the 1080 variants in these samples, 414 and 385 were shared in tumors of either side, approximately 37%. Four heterozygous mutations were identified in blood samples from 3 patients, affecting TP53 (p.Phe341Val), FOXM1, APOBEC3B and EGFR, at the expected germline VAF, around 50% (Table 3). TP53 Phe341Val was present in tumors bilaterally. The EGFR mutation was not informative in the matched tumors, while FOXM1 and APOBEC3B mutations were not preserved in the tumors of the third patient.
As previously described, 29 patients had been tested within the frameworks of a separate protocol for germline mutations in cancer predisposing genes. Among them 9 harbored BRCA1 germline mutations, all described as pathogenic, while one patient harbored the aforementioned cross-validated TP53 p.Phe341Val (germline genotype (details in Table S1 in file: BBC supplementary data; https://figshare.com/s/39adae1d323bde0884d5). Seven out of 10 germline mutation carriers had tumors with mutations (Figure 3B and 3C). Six out of the 9 BRCA1 mutation carriers developed metachronous tumors with a median time interval of 6 years between diagnoses. Median age at diagnosis was 37 years. The majority of the tumors were high-grade ductal carcinomas (NST), 2 were medullary and 1 was sarcomatous carcinoma. BRCA1 carriers had an increased rate of TNBC (8 out of 17 comparable tumors), 8 tumors were Luminal B and one was Luminal-HER2. Two out of the five hypermutated tumors were identified in BRCA1 mutation carriers. The rest of the patients with such tumors were not tested for germline mutations in BRCA1.
Discussion
To date contralateral tumors are treated as independent primary tumors. It is of great clinical importance to ascertain whether the contralateral tumor is indeed independent or whether it represents a metastatic lesion, since appropriate treatment modalities would be modified. To approach this issue, we compared genomic data obtained by multigene sequencing in bilateral tumors. The number of the examined patients may appear small but the series is currently the largest that has been extensively genotyped for this disease.
The majority of bilateral breast tumors did not share common mutations that would indicate a common origin for the development of the second tumor, as has been shown for metastatic lesions [29] and for the clonal evolution of TNBC [30]. A limitation of our approach is that targeted NGS used for genotyping may have missed common origin mutations in genes not included in the panel. As shown for multifocal breast cancer [31], wide genome sequencing revealed common unique structural alterations in multiple cancer foci from the same patient, indicating that they developed on the basis of a common defective genetic background. These lesions did not share common mutations when assessed by targeted NGS. As reported, the higher the distance between multifocal lesions, the higher the genomic diversity. Our findings in bilateral breast cancer fit this model, based on the mutation diversity in the majority of cases. Nevertheless, at least 15% of the examined bilateral tumors shared common mutations indicative of clonal origin in both sides. The affected genes were previously reported as mutated in breast cancer [22] and were also found in the clonal trunk in breast tumor phylogenetics [30,32]. The rate of such clonal tumors was 4:1 in concurrent compared to metachronous disease. The high mutant allele frequency in concurrent tumors and the increased such frequency in metachronous tumors that developed years later indicate a driver role for these shared mutations. Whether bilateral tumors with shared mutations should be considered as metastatic to the contralateral breast remains questionable. Such tumors may also develop independently as a result of locally operating mutational processes on a cancer predisposing genetic background [33]. Whether individualized treatments would benefit this minority of patients is still unexplored.
Bilateral breast cancers have classically been associated with genetic predisposition [34,35]. Correspondingly, 1/3 of our patients tested for mutations in cancer predisposing genes carried pathogenic BRCA1 and, in one case, TP53 mutations. All were younger than 50 years at first diagnosis. The incidence of BRCA1 carriers was higher in metachronous cases as reported in large series, with a rate higher than 2:1 for metachronous vs. concurrent disease [35]. TP53 p.Phe341Val is a novel germline mutation, since it has not been reported before either in IARC TP53 or COSMIC databases. This mutation was predicted as benign by all pathogenicity prediction algorithms used by ANNOVAR. However, based on the functional aspects of TP53 codon 341 [36], a disruptive effect cannot be excluded. TP53 p.Phe341Val was preserved in the metachronous tumor that developed years apart in the same patient, further supporting its possible pathogenic role.
Beyond the above mutations in traditional cancer predisposing genes we also observed changes in blood samples at heterozygote germline frequencies, in genes usually reported as somatically mutated, such as FOXM1, APOBEC3B and EGFR. All these mutations were predicted as benign by ANNOVAR and older patient age at first diagnosis in these cases may support this feature. However, since none of the three genes is included in cancer predisposition panels, the role of the corresponding mutations in the development of the bilateral tumors in these patients remains unknown. To this end, such mutations are useful to report for future comparisons.
Three main genotype features characterized metachronous as opposed to concurrent disease: prevalence of TP53 mutations; presence of hypermutated tumors; and altered zygosity for common SNPs in the 2nd occurrence. Starting with the latter, altered zygosity status at SNP positions and genomic stability of metachronous tumors may just reflect the temporal instability changes that are established in cells following a greater number of divisions as revealed with mathematical models [37].
The prevalence of TP53 mutations may be related to the TNBC phenotypes more frequently observed in metachronous tumors. Compared to concurrent tumors, which were mostly ER/PgR-positive, metachronous tumors were significantly more frequently hormone receptor negative demonstrating significantly lower ER/PgR concordance, as previously described [2]. Concurrent and metachronous tumors demonstrated luminal-like and TNBC-like mutation patterns, respectively, in accordance with TCGA data [22,38]. The luminal-like profile included a high rate of PIK3CA mutations [38,39] in the usually described hotspots [27], followed by TP53 and GATA3. The TNBC-like profile of metachronous tumors was marked by TP53 followed by PIK3CA and CDH1 mutations [40-42].
The high rate of TP53 mutations in metachronous tumors may be related to mutations in cancer predisposing genes in such cases. Germline BRCA1 mutations are associated with tumor TP53 mutations in TNBC [43]. This condition may also be related to the hypermutated tumors that we only observed in patients with metachronous disease. Hypermutated tumors develop in BRCA1 mutation carriers [44], while the combination of inherited BRCA1 and acquired replication repair defects may result in ultramutated tumors [45]. The present hypermutated tumors had TP53 mutations, while three of them also had germline BRCA1 and/or somatic APOBEC3B mutations. Whether these APOBEC3B mutations contributed to the high mutation load in the two affected tumors, as published for this deaminase [46], needs functional proof. Intriguingly, in the 5 patients with hypermutated tumors, we observed 0-1 mutations in the contralateral tumor that developed years later. This is of clinical importance, since it indicates that whatever the cause releasing hypermutation, it may have acted locally and perhaps temporarily.
Alterations in normal and pre-invasive lesions in the breast have been described at the genomic level [47-49], as has a shared susceptibility for infiltrative carcinomas and DCIS [50]. Our findings are similar to those reports except that we did not observe any PIK3CA mutations in DCIS, probably because of the small sample number. Mutations in the normal or hyperplastic breast or in DCIS were not necessarily preserved in the infiltrative tumor in the ipsilateral or contralateral breast. A characteristic example was the COSMIC registered TP53 p.Arg273Pro mutation that was present bilaterally in the normal breast of our male patient but in none of the corresponding tumors; the same mutation was present in both tumors of a female patient in the present series. These data are in line with recently proposed models on the genomic dynamics during cancer development and evolution that have been described for different types of cancer [51] and on the selective pressure for the preservation of mutations during tumor evolution [32].
Conclusions
Based on their histopathological and particularly genomic characteristics, bilateral breast carcinomas can be considered as two separate primary tumors arising in the same environmental and genetic background. Bilateral breast cancer may represent a model for studying the development of different cancers in the same organ and individual. In the majority of cases, bilateral breast tumors do not share mutations in recurrently mutated breast cancer related genes. Until the common genetic and/or environmental basis of these cancers is fully revealed it seems appropriate to consider the characteristics of both sides for clinical decision making. Assessing the clinicopathological and genetic characteristics of one side does not necessarily reflect those in the contralateral side, in either concurrent or metachronous disease, the latter more often exhibiting tumor-private mutation profiles. These findings support the management of bilateral breast cancers as independent tumors, as currently practiced.
Disclosure of conflict of interest
None.
Authors’ contribution
Elena Fountzilas and Vassiliki Kotoula designed experiments, analyzed data and wrote the paper. Eleni Giannoulatou and George Kouvatseas: provided technical support and aided the statistical analysis, data analysis and interpretation. Triantafyllia Koletsa and Mattheos Bobos reviewed all histology parameters. Kyriaki Papadopoulou performed NGS experiments. Florentia Fostira performed germline testing for cancer susceptibility genes. Epaminontas Samantas, Efterpi Demiri, Spyros Miliaras, Christos Christodoulou, Evangelia Razis, Dimitrios Pectasides, George Zografos, Flora Zagouri, George Pentheroudakis provided patient samples and clinical data. Sofia Chrisafi collected the data. George Fountzilas had the conception and design of this project, provided the majority of patient samples and clinical data, and the funding. All authors commented on the manuscript and accepted it in its final form.
Supporting Information
References
- 1.SEER Database: Surveillance EaER, (SEER) Available from: http://seer.cancer.gov/
- 2.Gong SJ, Rha SY, Jeung HC, Roh JK, Yang WI, Chung HC. Bilateral breast cancer: differential diagnosis using histological and biological parameters. Jpn J Clin Oncol. 2007;37:487–492. doi: 10.1093/jjco/hym056. [DOI] [PubMed] [Google Scholar]
- 3.Narod SA. Bilateral breast cancers. Nat Rev Clin Oncol. 2014;11:157–166. doi: 10.1038/nrclinonc.2014.3. [DOI] [PubMed] [Google Scholar]
- 4.Hartman M, Czene K, Reilly M, Adolfsson J, Bergh J, Adami HO, Dickman PW, Hall P. Incidence and prognosis of synchronous and metachronous bilateral breast cancer. J. Clin. Oncol. 2007;25:4210–4216. doi: 10.1200/JCO.2006.10.5056. [DOI] [PubMed] [Google Scholar]
- 5.Intra M, Rotmensz N, Viale G, Mariani L, Bonanni B, Mastropasqua MG, Galimberti V, Gennari R, Veronesi P, Colleoni M, Tousimis E, Galli A, Goldhirsch A, Veronesi U. Clinicopathologic characteristics of 143 patients with synchronous bilateral invasive breast carcinomas treated in a single institution. Cancer. 2004;101:905–912. doi: 10.1002/cncr.20452. [DOI] [PubMed] [Google Scholar]
- 6.Huo D, Melkonian S, Rathouz PJ, Khramtsov A, Olopade OI. Concordance in histological and biological parameters between first and second primary breast cancers. Cancer. 2011;117:907–915. doi: 10.1002/cncr.25587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibata A, Tsai YC, Press MF, Henderson BE, Jones PA, Ross RK. Clonal analysis of bilateral breast cancer. Clin Cancer Res. 1996;2:743–748. [PubMed] [Google Scholar]
- 8.Tse GM, Kung FY, Chan AB, Law BK, Chang AR, Lo KW. Clonal analysis of bilateral mammary carcinomas by clinical evaluation and partial allelotyping. Am J Clin Pathol. 2003;120:168–174. doi: 10.1309/6YEP-MCHA-CPG2-BD15. [DOI] [PubMed] [Google Scholar]
- 9.Janschek E, Kandioler-Eckersberger D, Ludwig C, Kappel S, Wolf B, Taucher S, Rudas M, Gnant M, Jakesz R. Contralateral breast cancer: molecular differentiation between metastasis and second primary cancer. Breast Cancer Res Treat. 2001;67:1–8. doi: 10.1023/a:1010661514306. [DOI] [PubMed] [Google Scholar]
- 10.Teixeira MR, Ribeiro FR, Torres L, Pandis N, Andersen JA, Lothe RA, Heim S. Assessment of clonal relationships in ipsilateral and bilateral multiple breast carcinomas by comparative genomic hybridisation and hierarchical clustering analysis. Br J Cancer. 2004;91:775–782. doi: 10.1038/sj.bjc.6602021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brommesson S, Jonsson G, Strand C, Grabau D, Malmstrom P, Ringner M, Ferno M, Hedenfalk I. Tiling array-CGH for the assessment of genomic similarities among synchronous unilateral and bilateral invasive breast cancer tumor pairs. BMC Clin Pathol. 2008;8:6. doi: 10.1186/1472-6890-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banelli B, Casciano I, Di Vinci A, Gatteschi B, Levaggi A, Carli F, Bighin C, Salvi S, Allemanni G, Ghiorzo P, Pronzato P, Venturini M, Romani M, Del Mastro L. Pathological and molecular characteristics distinguishing contralateral metastatic from new primary breast cancer. Ann Oncol. 2010;21:1237–1242. doi: 10.1093/annonc/mdp470. [DOI] [PubMed] [Google Scholar]
- 13.Fountzilas G, Giannoulatou E, Alexopoulou Z, Zagouri F, Timotheadou E, Papadopoulou K, Lakis S, Bobos M, Poulios C, Sotiropoulou M, Lyberopoulou A, Gogas H, Pentheroudakis G, Pectasides D, Koutras A, Christodoulou C, Papandreou C, Samantas E, Papakostas P, Kosmidis P, Bafaloukos D, Karanikiotis C, Dimopoulos MA, Kotoula V. TP53 mutations and protein immunopositivity may predict for poor outcome but also for trastuzumab benefit in patients with early breast cancer treated in the adjuvant setting. Oncotarget. 2016;7:32731–53. doi: 10.18632/oncotarget.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotoula V, Lyberopoulou A, Papadopoulou K, Charalambous E, Alexopoulou Z, Gakou C, Lakis S, Tsolaki E, Lilakos K, Fountzilas G. Evaluation of two highly-multiplexed custom panels for massively parallel semiconductor sequencing on paraffin DNA. PLoS One. 2015;10:e0128818. doi: 10.1371/journal.pone.0128818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, Wienert S, Van den Eynden G, Baehner FL, Penault-Llorca F, Perez EA, Thompson EA, Symmans WF, Richardson AL, Brock J, Criscitiello C, Bailey H, Ignatiadis M, Floris G, Sparano J, Kos Z, Nielsen T, Rimm DL, Allison KH, Reis-Filho JS, Loibl S, Sotiriou C, Viale G, Badve S, Adams S, Willard-Gallo K, Loi S International TILsWorking Group 2014. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol. 2015;26:259–271. doi: 10.1093/annonc/mdu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotoula V, Chatzopoulos K, Lakis S, Alexopoulou Z, Timotheadou E, Zagouri F, Pentheroudakis G, Gogas H, Galani E, Efstratiou I, Zaramboukas T, Koutras A, Aravantinos G, Samantas E, Psyrri A, Kourea H, Bobos M, Papakostas P, Kosmidis P, Pectasides D, Fountzilas G. Tumors with high-density tumor infiltrating lymphocytes constitute a favorable entity in breast cancer: a pooled analysis of four prospective adjuvant trials. Oncotarget. 2016;7:5074–87. doi: 10.18632/oncotarget.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fountzilas G, Dafni U, Bobos M, Batistatou A, Kotoula V, Trihia H, Malamou-Mitsi V, Miliaras S, Chrisafi S, Papadopoulos S, Sotiropoulou M, Filippidis T, Gogas H, Koletsa T, Bafaloukos D, Televantou D, Kalogeras KT, Pectasides D, Skarlos DV, Koutras A, Dimopoulos MA. Differential response of immunohistochemically defined breast cancer subtypes to anthracycline-based adjuvant chemotherapy with or without paclitaxel. PLoS One. 2012;7:e37946. doi: 10.1371/journal.pone.0037946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harbeck N, Thomssen C, Gnant M. St. Gallen 2013: brief preliminary summary of the consensus discussion. Breast Care (Basel) 2013;8:102–109. doi: 10.1159/000351193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konstantopoulou I, Tsitlaidou M, Fostira F, Pertesi M, Stavropoulou AV, Triantafyllidou O, Tsotra E, Tsiftsoglou AP, Tsionou C, Droufakou S, Dimitrakakis C, Fountzilas G, Yannoukakos D. High prevalence of BRCA1 founder mutations in Greek breast/ovarian families. Clin Genet. 2014;85:36–42. doi: 10.1111/cge.12274. [DOI] [PubMed] [Google Scholar]
- 20.Stavropoulou AV, Fostira F, Pertesi M, Tsitlaidou M, Voutsinas GE, Triantafyllidou O, Bamias A, Dimopoulos MA, Timotheadou E, Pectasides D, Christodoulou C, Klouvas G, Papadimitriou C, Makatsoris T, Pentheroudakis G, Aravantinos G, Karydakis V, Yannoukakos D, Fountzilas G, Konstantopoulou I. Prevalence of BRCA1 mutations in familial and sporadic greek ovarian cancer cases. PLoS One. 2013;8:e58182. doi: 10.1371/journal.pone.0058182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerød A Oslo Breast Cancer Consortium (OSBREAC) Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van ‘t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Børresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward EM, DeSantis CE, Lin CC, Kramer JL, Jemal A, Kohler B, Brawley OW, Gansler T. Cancer statistics: Breast cancer in situ. CA Cancer J Clin. 2015;65:481–495. doi: 10.3322/caac.21321. [DOI] [PubMed] [Google Scholar]
- 24.Rogozinska-Szczepka J, Utracka-Hutka B, Grzybowska E, Maka B, Nowicka E, Smok-Ragankiewicz A, Zientek H, Steffen J, Wojciechowska-Lacka A. BRCA1 and BRCA2 mutations as prognostic factors in bilateral breast cancer patients. Ann Oncol. 2004;15:1373–1376. doi: 10.1093/annonc/mdh352. [DOI] [PubMed] [Google Scholar]
- 25.Loi S, Michiels S, Lambrechts D, Fumagalli D, Claes B, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Piccart MJ, Joensuu H, Sotiriou C. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. 2013;105:960–967. doi: 10.1093/jnci/djt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arthur LM, Turnbull AK, Renshaw L, Keys J, Thomas JS, Wilson TR, Lackner MR, Sims AH, Dixon JM. Changes in PIK3CA mutation status are not associated with recurrence, metastatic disease or progression in endocrine-treated breast cancer. Breast Cancer Res Treat. 2014;147:211–219. doi: 10.1007/s10549-014-3080-x. [DOI] [PubMed] [Google Scholar]
- 28.Kang HJ, Chun SM, Kim KR, Sohn I, Sung CO. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS One. 2013;8:e72609. doi: 10.1371/journal.pone.0072609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, Koboldt DC, Schmidt H, Kalicki J, Zhang Q, Chen L, Lin L, Wendl MC, McMichael JF, Magrini VJ, Cook L, McGrath SD, Vickery TL, Appelbaum E, Deschryver K, Davies S, Guintoli T, Lin L, Crowder R, Tao Y, Snider JE, Smith SM, Dukes AF, Sanderson GE, Pohl CS, Delehaunty KD, Fronick CC, Pape KA, Reed JS, Robinson JS, Hodges JS, Schierding W, Dees ND, Shen D, Locke DP, Wiechert ME, Eldred JM, Peck JB, Oberkfell BJ, Lolofie JT, Du F, Hawkins AE, O’Laughlin MD, Bernard KE, Cunningham M, Elliott G, Mason MD, Thompson DM Jr, Ivanovich JL, Goodfellow PJ, Perou CM, Weinstock GM, Aft R, Watson M, Ley TJ, Wilson RK, Mardis ER. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desmedt C, Fumagalli D, Pietri E, Zoppoli G, Brown D, Nik-Zainal S, Gundem G, Rothe F, Majjaj S, Garuti A, Carminati E, Loi S, Van Brussel T, Boeckx B, Maetens M, Mudie L, Vincent D, Kheddoumi N, Serra L, Massa I, Ballestrero A, Amadori D, Salgado R, de Wind A, Lambrechts D, Piccart M, Larsimont D, Campbell PJ, Sotiriou C. Uncovering the genomic heterogeneity of multifocal breast cancer. J Pathol. 2015;236:457–466. doi: 10.1002/path.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, Li Y, Ju YS, Ramakrishna M, Haugland HK, Lilleng PK, Nik-Zainal S, McLaren S, Butler A, Martin S, Glodzik D, Menzies A, Raine K, Hinton J, Jones D, Mudie LJ, Jiang B, Vincent D, Greene-Colozzi A, Adnet PY, Fatima A, Maetens M, Ignatiadis M, Stratton MR, Sotiriou C, Richardson AL, Lønning PE, Wedge DC, Campbell PJ. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev. 2014;24:52–60. doi: 10.1016/j.gde.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gershoni-Baruch R, Dagan E, Fried G, Kepten I, Robinson E. BRCA1 and BRCA2 founder mutations in patients with bilateral breast cancer. Eur J Hum Genet. 1999;7:833–836. doi: 10.1038/sj.ejhg.5200371. [DOI] [PubMed] [Google Scholar]
- 35.Kast K, Rhiem K, Wappenschmidt B, Hahnen E, Hauke J, Bluemcke B, Zarghooni V, Herold N, Ditsch N, Kiechle M, Braun M, Fischer C, Dikow N, Schott S, Rahner N, Niederacher D, Fehm T, Gehrig A, Mueller-Reible C, Arnold N, Maass N, Borck G, de Gregorio N, Scholz C, Auber B, Varon-Manteeva R, Speiser D, Horvath J, Lichey N, Wimberger P, Stark S, Faust U, Weber BH, Emons G, Zachariae S, Meindl A, Schmutzler RK, Engel C German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC) Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet. 2016;53:465–71. doi: 10.1136/jmedgenet-2015-103672. [DOI] [PubMed] [Google Scholar]
- 36.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenman CD, Cooke SL, Marshall J, Stratton MR, Campbell PJ. Modeling the evolution space of breakage fusion bridge cycles with a stochastic folding process. J Math Biol. 2016;72:47–86. doi: 10.1007/s00285-015-0875-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papaxoinis G, Kotoula V, Alexopoulou Z, Kalogeras KT, Zagouri F, Timotheadou E, Gogas H, Pentheroudakis G, Christodoulou C, Koutras A, Bafaloukos D, Aravantinos G, Papakostas P, Charalambous E, Papadopoulou K, Varthalitis I, Efstratiou I, Zaramboukas T, Patsea H, Scopa CD, Skondra M, Kosmidis P, Pectasides D, Fountzilas G. Significance of PIK3CA Mutations in Patients with Early Breast Cancer Treated with Adjuvant Chemotherapy: A Hellenic Cooperative Oncology Group (HeCOG) Study. PLoS One. 2015;10:e0140293. doi: 10.1371/journal.pone.0140293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, Turpin E, Plassa LF, de Roquancourt A, Bourstyn E, de Cremoux P, Janin A, Giacchetti S, Espié M, de Thé H. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013;22(Suppl 2):S27–29. doi: 10.1016/j.breast.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Yang P, Du CW, Kwan M, Liang SX, Zhang GJ. The impact of p53 in predicting clinical outcome of breast cancer patients with visceral metastasis. Sci Rep. 2013;3:2246. doi: 10.1038/srep02246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Severson TM, Peeters J, Majewski I, Michaut M, Bosma A, Schouten PC, Chin SF, Pereira B, Goldgraben MA, Bismeijer T, Kluin RJ, Muris JJ, Jirström K, Kerkhoven RM, Wessels L, Caldas C, Bernards R, Simon IM, Linn S. BRCA1-like signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol Oncol. 2015;9:1528–1538. doi: 10.1016/j.molonc.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang D, Khan S, Sun Y, Hess K, Shmulevich I, Sood AK, Zhang W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306:1557–1565. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, Van Loo P, Tarpey PS, Coupland P, Behjati S, Pollett A, Lipman T, Heidari A, Deshmukh S, Avitzur N, Meier B, Gerstung M, Hong Y, Merino DM, Ramakrishna M, Remke M, Arnold R, Panigrahi GB, Thakkar NP, Hodel KP, Henninger EE, Göksenin AY, Bakry D, Charames GS, Druker H, Lerner-Ellis J, Mistry M, Dvir R, Grant R, Elhasid R, Farah R, Taylor GP, Nathan PC, Alexander S, Ben-Shachar S, Ling SC, Gallinger S, Constantini S, Dirks P, Huang A, Scherer SW, Grundy RG, Durno C, Aronson M, Gartner A, Meyn MS, Taylor MD, Pursell ZF, Pearson CE, Malkin D, Futreal PA, Stratton MR, Bouffet E, Hawkins C, Campbell PJ, Tabori U Biallelic Mismatch Repair Deficiency Consortium. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet. 2015;47:257–262. doi: 10.1038/ng.3202. [DOI] [PubMed] [Google Scholar]
- 46.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jönsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerød A, Tutt A, Martens JW, Aparicio SA, Borg Å, Salomon AV, Thomas G, Børresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR Breast Cancer Working Group of the International Cancer Genome Consortium. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abba MC, Gong T, Lu Y, Lee J, Zhong Y, Lacunza E, Butti M, Takata Y, Gaddis S, Shen J, Estecio MR, Sahin AA, Aldaz CM. A Molecular Portrait of High-Grade Ductal Carcinoma In Situ. Cancer Res. 2015;75:3980–3990. doi: 10.1158/0008-5472.CAN-15-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, Chung YJ, Lee SH. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6:7597–7607. doi: 10.18632/oncotarget.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ang DC, Warrick AL, Shilling A, Beadling C, Corless CL, Troxell ML. Frequent phosphatidylinositol-3-kinase mutations in proliferative breast lesions. Mod Pathol. 2014;27:740–750. doi: 10.1038/modpathol.2013.197. [DOI] [PubMed] [Google Scholar]
- 50.Petridis C, Brook MN, Shah V, Kohut K, Gorman P, Caneppele M, Levi D, Papouli E, Orr N, Cox A, Cross SS, Dos-Santos-Silva I, Peto J, Swerdlow A, Schoemaker MJ, Bolla MK, Wang Q, Dennis J, Michailidou K, Benitez J, González-Neira A, Tessier DC, Vincent D, Li J, Figueroa J, Kristensen V, Borresen-Dale AL, Soucy P, Simard J, Milne RL, Giles GG, Margolin S, Lindblom A, Brüning T, Brauch H, Southey MC, Hopper JL, Dörk T, Bogdanova NV, Kabisch M, Hamann U, Schmutzler RK, Meindl A, Brenner H, Arndt V, Winqvist R, Pylkäs K, Fasching PA, Beckmann MW, Lubinski J, Jakubowska A, Mulligan AM, Andrulis IL, Tollenaar RA, Devilee P, Le Marchand L, Haiman CA, Mannermaa A, Kosma VM, Radice P, Peterlongo P, Marme F, Burwinkel B, van Deurzen CH, Hollestelle A, Miller N, Kerin MJ, Lambrechts D, Floris G, Wesseling J, Flyger H, Bojesen SE, Yao S, Ambrosone CB, Chenevix-Trench G, Truong T, Guénel P, Rudolph A, Chang-Claude J, Nevanlinna H, Blomqvist C, Czene K, Brand JS, Olson JE, Couch FJ, Dunning AM, Hall P, Easton DF, Pharoah PD, Pinder SE, Schmidt MK, Tomlinson I, Roylance R, García-Closas M, Sawyer EJ. Genetic predisposition to ductal carcinoma in situ of the breast. Breast Cancer Res. 2016;18:22. doi: 10.1186/s13058-016-0675-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.