

Abstract
Cell death and survival signaling pathways have opposed but fundamental functions for various cellular processes and maintain cell homeostasis through cross talk. Here we report a novel mechanism of interaction between these two pathways through the cleavage of RNF31 by caspases. RNF31, a component of the linear ubiquitin chain assembly complex (LUBAC), regulates cell survival by inducing linear ubiquitination of NF-κB signaling components. We found that RNF31 is cleaved under apoptosis conditions through various stimulations. The effector caspases caspase 3 and caspase 6 are responsible for this event, and aspartates 348, 387, and 390 were identified as target sites for this cleavage. Cleavage of RNF31 suppressed its ability to activate NF-κB signaling; thus, mutation of cleavage sites inhibited the induction of apoptosis by treatment with tumor necrosis factor alpha (TNF-α). Our findings elucidate a novel regulatory loop between cell death and the survival signal and may provide guidance for the development of therapeutic strategies for cancers through the sensitization of tumor cells to death-inducing drugs.
INTRODUCTION
The nuclear factor κB (NF-κB) signaling pathway plays a critical role in various cellular processes, including proliferation, differentiation, survival, and death. In the resting state, inhibitor of κB-α (IκB-α) sequesters the NF-κB complex in the cytoplasm by interacting with it. Through the activation of the IκB kinase (IKK) complex (composed of IKKα, -β, and -γ), followed by the phosphorylation and consequent degradation of IκB-α, free NF-κB complex acquires the ability to enter the nucleus and induce target gene expression (1). Previous studies have revealed that K63-linked polyubiquitination of IKKγ (also called NEMO) is critical for NF-κB activation (2). Recently, linear ubiquitination was identified as a novel type of ubiquitination that forms the ubiquitin linkage between the N-terminal Met of one ubiquitin and the C-terminal Gly of another (3–5). To date, the linear ubiquitin chain assembly complex (LUBAC), which is composed of one main E3 ligase, ring finger protein 31 (RNF31; also known as HOIP), and two associated proteins, HOIL-1 and Sharpin, is the only E3 ligase complex for linear ubiquitination. Upstream activation leads to the linear ubiquitination of NEMO. Then these modified molecules function as a bridge between the receptor complex and the downstream IKK complex to activate NF-κB signaling (6). Genetic studies have shown that defects in HOIL-1 or Sharpin result in reduced phosphorylation and degradation of IκB-α, impaired and delayed nuclear translocation of the NF-κB subunit p65, diminished overall gene induction, and increased tumor necrosis factor (TNF)-induced cell death (3–5).
The activation of NF-κB signaling not only directly prompts cell growth and proliferation but also suppresses cell death by upregulating antiapoptotic molecules that inhibit the function of caspases. Caspases are regulatory proteases that are essential for apoptosis activation. Briefly, diverse factors, including TNF-α, TNF-related apoptosis-inducing ligand (TRAIL), and Fas ligand (FasL), promote the cleavage of initiator caspases (caspase 8 or 9), thereby activating them. Then these active initiators further process effector caspases (caspases 3, 6, and 7), which, in turn, execute cell death by processing cellular proteins (7). To balance cell death and survival, diverse inhibitory mechanisms that are regulated by survival signaling participate in the suppression of the caspase cascade. For example, the activation of NF-κB promotes the expression of inhibitors of apoptosis proteins (IAPs), cellular FLICE-inhibitory protein (cFLIP), and B-cell lymphoma 2 (BCL2) family members, and these molecules suppress the function of caspases (8). Reversely, active apoptosis signaling dampens survival signaling by the cleavage of its components. Critical components of NF-κB signaling, such as p65, receptor-interacting protein 1 (RIP1), and NEMO, are the substrates of caspases, and the cleavage of these molecules results in the suppression of survival signaling (9–11). Although the NF-κB and apoptosis pathways exhibit active interplay that balances death and survival, the mechanism of reciprocal regulation between these two pathways is not completely established.
Here we present the novel cross talk between cell death signaling and the survival pathway. RNF31, a major E3 ligase in the LUBAC for linear ubiquitination, is cleaved in an effector caspase-dependent manner under apoptotic conditions. This cleavage event attenuates the ability of RNF31 to activate downstream signaling, thereby leading to the sensitization of resistant cells to TNF-α-induced apoptosis.
MATERIALS AND METHODS
Cell cultures and transfection.
HEK293T, Phoenix, HeLa, BxPC-1, Panc-1, A549, HT29, and HCT116 cells were purchased from the ATCC and were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. Jurkat cells were obtained from the ATCC, and caspase 8-deficient and FADD-deficient Jurkat cells were kindly provided by Jianke Zhang (Thomas Jefferson University). Wild-type (WT) and mutant Jurkat cells were maintained in RPMI 1640 medium supplemented with FBS and antibiotics. Cells were starved for 16 h with DMEM or RPMI medium containing 0.5% FBS before the induction of apoptosis by treatment with various inducers. For overexpression experiments, 293T cells were transfected using the calcium phosphate transfection method. Briefly, 7.5 × 105 cells were plated onto 6-well plates, and a CaCl2–Hanks balanced salt solution (HBSS)–DNA precipitate (1 to 4 μg of DNA per well) was added the next day. After 24 h, cells were lysed for further experiments.
Reagents and plasmid.
Human TNF-α recombinant protein (RTNFAI; Thermo Scientific), a de novo protein synthesis inhibitor (cycloheximide [CHX]; Enzo), a proteasome inhibitor (MG132; Enzo), a pancaspase inhibitor (Z-VAD-FMK; Enzo), fluorouracil (5-FU; Sigma-Aldrich), and doxorubicin (Dox; Enzo) were used to stimulate cells. Antibodies against poly(ADP-ribose) polymerase (PARP) (catalog no. 9542), caspase 3 (catalog no. 9665), cleaved caspase 3 (catalog no. 9661), caspase 8 (catalog no. 9746), caspase 9 (catalog no. 9508), IκB-α (catalog no. 4814), pIκB-α (catalog no. 9246), pIKKα/β (catalog no. 2697), pJNK (catalog no. 9255), and Jun N-terminal protein kinase (JNK) (catalog no. 4672) were obtained from Cell Signaling Technology. Antibodies against cFLIPS/L (sc-5276), actin (sc-8432), TNF-α receptor 1 (TNFR1; sc-8436), IKKα (sc-7218), p65 (sc-109), lamin B (SC-6219), β-tubulin (sc-5274), Myc (sc-7899), Flag (sc-807), and ubiquitin (sc-9133) from Santa Cruz Biotechnology, an antibody for RNF31 (ab85294; Abcam), and anti-FADD (610400; BD Biosciences) were used for immunoblotting. An anti-linear polyubiquitin antibody was provided by Genentech. Wild-type RNF31, HOIL-1, and Sharpin were cloned from Jurkat cell cDNA. WT RNF31 and all RNF31 mutants (including the C885S, N-terminal [NT], C-terminal [CT], D390A, D348A D390A [D348/390A], and D348/387/390A mutants) were generated by overlapping PCR using WT cDNA and were verified by sequencing.
Virus production and infection for knockdown.
Retroviral supernatant was collected 48 h after the transfection of plasmid pMX into Phoenix cells. Then target cells were incubated with the supernatant in the presence of Polybrene (2 μg/ml) for 8 to 12 h. After infection, viral supernatants were replaced with flesh medium. After 48 h, infected cells were selected with puromycin (2 μg/ml; Invivogen), and the efficiency of infection was determined by Western blot (WB) analysis.
In vitro cleavage assay.
Myc-tagged WT human recombinant RNF31 and its D348/387/390A mutant were expressed in 293T cells. After 24 h, cell lysates were prepared with lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 1% NP-40, 1 mM EDTA) containing 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Roche). Then overexpressed RNF31 protein was precipitated with the EZview Red anti-c-Myc affinity gel (Sigma). Recombinant caspases were obtained from Biovision (active recombinant caspase set III; catalog no. K232-8-25) and were prepared according to the manufacturer's recommendation. Two units of recombinant caspases and precipitated RNF31 protein were incubated at 37°C in a reaction solution comprising 50 mM HEPES (pH 7.2), 50 mM NaCl, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 10 mM EDTA, 5% glycerol, and 10 mM dithiothreitol (DTT). After 1 to 2 h, the reaction was terminated by the addition of 4× loading buffer, followed by boiling for 5 min. Then the cleavage band was analyzed by a Western blot assay.
Western blot analysis.
Western blot assays were performed as described previously. Briefly, cells were lysed in lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 1% NP-40, 1 mM EDTA) containing 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Roche). After a 20-min incubation, samples were centrifuged at 13,000 rpm for 10 min at 4°C. The supernatant was transferred and was mixed with 4× loading buffer. For immunoprecipitation experiments, lysates from transfected 293 cells were incubated with an anti-FLAG M2 affinity gel for 16 h. After four washes with lysis buffer, the proteins eluted with 2× SDS loading buffer. Then the cell lysates or immunoprecipitates were separated by SDS-PAGE and were transferred to a nitrocellulose membrane (Bio-Rad). The membrane was probed with primary antibodies, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. The bands were then visualized with ECL substrates (Pierce).
Ubiquitination assay.
After the transfection of plasmids expressing NEMO and RIP1, cells were lysed with 2% sodium dodecyl sulfate (SDS) lysis buffer (2% SDS, 150 mM NaCl, 10 mM Tris-HCl [pH 8.0]) containing 2 mM sodium orthovanadate, 5 mM sodium fluoride, and 1 mM N-ethylmaleimide (NEM). After boiling and sonication, samples were diluted with dilution buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) up to 10-fold. After incubation on ice for 30 min, immunoprecipitation and WB analysis were performed.
Luciferase assay.
HEK293T cells were transfected with reporter plasmids encoding NF-κB-luc and with pEF-Renilla-luc together with expression vectors. Each transfection was performed in triplicate. Cells were lysed 24 h after transfection, and luciferase activities were measured with Dual-Luciferase assay kits (product no. E1980; Promega). NF-κB activities in each lysate were determined by the ratios of Renilla luciferase readings to firefly luciferase readings, and the average of the activity measured in each group was normalized to the activity of the empty construct.
MTT assay.
A total of 1 × 103 to 3 × 103 cells were plated on 96-well plates with 100 μl of complete medium. After 16 h of starvation (0.5% FBS), cells were treated with each agent, and MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] solution was added to each well to a final concentration of 0.5 mg/ml (Sigma). After 2 h, the medium was aspirated, and formazan was dissolved with dimethyl sulfoxide (DMSO). The absorbance was measured at 570 nm by using a spectrophotometric microplate reader (BioTek), and the value measured for each group was normalized to the value for untreated cells. All experiments were performed in triplicate.
Statistical analysis.
Statistical significance was analyzed using Prism 6 software (GraphPad) with the independent two-sample Student t test.
RESULTS
RNF31 is cleaved under apoptotic conditions.
To determine whether LUBAC components are regulated during apoptosis, we monitored the level of RNF31 under apoptotic conditions. Upon treatment with TNF-α and cycloheximide (CHX), apoptosis signaling was activated, as evidenced by cleavage of caspases 3 and 9 and PARP. Along with these indicators, we observed significant decreases in the levels of full-length RNF31 (about 110 to 120 kDa) and inducible bands (70 to 75 kDa) (Fig. 1A). Since TNF-α receptor (TNFR) and other death receptors share signaling components to promote cell death, we then induced apoptosis by using TRAIL, which activates death receptors 4 and 5 (DR4 and DR5). The cleavage of RNF31 in TRAIL-stimulated cells indicated that RNF31 is cleaved under apoptotic conditions induced by the activation of other DRs (Fig. 1B). To generalize this phenomenon, we treated different kinds of cancer cells with TNF-α and CHX. Cleaved RNF31 could be seen in every cell line examined (Fig. 1C), indicating that RNF31 cleavage is a general process in a wide range of cells.
FIG 1.

RNF31 is cleaved during the process of apoptosis. (A and B) WB analysis of the indicated proteins in HeLa cells treated either with TNF-α (40 ng/ml) and CHX (10 μg/ml) (A) or with TRAIL (100 ng/ml) (B). Ex., exposure. (C) WB analysis of lysates of BxPC-1, Panc-1, A549, HCT116, HT29, and HeLa cells stimulated with TNF-α (20 ng/ml) and CHX (10 μg/ml) for 6 h. (D) WB analysis of HeLa cells exposed to Dox (3 μg/ml) or CPT (20 μM). (E) WB analysis of HeLa cells treated with a Smac mimetic (20 μM).
The apoptosis pathway is triggered by two major sources, extrinsic and intrinsic inducers (7). To determine whether RNF31 is cleaved under both conditions, we treated cells with a DNA damage inducer, either doxorubicin (Dox) or camptothecin (CPT). Since caspase 9 is the initiator caspase in the intrinsic pathway, we monitored the cleavage of caspase 9. In agreement with the observation for extrinsic activation, cleaved bands of RNF31 were detected upon treatment with Dox or CPT (Fig. 1D). Additionally, we observed RNF31 cleavage under conditions of apoptosis induced by a Smac mimetic, which directly activates initiator and effector caspases (Fig. 1E). Taken together, these findings suggest that RNF31 is cleaved upon the activation of extrinsic and intrinsic apoptosis pathways.
RNF31 is cleaved by caspases during apoptosis, while necroptosis does not induce cleavage of RNF31.
To determine whether caspases are responsible for the cleavage of RNF31, we used the pancaspase inhibitor Z-VAD-FMK to block general caspase function before TNF-α and CHX stimulation. Treatment of HeLa cells with the de novo protein synthesis inhibitor CHX or TNF-α alone failed to induce the cleavage of either PARP or RNF31 (Fig. 2A), since HeLa cells are resistant to TNF-α-induced apoptosis. However, treatment with TNF-α plus CHX activated apoptosis, as indicated by the cleavage of PARP, and induced the cleavage of RNF31 4 h after stimulation (Fig. 2A). Notably, RNF31 cleavage was completely blocked by 1 h of Z-VAD-FMK pretreatment (Fig. 2A). The degradation of IκB-α in TNF-α- and CHX-treated cells with or without Z-VAD-FMK pretreatment indicated that stimulation was intact.
FIG 2.
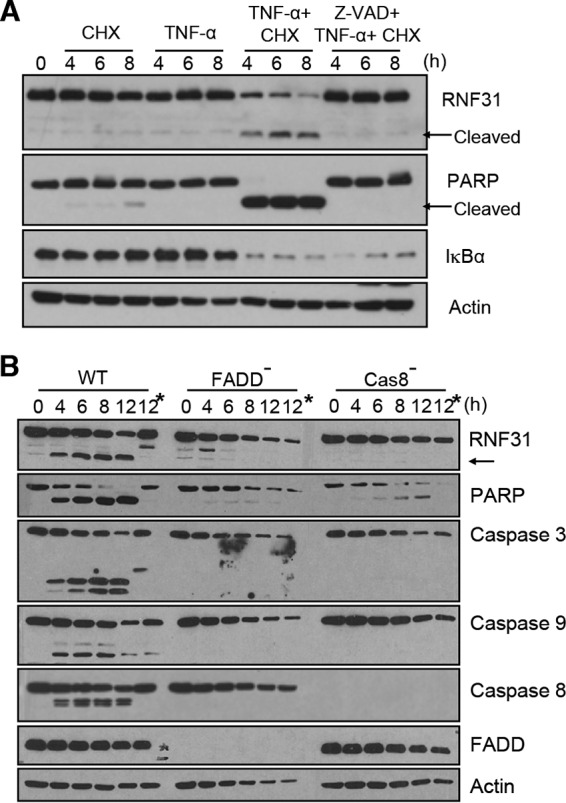
RNF31 is cleaved by caspases during apoptosis but not during necroptosis. (A) Nonpretreated HeLa cells or HeLa cells pretreated with Z-VAD-FMK (20 μM) were treated with TNF-α (20 ng/ml) alone, CHX (10 μg/ml) alone, or both TNF-α and CHX and were then subjected to WB analysis. (B) WB analysis of WT, FADD-deficient, and caspase 8-deficient Jurkat cells stimulated with TNF-α (20 ng/ml) and CHX (10 μg/ml). Asterisks indicate pretreatment with the pancaspase inhibitor Z-VAD-FMK.
Since FADD and caspase 8 are critical for triggering cell death under TNFR activation, we next examined the functions of FADD and caspase 8 in RNF31 cleavage upon treatment with TNF-α and CHX by using FADD- or caspase 8-deficient Jurkat cells. Moreover, treatment of these modified cells with TNF-α and CHX activated necroptosis. Therefore, observation of these deficient cells would reveal whether necroptosis could induce the cleavage of RNF31. Upon treatment with TNF-α and CHX, cleaved RNF31 was observed 4 h after stimulation, and this cleavage event was completely blocked by treatment with Z-VAD-FMK in WT Jurkat cells. However, the cleaved band was not detected in FADD- or caspase 8-deficient Jurkat cells (Fig. 2B), suggesting that FADD and caspase 8 are vital for the cleavage of RNF31 in TNF-α- and CHX-induced apoptosis and that RNF31 is not cleaved in necroptosis.
The effector caspases, caspase 3 and caspase 6, are responsible for RNF31 cleavage.
Each caspase recognizes a specific sequence in its targets, and this specificity allows the caspases to have different roles in cellular processes (12). Therefore, we sought to determine which caspase is responsible for RNF31 cleavage. Since the results presented above showed that caspase 8 is essential for the cleavage of RNF31 under TNF-α stimulation, we induced apoptosis in A431 epidermal carcinoma cells, which have undetectable levels of caspase 8 due to the mutation. Because the caspase 8 deficiency leads to resistance to extrinsic inducers, we treated the cells with Dox or CPT to activate apoptosis. These agents induced the cleavage of RNF31 as well as those of PARP and caspase 9 (Fig. 3A). The cleavage of RNF31 in caspase 8-deficient Jurkat cells treated with an intrinsic inducer (Dox, CPT, or 5-FU) further supported the notion that caspase 8 is dispensable for RNF31 cleavage (Fig. 3B and C). Thus, we performed an in vitro cleavage assay to identify the caspase responsible for the cleavage of RNF31. Incubation of immunoprecipitated RNF31 with various recombinant caspases indicated that caspase 3 or caspase 6 is able to process RNF31 (Fig. 3D). Although a weak signal in the sample incubated with caspase 8 suggested that RNF31 is partially cleaved by caspase 8 in vitro, the data indicated that caspases 3 and 6 are the major caspases inducing RNF31 cleavage. The identical level of heavy chain in each sample suggested that equivalent levels of recombinant RNF31 were presented before the reaction (Fig. 3D). These data suggest that the effector caspases, caspase 3 and caspase 6, are responsible for the cleavage of RNF31.
FIG 3.

RNF31 is cleaved by the effector caspases caspase 3 and caspase 6. (A) WB analysis of A431 cells treated with Dox (3 μg/ml) or CPT (20 μM). (B and C) WB analysis of caspase 8-deficient Jurkat cells stimulated with Dox (3 μg/ml), CPT (20 μM), or 5-FU (20 μg/ml). (D) Results of an in vitro cleavage assay in which Myc-tagged RNF31 proteins were incubated with or without the indicated recombinant caspases for 2 h. C, caspase; IB, immunoblot.
RNF31 cleavage is dependent on Asp348, Asp387, and Asp390.
Since the balance between the apoptosis pathway and survival signaling governs the physiological features of cells, we hypothesized that the apoptosis pathway suppresses the function of the LUBAC in survival signaling via RNF31 cleavage. To test this hypothesis, we first identified the cleavage sites in RNF31. Treatment of 293T cells expressing N-terminally Myc-tagged RNF31 with TNF-α and CHX generated cleaved-RNF31 bands of ∼40 kDa in a time-dependent manner (Fig. 4A). On the basis of the Web-based prediction software Cascleave (13), we found that Asp348, Asp387, and Asp390 are potential cleavage sites (Fig. 4B). Since Asp390 has the highest probability score, we first generated a D390A mutant of RNF31 and then also generated a D348/390A RNF31 mutant. However, cleavage of these RNF31 mutants was still observed under apoptotic conditions (through treatment with TNF-α and CHX or cFLIP expression) (Fig. 4C), whereas the triple mutation of RNF31, D348/387/390A, completely blocked cleavage (Fig. 4C). Furthermore, the in vitro cleavage assay with recombinant WT and mutant RNF31 proteins showed that caspase 3 and caspase 6 were not able to process RNF31 D348/387/390A (Fig. 4D), indicating that Asp348, Asp387, and Asp390 in RNF31 are sites at which cleavage is initiated by effector caspases.
FIG 4.
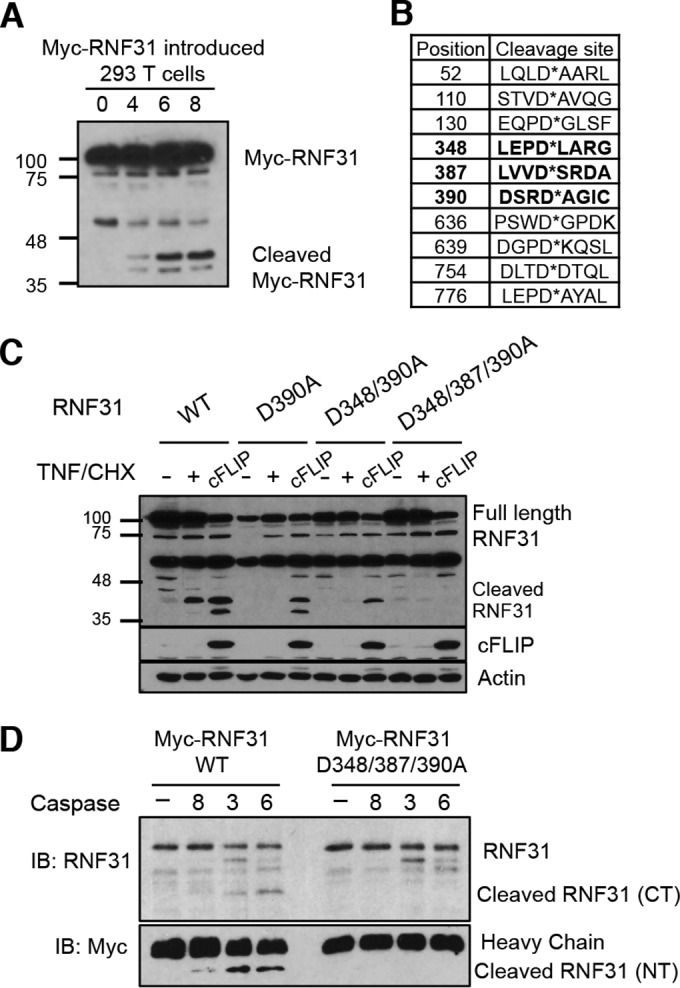
RNF31 is cleaved at aspartates 348, 387, and 390. (A) WB analysis of 293T cells transfected with RNF31 tagged with Myc at the N terminus, followed by treatment with TNF-α (40 ng/ml) and CHX (10 μg/ml). (B) Estimated sites of RNF31 cleavage by caspases. (C) WB analysis of 293T cells transfected with a plasmid encoding Myc-conjugated WT, D390A, D348/390A, or D348/387/390A RNF31. (D) Results of an in vitro cleavage assay in which recombinant WT or D348/387/390A mutant RNF31 was incubated with or without caspase 8, caspase 3, or caspase 6 for 1 h.
Cleavage of RNF31 suppresses its function in the NF-κB pathway.
Next, we examined the role of RNF31 cleavage in NF-κB activation. Previous studies have shown that full-length RNF31 (together with HOIL-1 and Sharpin) can activate NF-κB, while deletion of the ZF domain resulted in a partial defect in NF-κB activation (4). Therefore, we hypothesized that cleavage of RNF31 represses its ability to activate the NF-κB pathway. To test this hypothesis, we generated constructs expressing the N-terminal (residues 1 to 389) and C-terminal (residues 390 to 1072) fragments of RNF31 (referred to below as RNF31 NT and RNF31 CT, respectively) (Fig. 5A). A luciferase assay with full-length RNF31 and cleaved fragments of RNF31 demonstrated that neither of the cleaved fragments could fully activate NF-κB, even when expressed together with HOIL-1 and Sharpin (Fig. 5B). Specifically, RNF31 CT only partially induced NF-κB activation (Fig. 5B), although it still induced linear ubiquitination (Fig. 5C). The RNF31 C885S mutant, which lost its catalytic activity, exhibited defective NF-κB activation, indicating that NF-κB activation by the LUBAC depends on the catalytic activity of RNF31 (14). Moreover, simultaneous expression of both the RNF31 fragments, RNF31 NT and RNF31 CT, partially induced NF-κB activation, at a level similar to that with RNF31 CT alone (Fig. 5D). This set of data indicates that RNF31 cleavage inhibits NF-κB activation.
FIG 5.

Activation of the NF-κB pathway is suppressed by RNF31 cleavage. (A) Schematic diagram of RNF31 domains. PUB, PNGase/UBA- or UBX-containing proteins; UBA, ubiquitin associated; IBR, in between Ring fingers; LDD, linear ubiquitin chain-determining domain. (B) Luciferase analysis of lysates from 293T cells transfected with NF-κB luciferase reporter genes and the indicated RNF31 mutants with or without HOIL-1 and Sharpin. FL, full length. (C) WB analysis of lysates from the cells analyzed in panel B. ubi, ubiquitination. (D) Luciferase analysis of lysates from 293T cells transfected with NF-κB luciferase reporter genes and the indicated RNF31 mutants with HOIL-1 and Sharpin. Con, control; n.s., not significant. Triple asterisks indicate significant differences (***, P < 0.001.).
The C-terminal RNF31 fragment is able to induce linear ubiquitination of NEMO and RIP1.
We then examined the functional capacity of each fragment to bind with NEMO, because this interaction is essential for NF-κB activation. In agreement with previous reports (15), NEMO was able to interact with full-length RNF31 (RNF31 FL) and RNF31 NT (containing the ZF domain) but not with RNF31 CT (Fig. 6A, top). Binding ability was recovered when RNF31 CT was expressed together with HOIL and Sharpin (Fig. 6A). The ubiquitination assay, assessing the capabilities of the cleaved fragments and conjugating ubiquitin chains, showed that RNF31 CT still initiated the linear ubiquitination of NEMO in the presence of HOIL-1 and Sharpin (Fig. 6B). In agreement with the findings for NEMO, not only RNF31 FL but also RNF31 CT together with HOIL-1 and Sharpin generated the linear ubiquitination of RIP1 (Fig. 6D), another substrate of the LUBAC for linear ubiquitination, although we could not detect any interaction between RIP1 and RNF31, even in the presence of HOIL-1 and Sharpin (Fig. 6C). This series of data suggests that cleaved fragments of RNF31 are less potent NF-κB activators than full-length RNF31 but that the CT fragment can still generate the linear ubiquitination of NEMO and RIP in the presence of HOIL-1 and Sharpin.
FIG 6.

NEMO and RIP1 are conjugated with linear ubiquitination chains by the C-terminal RNF31 fragment. (A and C) WB analysis of immunoprecipitates (IP) from 293T cells transfected with FLAG-NEMO (A) or FLAG-RIP1 (C) and the indicated LUBAC components using anti-FLAG beads. (B and D) WB analysis of immunoprecipitates from lysates (prepared with 2% SDS lysis buffer and boiling) of 293T cells transiently transfected with the indicated plasmids using anti-FLAG beads. HS, HOIL-1 and Sharpin.
Mutation of cleavage sites suppresses the induction of apoptosis.
To determine the physiological role of RNF31 cleavage in the cell death process, we generated RNF31 knockdown (KD) HeLa cells, in which silenced RNF31 is reconstituted with WT RNF31 or D348/387/390A mutant RNF31, and performed an MTT assay (Fig. 7A). To exclude interference from knockdown efficiency, we generated RNF31-deficient Jurkat T cells using the clustered regularly interspaced short palindromic repeat (CRISPR) system (16) and reconstituted these cells with WT RNF31 or the D348/387/390A mutant of RNF31 (referred to below as Jurkat RKO-WT and Jurkat RKO-MT134 cells, respectively) (Fig. 7B). First, we monitored the levels of translocated p65 in TNF-α-treated Jurkat RKO-WT and Jurkat RKO-MT134 cells in order to check NF-κB activation and found that p65 translocation is enhanced in TNF-α-stimulated Jurkat RKO-MT134 cells relative to Jurkat RKO-WT cells (Fig. 7C). Next, to test the sensitivity of these modified cells to TNF-α induced apoptosis, we treated Jurkat RKO-WT and Jurkat RKO-MT134 cells with TNF-α. Flow cytometry analysis after annexin V staining revealed that Jurkat RKO-WT cells were more sensitive to TNF-α-induced apoptosis than Jurkat RKO-MT134 cells (Fig. 7D). We observed a higher percentage of annexin V-stained cells in TNF-α-treated Jurkat RKO-WT cells than in TNF-α-treated Jurkat RKO-MT134 cells (45.1% versus 36.7% [Fig. 7D]), and these different sensitivities to apoptosis were monitored at different time points (Fig. 7E) and were found to be statistically significant (Fig. 7F). We also confirmed that the MT134 mutant of RNF31 is resistant to TNF-α-induced cleavage and protects Jurkat cells from apoptosis more efficiently than WT RNF31 (Fig. 7G). Although we observed a higher level of NF-κB activation in TNF-α-treated Jurkat RKO-MT134 cells than in TNF-α-treated Jurkat RKO-WT cells, we also found that the sensitivities of these cells to TNF-α- and CHX-induced apoptosis are different when the effect of NF-κB on apoptosis is inhibited by CHX treatment (Fig. 7H). These data indicate that RNF31 regulates apoptosis in both NF-κB-dependent and NF-κB-independent ways.
FIG 7.

A cleavage-resistant RNF31 mutant enhances resistance to apoptosis. (A) MTT analysis of lysates from control KD (shCon), RNF31 KD (shRNF31), WT RNF31-reconstituted RNF31 KD (SR WT), and cleavage-resistant RNF31 mutant-reconstituted RNF31 KD (SR MT134) HeLa cells treated with TNF-α (100 ng/ml). Single and double asterisks indicate significant differences at P values of <0.05 and <0.01, respectively. OD, optical density. (B) WB analysis of control, RNF31-deficient, RKO-WT, and RKO-MT134 Jurkat cells. (C) WB analysis of nuclear lysates from RKO-WT and RKO-MT134 Jurkat cells treated with TNF-α (20 ng/ml). (D) Flow cytometry analysis of Jurkat RKO-WT and Jurkat RKO-MT134 cells treated with TNF-α (200 ng/ml) for 24 h. The percentage of annexin V-stained cells is given in each panel. (E) Sensitivities to apoptosis of Jurkat RKO-WT and Jurkat RKO-MT134 cells treated with TNF-α (200 ng/ml) for the indicated periods. (F) Flow cytometry analysis of Jurkat RKO-WT and Jurkat RKO-MT134 cells treated with TNF-α (200 ng/ml) plus cycloheximide for the times shown. ns, not significant. Double and triple asterisks indicate significant differences at P values of <0.01 and <0.001, respectively. (G) WB analysis of RKO-WT and RKO-MT134 Jurkat cells stimulated with TNF-α (50 ng/ml). (H) WB analysis of control, RNF31-deficient, RKO-WT, and RKO-MT134 Jurkat cells treated with TNF-α (10 ng/ml) and CHX (10 μg/ml). (I) WB analysis of Jurkat RKO-Mock, Jurkat RKO-FL, Jurkat RKO-NT, and Jurkat RKO-CT cells stimulated with TNF-α (10 ng/ml). (J) Flow cytometry analysis of Jurkat RKO, Jurkat RKO-FL, Jurkat RKO-CT, and Jurkat RKO-NT cells treated with TNF-α (100 ng/ml) for 24 h.
Additionally, we examined NF-κB activation in Jurkat RNF31 knockout cells reconstituted with RNF31 CT or RNF31 NT by monitoring nuclear p65 levels after TNF-α stimulation. We found higher levels of p65 translocation in these cells than in Jurkat RNF31 knockout cells reconstituted with full-length RNF31 (Fig. 7I). Moreover, we observed that reconstitution with the CT or NT fragment could not recover cell viability after TNF-α treatment (Fig. 7J), indicating that reconstitution of cleaved fragments does not rescue the function of full-length RNF31 in RNF31-deficient Jurkat cells. These results demonstrate that the cleavage of RNF31 sensitizes cells to TNF-α-induced death by suppressing survival signaling.
DISCUSSION
Caspases directly execute cell death by destroying fundamental proteins and indirectly restrict survival signaling by cleaving the cell survival mediators. Here we present a novel mechanism whereby caspases regulate LUBAC function. Once effector caspases are activated, they suppress the functions of RNF31 and the LUBAC in NF-κB signaling through the cleavage process. This negative regulation attenuates the inhibitory role of NF-κB in death signaling and further accelerates cell death or sensitizes cells to cell death (Fig. 8). Although it has been reported that the LUBAC plays a critical role in the activation of NF-κB signaling, previous studies have suggested that the LUBAC plays a role in the process of apoptosis. Sharpin-deficient cpdm mouse embryonic fibroblasts (MEFs) have increased sensitivity to TNF-α-induced apoptosis (3, 5), and RNF31-silenced ovarian cancer cells are more sensitive to cisplatin-induced death (17). Although the mechanism of apoptosis regulation has not been completely demonstrated, these previous studies support our finding that LUBAC inhibition by caspase-dependent RNF31 cleavage sensitizes cancer cells to apoptosis.
FIG 8.
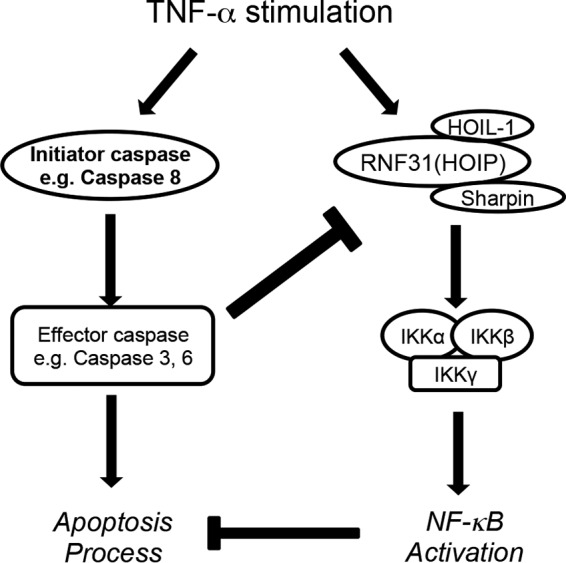
Proposed model of the current study. Upon TNF-α stimulation, activation of the caspase cascade leads to the initiation of apoptosis. Simultaneously, NF-κB signaling is activated through the activation of the LUBAC/IKKs, which promotes cell survival by regulating gene expression. Here we report the negative regulatory loop from apoptosis to NF-κB signaling. Activated effector caspases cleave RNF31, suppressing NF-κB activation and accelerating the apoptosis process.
RNF31 contains two functional domains to activate the NF-κB pathway: the catalytic domain (RBR [RING between RING] domain) and the interacting domain (ZF [zinc finger] domain) (6). Of note, the cleavage site that we discovered in this study is located between NZF1 and NZF2. On the basis of previous studies showing that ΔZF or NZF1 mutants have decreased ability to activate NF-κB signaling (4, 18), we speculated that RNF31 cleavage results in the separation of two functional domains (the RBR catalytic domain and the NZF1 domain) and that therefore, cleaved fragments are not able to fully induce NF-κB activation. However, we propose this model based on the interaction of RNF31 with NEMO and RIP1 and the effect of RNF31 on the ubiquitination of these proteins. Previous studies (15) and our data showed that a ΔZF or CT fragment could still bind with NEMO in the presence of HOIL-1 and Sharpin but that the ΔZF or CT fragment alone was not able to. Moreover, the CT fragment is capable of conjugating linear ubiquitination chains to its substrates, NEMO and RIP1, in the presence of Sharpin and HOIL and its ability to induce linear ubiquitination is even stronger than that of RNF31 FL. Since the expression of linearly ubiquitinated NEMO is adequate for the activation of NF-κB signaling (15), these data suggest that RNF31 has an additional function in the regulation of the NF-κB pathway downstream of ubiquitinated NEMO. Thus, RNF31 cleavage inhibits this additional function, other than linear ubiquitination, to suppress NF-κB activation and regulate cell death. In fact, LUBAC-deficient cells showed a delay, not a defect, in NF-κB activation, and deregulation of linear ubiquitination did not affect NF-κB activation in certain cell types (19, 20), so apoptosis may not be regulated solely by RNF31-mediated NF-κB signaling but may be controlled by other signaling pathways as well, such as direct regulation of apoptosis. To determine the mechanism by which RNF31 regulates NF-κB activation and cell death, further investigations are required. Additionally, the deubiquitinase enzyme OTULIN has been reported to play an essential role in NF-κB signaling pathways and inflammation by deconjugating linear ubiquitination from substrates (21, 22), and recently, clinical evidence from ORAS (OTULIN-related autoinflammatory syndrome) patients supported the fundamental role of OTULIN in human disease (19, 23). Since OTULIN binds with the PUB domain of RNF31 (located in the RNF31 NT mutant), further investigation on the regulation of OTULIN function by RNF31 cleavage will elucidate the mechanism of linear ubiquitination-mediated signaling and its biological significance.
Not only TNF-α but also many death-inducing agents, such as TRAIL, FasL, and DNA damage inducers, simultaneously activate LUBAC function and/or NF-κB signaling, which often leads to resistance to treatment (24, 25). Thus, the disruption of LUBAC or RNF31 activity could disrupt the balance between NF-κB and apoptosis signaling, which may be a promising target for treating diseases resulting from deregulated cell death. Our study not only expands our knowledge on the cross talk between cell death and survival but also provides a possible mechanism to treat diseases resulting from the imbalance between death and survival signals. Specifically, the model presented—the sensitization of cells by RNF31 cleavage—might represent a therapeutic strategy to increase the efficacy of drugs by providing advantageous conditions for combination cancer therapy.
ACKNOWLEDGMENTS
We thank Vishva Dixit (Genentech Corporation) for providing anti-linear ubiquitin antibodies.
This work was partially supported by grant R01AI116722 from the National Institutes of Health (NIH) to X.L. and a pilot grant from the Center for Inflammation and Cancer (CIC) in the University of Texas MD Anderson Cancer Center to X.L.
We declare no conflict of interest.
D.J., M.B., X.Z., and X.L. designed experiments; D.J. and Y.T. performed experiments; J.J. contributed new reagents; D.J. and X.L. analyzed the data and wrote the manuscript.
REFERENCES
- 1.Oeckinghaus A, Ghosh S. 2009. The NF-κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. 2006. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation [corrected]. Nat Cell Biol 8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, van Wijk SJ, Goswami P, Nagy V, Terzic J, Tokunaga F, Androulidaki A, Nakagawa T, Pasparakis M, Iwai K, Sundberg JP, Schaefer L, Rittinger K, Macek B, Dikic I. 2011. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471:637–641. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, Tanaka K, Nakano H, Iwai K. 2011. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471:633–636. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 5.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. 2011. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 6.Iwai K, Fujita H, Sasaki Y. 2014. Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat Rev Mol Cell Biol 15:503–508. doi: 10.1038/nrm3836. [DOI] [PubMed] [Google Scholar]
- 7.Strasser A, O'Connor L, Dixit VM. 2000. Apoptosis signaling. Annu Rev Biochem 69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 8.Karin M, Lin A. 2002. NF-κB at the crossroads of life and death. Nat Immunol 3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 9.Frelin C, Imbert V, Bottero V, Gonthier N, Samraj AK, Schulze-Osthoff K, Auberger P, Courtois G, Peyron JF. 2008. Inhibition of the NF-κB survival pathway via caspase-dependent cleavage of the IKK complex scaffold protein and NF-κB essential modulator NEMO. Cell Death Differ 15:152–160. doi: 10.1038/sj.cdd.4402240. [DOI] [PubMed] [Google Scholar]
- 10.Lin Y, Devin A, Rodriguez Y, Liu ZG. 1999. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravi R, Bedi A, Fuchs EJ, Bedi A. 1998. CD95 (Fas)-induced caspase-mediated proteolysis of NF-κB. Cancer Res 58:882–886. [PubMed] [Google Scholar]
- 12.Crawford ED, Wells JA. 2011. Caspase substrates and cellular remodeling. Annu Rev Biochem 80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- 13.Song J, Tan H, Shen H, Mahmood K, Boyd SE, Webb GI, Akutsu T, Whisstock JC. 2010. Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 26:752–760. doi: 10.1093/bioinformatics/btq043. [DOI] [PubMed] [Google Scholar]
- 14.Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P. 2013. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A 110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. 2009. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol 11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 16.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackay C, Carroll E, Ibrahim AF, Garg A, Inman GJ, Hay RT, Alpi AF. 2014. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res 74:2246–2257. doi: 10.1158/0008-5472.CAN-13-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita H, Rahighi S, Akita M, Kato R, Sasaki Y, Wakatsuki S, Iwai K. 2014. Mechanism underlying IκB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol Cell Biol 34:1322–1335. doi: 10.1128/MCB.01538-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, McHale D, Maher ER, McKenzie AN, Komander D. 2016. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166:1215–1230.e20. doi: 10.1016/j.cell.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park Y, Jin HS, Lopez J, Lee J, Liao L, Elly C, Liu YC. 2016. SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat Immunol 17:286–296. doi: 10.1038/ni.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu Y, Taraborrelli L, Walczak H. 2015. Linear ubiquitination in immunity. Immunol Rev 266:190–207. doi: 10.1111/imr.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott PR, Komander D. 2016. Regulation of Met1-linked polyubiquitin signalling by the deubiquitinase OTULIN. FEBS J 283:39–53. doi: 10.1111/febs.13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, Kuehn HS, Wang H, Yang D, Park YH, Ombrello AK, Blake M, Romeo T, Remmers EF, Chae JJ, Mullikin JC, Guzel F, Milner JD, Boehm M, Rosenzweig SD, Gadina M, Welch SB, Ozen S, Topaloglu R, Abinun M, Kastner DL, Aksentijevich I. 2016. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci U S A 113:10127–10132. doi: 10.1073/pnas.1612594113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakanishi C, Toi M. 2005. Nuclear factor-κB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 25.Niu J, Shi Y, Iwai K, Wu ZH. 2011. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J 30:3741–3753. doi: 10.1038/emboj.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]