

Abstract
Cell death in MI is the most critical determinant of subsequent left ventricular remodeling and heart failure. Besides apoptosis, autophagy and necroptosis have been recently found to be another two regulated cell death styles. HGF has been reported to have a protective role in MI, but its impact on the three death styles remains unclear. Thus, our study was performed to investigate the distribution of autophagy, apoptosis and necroptosis in cardiac tissues after MI and explore the role and mechanism of Ad-HGF on cardiac remodeling by regulating the three death styles. We firstly showed the distribution of autophagy, apoptosis and necroptosis differs in temporal and spatial context after MI using immunofluorescence. Notably, Ad-HGF treatment improves the cardiac remodeling of SD rats following MI by preserving the heart function, reducing the scar size and aggresomes. Further mechanism study reveals Ad-HGF promotes autophagy and necroptosis and inhibits apoptosis in vivo and in vitro. Co-immunoprecipitation assays showed Ad-HGF treatment significantly decreased the binding of Bcl-2 to Beclin1 but enhanced Bcl-2 binding to Bax in H9c2 cells under hypoxia. Moreover, HGF-induced sequestration of Bax by Bcl-2 allows Bax to become inactive, thereby inhibiting apoptosis. In addition, Ad-HGF markedly increased the formation of Beclin1-Vps34-Atg14L complex, which accounted for promoting autophagy. Both the western blot and activity assay showed Ad-HGF significantly decreased the caspase 8 protein and activity levels, which obligated the cell to undergo necroptosis under hypoxia and block apoptosis. Thus, our findings offer new evidence and strategies for the treatment of MI and post-MI cardiac remodeling.
Keywords: Ad-HGF, myocardial infarction, cardiac remodeling, autophagy, apoptosis, necroptosis
Introduction
Myocardial infarction (MI) is one of the leading causes of morbidity and mortality worldwide and the incidence of which shows a rising trend in developing countries [1]. Advances in the treatment of MI, such as early reperfusion therapy, have notably reduced the mortality in patients with acute myocardial infarction. However, the parallel increase of the prevalence and mortality from left ventricular (LV) remodeling and heart failure after MI has emerged as a growing challenging health problem of concern [2]. Currently, therapeutic strategies to prevent adverse LV remodeling and heart failure after MI are still limited. Therefore, it’s highly needed to identify new therapeutic targets and treatment options to prevent post-MI adverse cardiac remodeling.
For many years, apoptosis was considered as the only form of regulated cell death and studies investigating myocyte cell death mainly focused on apoptosis [3,4]. Recent years, autophagy and necroptosis have been found to be another two regulated cell death styles existing in various diseases including MI [5-7]. However, the distribution and roles of the three types of cell death in different periods and different regions of the heart after MI have not been fully elucidated. Study of autophagy, apoptosis and necroptosis in the process of myocardial infarction may provide us new insights to the treatment of MI and post-MI adverse cardiac remodeling.
Autophagy is an intracellular degradation process in which cytosolic proteins and organelles are confiscated into autophagosomes and degraded by lysosomes [6,8]. Because of its large capacity, autophagy plays an important role in maintaining organelle functions and protein quality by degrading damaged organelles and protein aggregates. Suppression of autophagy below physiological levels through genetic deletion of Atg5 induces heart failure with enhanced protein aggregation, manifesting autophagy is actually required to maintain baseline heart function [9]. Autophagy is found to be upregulated in various pathological conditions, including myocardial ischemia and ischemia/reperfusion (I/R) [10-12]. It is generally compensatory for the upregulation of autophagy during myocardial stress, which on the one hand alleviating energy loss, on the other hand, scavenging damaged mitochondria and protein aggregates [10,13].
Recently, a new death style, named necroptosis, was identified and proposed as a form of ‘programmed necrosis’, which was introduced in 2005 and demonstrated in cultured cells [14]. In contrast to apoptosis or autophagy, necroptosis is characterized by swollen organelles, disintegrated plasma membrane and lack of nuclear fragmentation [15]. Under the condition of various stimuli including TNF-α, ischemia and ischemia/reperfusion (I/R), etc., receptor interacting protein kinase 1 (RIP1) and receptor interacting protein kinase 3 (RIP3) auto- and transphosphorylate each other, and form necrosome, which then phosphorylates the pro-necroptotic mixed lineage kinase domain-like (MLKL) protein. Necroptosis is initiated by phosphorylated MLKL through forming oligomerization which inserts itself into the membranes of organelles and plasma membrane [16,17]. However, caspase-8 is able to disrupt the necrosome by cleaving RIP1 and RIP3, thereby effectively terminating necroptosis [17]. Thus, for necroptosis to ensue, the caspase-8 function must be compromised. But the interplay among autophagy, apoptosis and necroptosis has not been clearly elaborated.
Hepatocyte growth factor (HGF) is a multifunctional cytokine and its c-Met receptor is widely expressed in cardiovascular system including cardiomyocytes, endothelial cells and smooth muscle cells. Through phosphorylating its receptor and activating the downstream signaling pathway, HGF plays a series of biological effects including morphogenesis, stem cell maintenance and immunomodulation [18]. Studies have demonstrated the heart protective effect of HGF, which includes promoting angiogenesis, anti-fibrosis, anti-inflammation, myocardium protection and regeneration [19,20]. Little evidence has shown that the protection of HGF is related with the myocytes adaptable autophagy, apoptosis and necroptosis after MI.
Thus, our study was performed to investigate the distribution of autophagy, apoptosis and necroptosis in cardiac tissues after MI and explore the role and mechanism of HGF on cardiac remodeling by regulating autophagy, apoptosis and necroptosis, which may offer new evidence and strategies for the treatment of MI and post-MI cardiac remodeling.
Materials and methods
Antibodies and reagents
The following antibodies were purchased from abcam: Anti-HGF antibody (ab83760), p62 (ab91526), RIP1 (ab72139), anti-beta actin antibody (ab8226), anti-ATG14L antibody (ab139727), rabbit anti-mouse IgG H&L (HRP) antibody (ab6728) and goat anti-rabbit IgG H&L (HRP) antibody (ab6721); from sigma: RIP3 (R4277); from Cell Signaling (USA): Phospho-Met (Tyr1234/1235) antibody (#3077), LC3A/B (#4108), Beclin-1 (#3495), cleaved caspase 3 (#9664), caspase 3 (#9662), Bcl-2 (#2870), Bax (#2772), Vps34 (#4263), GAPDH (#5174); from Santa Cruz Biotechnology: Troponin I antibody (sc-133117), Met (sc-8057), Bcl-2 (sc-7382), Bcl-XL (sc-8392), MLKL (sc-165025). FITC or CY3 labeled secondary antibodies were purchased from Jackson ImmunoResearch (USA). ProteoStat® Aggresome Detection Kit was purchased from Enzo Life Sciences, Switzerland. TUNEL Cell Death Detection kit (Cat. No. 11684795910) and propidium iodide (PI) was purchased from Sigma, USA. Cell counting kit-8 (CCK-8) and Hoechst 33342 staining kit were purchased from KeyGEN BioTECH, China. Ad-mRFP-GFP-LC3 was purchased from HanBio Technology Co. Ltd, China. Ad-HGF adenovirus vectors were constructed as previously described [21,22].
Animal model
SD rats (male, 180-220 g) were purchased from Nanjing Medical University Experimental Animal Center. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication No. 85-23, revised 1996) and approved by the Animal Care and Use Committee of Nanjing Medical University. Myocardial infarction was induced by permanent ligation of the left anterior descending (LAD) coronary artery. And the detailed operation procedure was as previously described [21]. A total of 50 microliter Ad-HGF (1.0×109 PFU) or Ad-null (1.0×109 PFU) was injected into the myocardium in the vicinity of the ischemic region (pale area) of the left ventricular wall immediately following ligation. Sham operations were carried out by the same method, but without tying the suture on the LAD. Rats were sacrificed using carbon dioxide (CO2) at day 7 and 28, respectively.
Echocardiography
Cardiac structure and function were evaluated by echocardiography with Vevo 2100-a high resolution imaging system (Visual Sonics, Canada) with a MS-250, 16.0-21.0 MHZ imaging transducer at baseline, week 1 and week 4. All of the SD rats were anesthetized by ether before the process of echocardiography examination.
Assessment of LV scar size after chronic MI
Hearts of SD rats were fixed in 4% paraformaldehyde, cut into four transverse slices and subjected to Masson’s Trichrome staining for later measurement of the LV scar 28 days after MI. The average infarct size was obtained by calculation of the mean length of the circumference in the infarct region and the normal area from the consecutive myocardial slices.
Cell culture and treatment
H9c2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Wisent Inc., Montreal, QC, Canada) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 100 U/ml of penicillin and 100 μg/ml of streptomycin. Cells were incubated at 37°C in a humidified air atmosphere supplemented with 5% CO2. The culture mediums were changed every two days. AnaeroPack-Anaero (MGC, Japan) and anaerobic jar (MGC, Japan) were used for H9c2 cells hypoxia. The AnaeroPack could consume all the oxygen and release equivalent carbon dioxide within thirty minutes and the mixed gases in the anaerobic jar were CO2 and N2. Culture medium was changed into DMEM (Gibco, USA) without glucose and serum before hypoxia. Ad-HGF (12.5, 25 or 50 MOI) or HGF (5, 20 or 80 ng/ml) or HGF receptor inhibitor SU11274 (10 μM) was added into medium before hypoxia in different groups. H9c2 cells, anaerobic indicator and AnaeroPack were placed into the anaerobic jar and the jar lid was immediately closed. After about thirty minutes, the oxygen concentration decreased to less than 0.1%. The whole anoxic process was performed at 37°C.
Transmission electron microscope examination
After treatment, the cells were collected using 0.25% trypsin (containing 0.02% EDTA) digestion, washed in fresh PBS (pH=7.4), and fixed with 2.5% glutaraldehyde. The cells were then washed with 0.1 M cacodylate buffer, post-fixed with 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h, stained with 1% aqueous uranyl acetate for 30 min, dehydrated through a graded series of ethanol to 100%. Cells were then infiltrated and embedded in TAAB Epon (Marivac Canada Inc., St. Laurent, Canada). Ultrathin section (60 nm) was cut on a Reichert Ultracut-S microtome, placed onto copper grids, stained with uranyl acetate and lead citrate, and observed on a transmission electron microscope (JEM-1010; JEOL, Tokyo, Japan) at an accelerating voltage of 80 kV.
Western blot
Protein was isolated using RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail, Beyotime, Shanghai, China) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF, Sigma-Aldrich, St. Louis, MO, USA), and was quantified using BCA Protein Assay (Pierce, Rockford, IL, USA). Electrophoresis was performed on SDS-PAGE gels. Separated proteins were transferred from the gels to PVDF membranes (Millipore, Billerica, MA, USA) and immunoblotting was performed as previously described [23].
TUNEL assay
TUNEL Cell Death Detection kit (Cat. No. 11684795910, Sigma, USA) for in situ apoptosis detection, was used for labeling DNA strand breaks, by terminal deoxynucleotidyl transferase (TdT), which catalyzes polymerization of labeled nucleotides to free 3’-OH DNA ends in a template-independent manner (TUNEL-reaction). The heart sections or the cells were treated according to the manufacturer’s instructions and co-stained with cardiac troponin I and DAPI. A fluorescence microscope (Nikon, Tokyo, Japan) was used to observe the TUNEL-positive cells, and Image J (NIH) software was used to analyze the images. Data presented in the following text are the averages calculated from five different fields.
Immunofluorescence
Rat Hearts were embedded in OCT (Sakura, Japan) and cut into 8 μm sections which were stored at -80°C. H9c2 cells were cultured on coverslips, and fixed in 4% paraformaldehyde after the indicated treatment. Immunofluorescence was conducted as briefly described below. Sections were fixed in cold acetone at -20°C and placed in the fume hood to ventilate for 20 minutes. The slides were gently rinsed twice with PBS and permeabilize with 0.1% Triton X-100 for 10 minutes. The slides were rinsed twice with PBS and then blocked in 2.5% BSA in PBS for 30 minutes. Sections were incubated with ProteoStat® aggresome detection reagent, p62 (1/300 dilution), RIP1 (1/200 dilution) or RIP3 (1/300 dilution) diluted in the blocking buffer at 4°C overnight. Prior to the addition of the secondary antibody, the slides were rinsed three times with PBS. Secondary antibodies were added in the same blocking buffer (for green light: use 1/200 dilution of fluorescein and for red light: use 1/400 dilution of CY3 or Rhodamine labeled secondary antibodies) for 1 hour in the dark, followed by another three washes in PBS. The sections were mounted with VECTASHIELD mounting medium with Diamidino-2-phenylindole (DAPI), (Vector Laboratories, Burlingame, CA, USA), and captured images under LSM 5 Live DuoScan Laser Scanning Microscope (Zeiss, Oberkochen, Germany).
Detection of aggresomes
The ProteoStat® Aggresome Detection Kit (Enzo Life Sciences, Switzerland) were used for the detection of aggresomes in cells. The ProteoStat® aggresome detection reagent is a kind of 488 nm excitable red fluorescent molecular rotor which binds selectively to protein cargo within aggresomes. All components of the ProteoStat® Aggresome Detection Kit were prepared and the procedures were conducted according to the manufacturer’s instructions.
CCK-8 assay
Cell viability was assessed by the cell counting kit-8 (CCK-8) (KeyGEN BioTECH, China). Cells were seeded into 96-well plates at 4×104 per well. After indicated treatment, a total of 10 μl CCK-8 was added to each well and incubated at 37°C in the dark for 2 hours and then the absorbance at 450 nm was measured with a microplate reader (BioTek, USA).
Hoechst 33342 staining
Typical morphological features of apoptotic cells were evaluated by Hoechst 33342 staining (KeyGEN BioTECH, China). After indicated treatment, the cells were washed twice with cold phosphate buffer saline (PBS) and fixed in 4% paraformaldehyde for 20 minutes. Cells were then washed with cold PBS again before incubated with 5 μg/ml Hoechst 33342 for 15 min at 37°C in the dark. Finally, cells were washed with PBS and apoptotic cells were identified under the fluorescence microscope (Nikon, Tokyo, Japan). Normal cells showed homogeneous blue chromatin with organized structure. By contrast, apoptotic cells showed bright blue chromatin which is highly condensed or fragmented.
Flow cytometry analysis
The necroptotic cells were identified by propidium iodide (PI) staining and flow cytometry analysis as previously described [7,24]. Briefly, the adherent cells were detached by 0.25% trypsin digestion and washed in fresh PBS (pH=7.4). After centrifugation at 1000 rpm, cells were resuspended in cold PBS with 2 μg/ml PI for 3 minutes before measurement of red fluorescence for 10000 cells using Flow cytometry.
RNA interference
Two different siRNAs targeted RIP1, RIP3 and MLKL, respectively, and a negative control siRNA were designed by GenePharma Co., Ltd. (Shanghai, China). Below were the siRNA sequences: RIP1-sh#1: 5’-CCA CUA GUC UGA CGG AUA A-3’, RIP1-sh#2: 5’-GCA CAA ATA CGA ACT TCA A-3’, RIP3-sh#1: 5’-UAA CUU GAC GCA CGA CAU CAG GCU GUU-3’, RIP3-sh#2: 5’-GCA GUU GUA UAU GUU AAC GAG CGG UCG-3’, MLKL-sh#1: 5’-CAA ACU UCC UGG UAA CUC A-3’ and MLKL-sh#2: 5’-GCG TAT ATT TGG GAT TTG CAT-3’. X-tremeGENE siRNA Transfection Reagent (invitrogen) was used to perform the siRNA knockdowns according to the manufacturer’s protocol. Briefly, 1.2×106 cells were seeded in 35-mm dishes to obtain approximately 80% confluence. A mixture containing 200 μl of siRNA (2.5 μg for each) and transfection reagent (14 μL for each) was added into the cells for 7 h at 37°C. Then change the old culture medium with fresh medium. The RIP1, RIP3 and MLKL expression levels were examined using western blot analysis.
Immunoprecipitation
Cells were lysed by IGEPAL CA-630 buffer (50 mM Tris-HCl (pH 7.4), 1% IGEPAL CA-630, 10 mM EDTA, 150 mM NaCl, 50 mM NaF, 1 μM leupeptin and 0.1 μM aprotinin). Primary antibody was covalently immobilized on protein A/G agarose using the Pierce® Crosslink Immunoprecipitation Kit according to the manufacturer’s instructions (Thermo Fisher Scientific, USA). Samples were incubated with immobilized antibody beads for at least two hours at 4°C. After IP, the samples were washed with TBS five times, then eluted with glycine-HCl (0.1 M, pH=3.5) and the immunoprecipitates were subjected to immunoblotting using specific primary antibodies and conformation-specific secondary antibody that recognizes only the native IgG (Cell Signaling, USA).
Statistical analysis
Data from at least three independent experiments were used to calculate the mean ± standard deviation (SD) using SPSS 18.0 statistical software. Statistical analyses between groups were performed by unpaired Student’s t test or one-way ANOVA followed by a post hoc Fisher’s comparison test. A value of less than 0.05 was considered significant.
Results
Autophagy, apoptosis and necroptosis occurred in the heart after myocardial infarction
Firstly, we investigated how the three types of cell death distributed in the heart after myocardial infarction. LC3-II was a widely used indicator to mark autophagy [13]. We found LC3-II staining significantly increased in the heart 7 days and 28 days after MI. Moreover, in 7 days’ heart of MI, LC3-II staining was mainly concentrated in the infarcted area, which indicating more autophagy happened in the infarct zones than the peri-infarct zones. Inversely, in 28 days’ heart autophagy in peri-infarct zones was significantly higher than the infarct zones, but both of which were lower than the 7 days’ heart with MI. And there was no difference of autophagy in the non-infarct zone among the three groups (Figure 1A). Next, we used TUNEL staining to examine the apoptosis level in the heart following MI. More cardiomyocytes apoptosis occurred after MI. In 7 days’ and 28 days’ cardiac tissues of MI, apoptosis mainly appeared in the peri-infarct zones rather than the infarct zones. When compared with the 7 days’ heart of MI, 28 days’ heart showed lower apoptosis level. No difference of apoptosis level was observed in the non-infarcted areas of the indicated three groups (Figure 1B). Necroptosis was evaluated using PI labeling by intraperitoneal injection of PI one hour before termination [25]. Cardiomyocyte necroptosis significantly increased after MI. Intriguingly, we found much more necroptosis appeared in the infarct zones than the other areas in both 7 days’ and 28 days’ hearts of MI. Furthermore, necroptosis in the infarct zones of the 28 days’ heart markedly declined as compared with the 7 days’ heart. Necroptosis level showed no difference in the non-infarcted areas of the indicated three groups (Figure 1C). These data confirmed that autophagy, apoptosis and necroptosis occurred in the heart after myocardial infarction.
Figure 1.
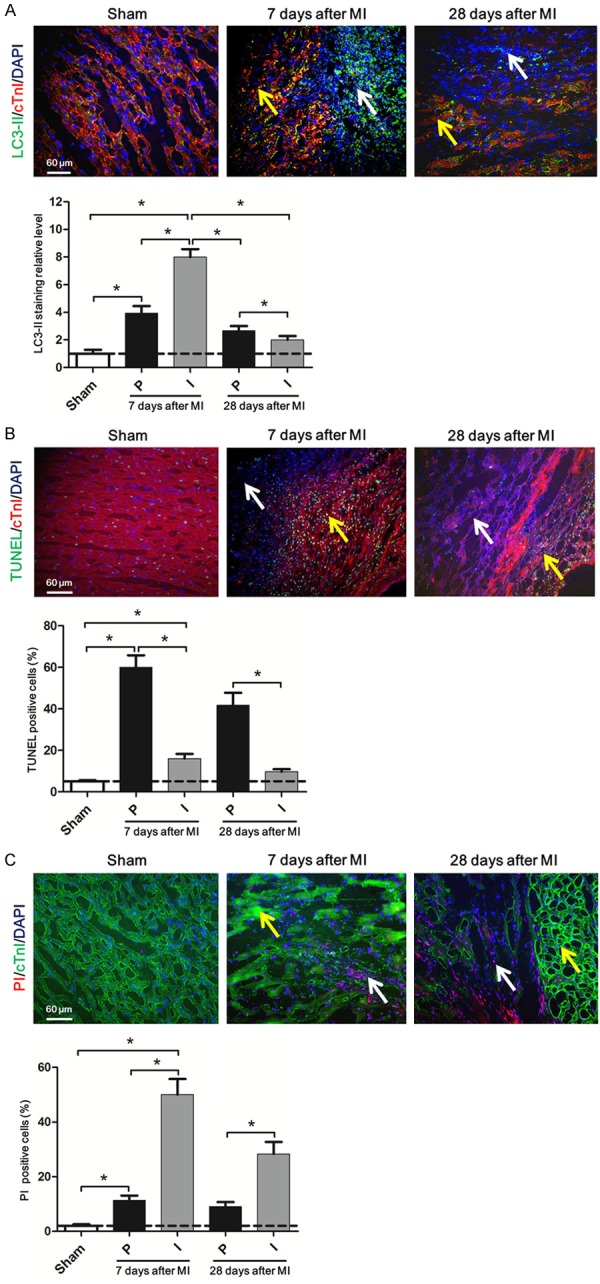
Autophagy, apoptosis and necroptosis occurred in the heart after myocardial infarction. A. Representative immunofluorescent images of staining with LC3-II (green), cTnI (red), and DAPI (blue) in the heart tissue of SD rats 7 days, 28 days after coronary artery ligation are shown. The white arrow indicates the infarct zones and yellow arrow indicates the peri-infarct zones. The scale bar in the lower left corner of image is 60 μm. Below is the same. The lower bar graph shows the quantitative analysis of the percent of LC3-II positive cells. B. Upper: Representative immunofluorescent images of staining with TUNEL (green), cTnI (red), and DAPI (blue) in the heart tissue of SD rats 7 days and 28 days after MI are shown. Lower: The bar graph shows quantitative analysis of the percent of TUNEL positive cells. C. Upper: Representative immunofluorescent images of staining with PI (red), cTnI (green), and DAPI (blue) in the heart tissue of SD rats 7 days and 28 days after MI are shown. Lower: The bar graph shows quantitative analysis of the percent of PI positive cells. Data are expressed as mean ± SD, n=4, *P<0.05.
Ad-HGF treatment improved the cardiac remodeling by upregulating autophagy and necroptosis and inhibiting apoptosis after MI
Ad-HGF and Ad-null vectors were injected into three sites of the myocardium in proximity to the ischemic area immediately after ligation of the LAD. One week after the injection, the expressions of HGF, Met and p-Met were analyzed by western blot. The results showed Ad-HGF treatment significantly increased the HGF and p-Met expression level in the cardiac peri-infarct zones than the Ad-null and sham groups (Figure 2A). The representative M-mode echocardiograms of SD rats showed that motion range of the left ventricular anterior wall weakened at 1 week after MI. After 4 weeks, the abnormality of anterior wall movement was more serious in MI groups. The motion range of the left ventricular anterior wall was much better in MI+Ad-HGF treatment group than MI+Ad-null group. LVEF and LVFS were evaluated by measurements of echocardiography. Compared with the sham group, LVEF and LVFS both significantly decreased in MI group 1 week after ligation. However, Ad-HGF treatment group showed significantly improvement in LVEF and LVFS than the MI+Ad-null group 4 weeks after MI (Figure 2B). Cardiac fibrosis is a well-known feature of cardiac remodeling after MI [26]. Masson-Trichrome staining revealed that the MI scar size and cardiac fibrosis were significantly attenuated in Rat receiving Ad-HGF treatment as compared to Ad-null group 28 days after MI (Figure 2C). Heart failure is often accompanied by accumulation of damaged proteins and organelles, in the form of aggresomes [8]. Microscopic analyses of the chronic MI rat heart showed that aggresomes accumulated in the perinucleus of border zone and remote area cardiomyocytes (Figure 2D). The aggresomes co-localized with p62/SQSTM1 (p62), a protein known to be degraded by autophagy, suggesting insufficient clearance of aggresomes by autophagy (Figure 2D). The accumulation of aggresomes and p62 was markedly attenuated in rat with Ad-HGF treatment as compared to the Ad-null group, suggesting that endogenous HGF positively affects protein and organelle quality control in post MI hearts. Autophagy is activated in the post MI heart, as evidenced by increases in LC3-II protein level. Beclin-1 and LC3-II protein level were further upregulated and p62 was downregulated in Ad-HGF-treated rat after MI, which proved the promoting autophagy effects of Ad-HGF treatment (Figure 2E). Cleaved caspase 3/caspase 3 protein level, indicator of apoptosis, significantly increased after MI and was markedly attenuated after Ad-HGF treatment which was related to the increase of the anti-apoptotic proteins, Bcl-2 and Bcl-xL, and the decrease of the pro-apoptotic protein, Bax level (Figure 2F). The protein levels of RIP1/3 are positively correlated with the extent of necroptosis [17]. Therefore, we examined the expression levels of RIP1 and RIP3 in the MI heart. Western blot analysis showed that the levels of these two proteins were significantly increased in cardiac tissues following MI. And Ad-HGF treatment further increased the level of RIP1 and RIP3 (Figure 2G). Taken together, these results suggest that Ad-HGF treatment improved the cardiac remodeling by upregulating autophagy and necroptosis and inhibiting apoptosis after MI.
Figure 2.
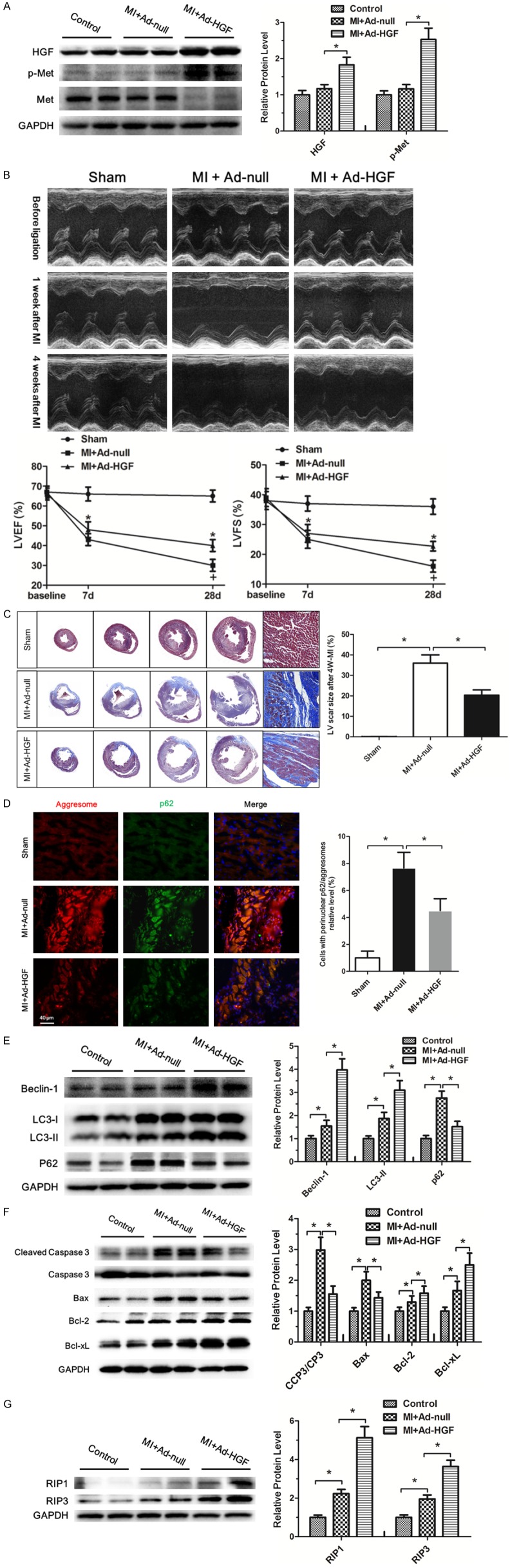
Ad-HGF treatment improved the cardiac remodeling by upregulating autophagy and necroptosis and inhibiting apoptosis after MI. A. Representative western blot images of heart homogenates with antibodies to HGF, p-Met, Met, and GAPDH are shown. The protein extraction was from sham, MI+Ad-null and MI+Ad-HGF rats after one week of MI. The bar graph shows the quantitative analysis of the western blot results. Data are expressed as mean ± SD, n=4, *P<0.05. B. Representative M-mode echocardiograms of SD rats before and after MI. The line charts shows the analysis of left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) in SD rats at 1 week and 4 week after MI, respectively. Data are expressed as mean ± SD, n=8 per group, *P<0.05. C. Representative images of four consecutive myocardial slices stained with Masson’s trichrome in sham, MI+Ad-null and MI+Ad-HGF groups after 4 weeks of MI are shown. The bar graph shows the quantitative analysis of the LV scar size. Data are shown as mean ± SD, n=4, *P<0.05. D. Representative immunofluorescent images of staining with ProteoStat® aggresome detection reagent (red), p62/SQSTM1 (green) and DAPI (blue) in sham, MI+Ad-null and MI+Ad-HGF groups after 4 weeks of MI are shown. The bar graph shows the quantitative analysis of the results. Data are represented as mean ± SD, n=4, *P<0.05. E-G. Western blot analysis of Beclin-1, LC3-I, LC3-II, p62, cleaved caspase 3, caspase 3, Bax, Bcl-2, Bcl-xL, RIP1, RIP3 and GAPDH levels in the sham, MI+Ad-null and MI+Ad-HGF groups, respectively. Protein expression was quantified with reference to GAPDH. The bar graph shows the quantitative analysis of the western blot results. Data are represented as mean ± SD, n=3, *P<0.05.
In vitro hypoxia treatment induced autophagy, apoptosis and necroptosis in H9c2 cells
The insufficient supply of oxygen and nutrients results in the damages to the heart after MI. To mimic the pathological process of MI, we treated rat myocardial cell line, H9c2 cell, with serum-free and glucose-free medium in a hypoxia chamber. To discern which type of cell death occurred in H9c2 cells after the insult, we used TEM to observe cellular morphology. After ischemia and hypoxia treatment, all types of cell death, including apoptosis, necroptosis, and necrosis, were observed in H9c2 cells (Figure 3A). Compared with the normal structure of H9c2 cell under normoxia (Figure 3Aa), TEM detected the formation of representative autophagosome in H9c2 cells at 3 h of hypoxia (Figure 3Ab). Apoptotic cell characterized by chromatin condensation and nuclear fragmentation is seen at 6 h of hypoxia (Figure 3Ac). The morphological changes of necroptosis cells are distinct from that of autophagy and apoptosis. In contrast to autophagic (Figure 3Ab) and apoptotic cells (Figure 3Ac), membrane leakiness, organelle and nuclear swelling, increased cell volume with translucent cytoplasm were observed to occur in necroptosis cells at 9 h of hypoxia (Figure 3Ad); but autophagosome and apoptotic bodies were not formed in these cells. We further investigated the changes of the relative proteins indicating autophagy, apoptosis and necroptosis under hypoxia, respectively. Previous studies have demonstrated that LC3 are autophagy-related proteins and enhanced autophagy accompanied by increased LC3-II expression [10]. The results of the western blot showed autophagy significantly increased soon after hypoxia and reached the summit at 3 h under hypoxia, then decreased. However, we intriguingly observed autophagy level Increased again at 15 h and reached the second summit at 18 h after hypoxia, and then declined (Figure 2B and 2C). The changes of cleaved caspase 3/caspase 3 protein level revealed that apoptosis process started after hypoxia and got to the peak at 6 h, and then slowly went down (Figure 2B and 2D). The protein levels of RIP1/3 are positively correlated with the extent of necroptosis [17]. The expressions of the two proteins markedly increased under hypoxia insult and lasted 3 hours before gradually falling down. Collectively, the results demonstrated that hypoxia treatment induced autophagy, apoptosis and necroptosis in H9c2 cell in vitro.
Figure 3.
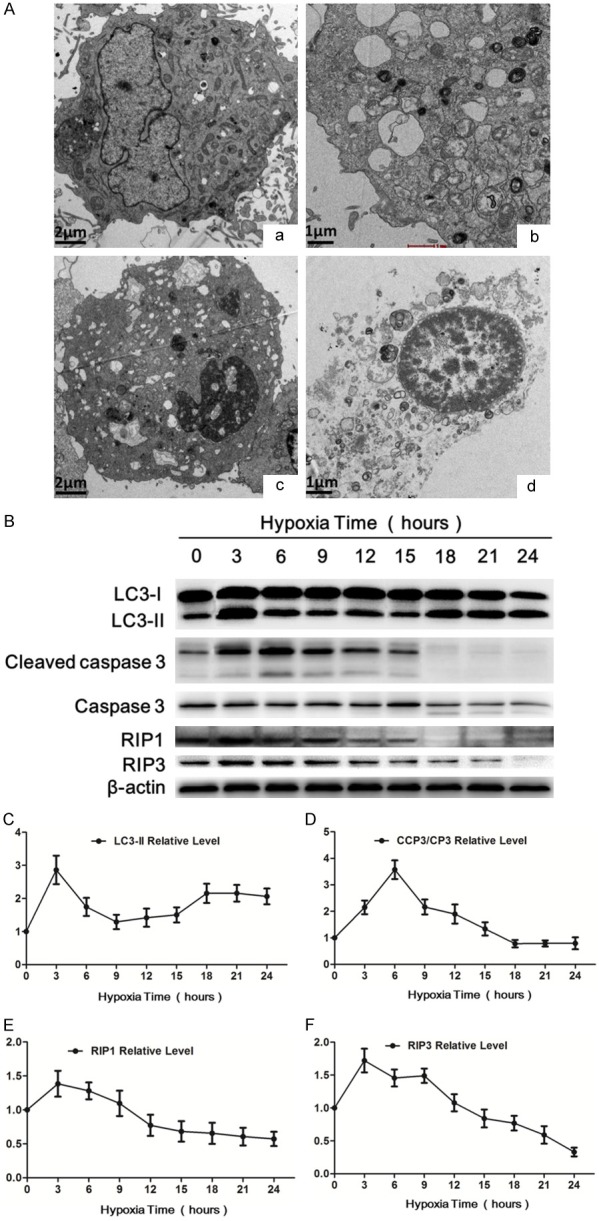
In vitro hypoxia treatment induced autophagy, apoptosis and necroptosis in H9c2 cells. (A) Transmission electron microscope (TEM) was used to observe cellular morphology changes. a. Control group showing the viable H9c2 cell morphology, (b-d) Hypoxia groups, (b) TEM detected autophagosomes in H9c2 cells after hypoxia for 3 h. (c) Apoptotic cell showing nuclear shrinkage and chromatin condensation was observed in H9c2 cells after hypoxia for 6 hours. (d) Necroptotic cell showing swollen nucleus, increased cell volume and disrupted cell membrane integrity with translucent cytoplasm was observed in H9c2 cell after hypoxia for 9 hours. (B) Western blot showing the LC3-I, LC3-II, cleaved caspase 3, caspase 3, RIP1, RIP3 protein levels in H9c2 cells after hypoxia for indicated hours. β-actin was used as the loading control. (C-F) Showing the quantitative analysis of the above protein levels after hypoxia for indicated hours, respectively. Data are expressed as mean ± SD, n=4.
HGF promotes autophagy in H9c2 cells under hypoxia
To explore the effect of HGF on autophagy in H9c2 cells under hypoxia, we used Ad-mRFP-GFP-LC3 adenovirus Infection. Only when autophagy occurs, mRFP and GFP tagged LC3 gathered together and formed fluorescence points which could be counted under fluorescence microscope. In merged images, red points represented autolysosome while yellow (merged by green and red points) represented autophagosome. As expected, both autophagosome and autolysosome were significantly increased in H9c2 cells after hypoxia. Moreover, the addition of HGF markedly promoted the formation of autophagosome and autolysosome in H9c2 cells under hypoxia. SU11274, inhibitor of HGF receptor, reversed the function of HGF on autophagy (Figure 4A). Because Ad-HGF was used in our study in vivo, we further investigated the impact of Ad-HGF infection with different multiplicity of infection (MOI) on the autophagy level in H9c2 cell under hypoxia. The appropriate MOI of Ad-HGF to infect the H9c2 cells had been explored and we chose the 12.5, 25, 50 MOI of Ad-HGF for the following experiments (Supplementary Figure 1). Ad-HGF (12.5, 25, 50 MOI) gradually increased the HGF and p-Met protein levels in H9c2 cells under hypoxia which could be blocked by SU11274. Our results demonstrated Ad-HGF worked well at the selected MOIs (Supplementary Figure 1). As expected, Ad-HGF (12.5, 25, 50 MOI) dose-dependently increased the LC3-II protein level in H9c2 cells under hypoxia, which was weakened by the SU11274 (10 μM) (Figure 4B). Beclin1 plays an important role in both autophagosome formation and autolysosome fusion [27]. Ad-HGF significantly increased Beclin-1 protein level in H9c2 cells under hypoxia in a dose-dependent fashion. Meanwhile, Ad-HGF markedly decreased p62 level by upregulating autophagy level. These impacts of Ad-HGF on autophagy could be reversed by SU11274 (Figure 4C). Heart failure is often accompanied by accumulation of damaged proteins and organelles, in the form of aggresomes [8]. Our study in vivo has confirmed Ad-HGF treatment could decrease the aggresomes accumulated in the cardiomyocytes after MI. Here, we showed the accumulation of aggresomes and p62 was markedly attenuated with Ad-HGF treatment (Figure 4D), which confirmed HGF promoted autophagy and reduced the accumulation of damaged proteins or organelles in H9c2 cells under hypoxia.
Figure 4.

HGF promotes autophagy in H9c2 cells under hypoxia. A. Confocal microscopy analysis of H9c2 cells transiently overexpressing mRFP-GFP-LC3. HGF (80 ng/mL), SU11274 (10 μM) or PBS was added into medium and hypoxia for 3 hours and normoxic group was shown as the control. The bar graph showed the statistical analysis of fluorescent points in H9c2 cells. B, C. Western blot analysis for LC3-I, LC3-II, Beclin-1 and p62/SQSTM1 protein levels in H9c2 cells after Ad-HGF (0, 12.5, 25, 50 MOI) overexpression or SU11274 (10 μM) treatment and hypoxia for 3 hours. The bar graph showed the quantitative analysis of the above protein levels. D. Fluorescent microscopy analysis of H9c2 cells staining with ProteoStat® aggresome detection reagent (red), p62/SQSTM1 (green) and DAPI (blue) in Control, Hypoxia, Hypoxia+Ad-HGF and Hypoxia+Ad-HGF+SU11274 groups. The bar chart showed the statistical analysis of cells with perinuclear p62/aggresomes relative level in the indicated groups. All results are expressed as mean ± SD, n=3, *P<0.05.
HGF inhibits apoptosis in H9c2 cells under hypoxia
Next, we assessed the impact of HGF on apoptosis in H9c2 cells under hypoxia. Hoechst staining of apoptosis showed hypoxia resulted in the significant increase of H9c2 cells apoptosis. Pre-infection of Ad-HGF markedly reduced the apoptosis percent of H9c2 cells under hypoxia, which could be blocked by SU11274 inhibitor (Figure 5A). The anti-apoptotic effect of HGF was further confirmed by the TUNEL assay (Supplementary Figure 2). Further western blot analysis revealed that Ad-HGF treatment significantly decreased the cleaved caspase 3/caspase 3 protein level in H9c2 cell under hypoxia in a dose-dependent manner (Figure 5B). To further explore the anti-apoptotic mechanism of HGF, we tested the pro-apoptotic Bax protein and the anti-apoptotic Bcl-2 and Bcl-xL protein levels. Ad-HGF overexpression significantly inhibited the rise of Bax protein and increased Bcl-2 and Bcl-xL protein levels in H9c2 cells. SU11274 could reverse the protective function of Ad-HGF on H9c2 cells under hypoxia insult (Figure 5C). These data confirmed HGF indeed inhibited apoptosis in H9c2 cells under hypoxia.
Figure 5.
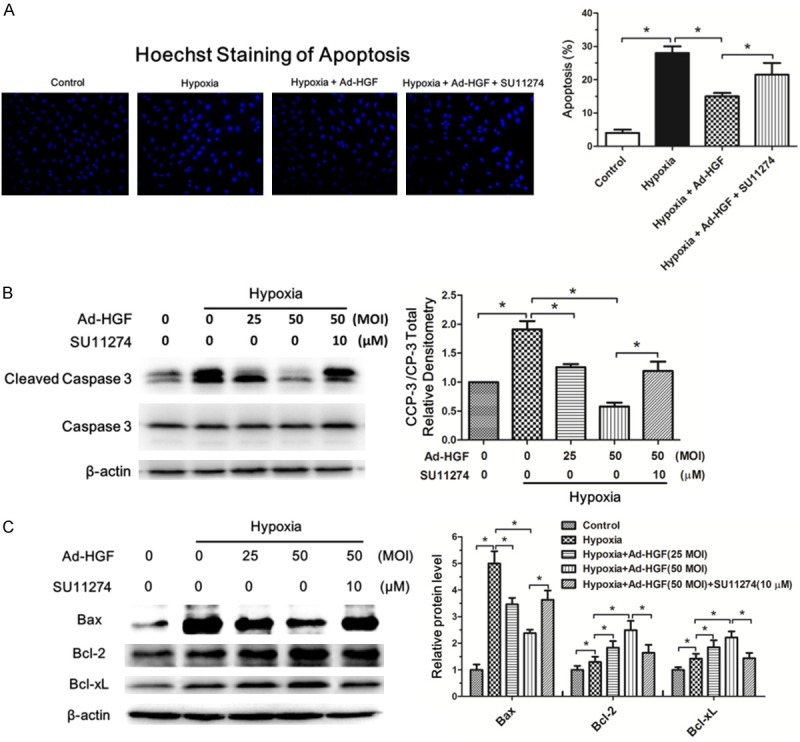
HGF inhibits apoptosis in H9c2 cells under hypoxia. A. Fluorescent microscopy analysis showed the Hoechst staining of apoptosis in H9c2 cells from the control, hypoxia, hypoxia+Ad-HGF and hypoxia+Ad-HGF+SU11274 groups, respectively. The bar chart described the statistical analysis of the percent of apoptotic cells. B, C. Western blot detection of cleaved caspase 3, caspase 3, Bax, Bcl-2 and Bcl-xL levels in H9c2 cells after the indicated treatment. β-actin is shown as a loading control. The bar graphs displayed the quantitative analysis of the protein levels. Data are expressed as mean ± SD, n=3, *P<0.05.
HGF promotes necroptosis in H9c2 cells under hypoxia
Necroptosis occurring in cardiac tissues of MI and H9c2 cells under hypoxia has been demonstrated in our foregoing study. Here, we mainly investigated the impact and mechanism of HGF on necroptosis in H9c2 cells under hypoxia. Propidium iodide (PI) staining has been widely used to mark necroptotic cells [7,24]. The flow cytometric analysis of PI staining showed HGF treatment significantly increased the percent of necroptotic cells under hypoxia, which was alleviated by c-Met receptor inhibitor, SU11274 (Figure 6A). Both the western blot and immunofluorescence analyses revealed the Ad-HGF treatment markedly enhanced the expressions of RIP1 and RIP3 proteins indicating the extent of necroptosis in H9c2 cell under hypoxia (Figure 6B and 6C). In addition, the immunofluorescence results showed RIP1 and RIP3 proteins displaying diffuse distribution but colocalization in the cells (Figure 6C). Previous studies have demonstrated RIP1/RIP3 complex, namely necrosome, plays a dominant role in mediating the downstream necroptosis process [17,28]. To verify the impact of HGF on the specific interaction between RIP1 and RIP3 in H9c2 cells under hypoxia, we used co-immunoprecipitation assay to detect the changes of RIP1/RIP3 complex in H9c2 cells. The RIP1/RIP3 complex was present in living H9c2 cells, but the amount of RIP1/RIP3 complex markedly increased by Ad-HGF intervention upon hypoxia induction, which was rescued by SU11274 (Figure 6D). MLKL has been reported to be the executioner of necroptosis signaling [14]. Our further study confirmed Ad-HGF overexpression dose-dependently enhanced MLKL protein level in H9c2 cells under hypoxia induction (Figure 6E). Knockdown of RIP3 or RIP1 in H9c2 cells decreased the MLKL protein level under hypoxia, but knockdown of MLKL has no impact on the protein level of RIP1 and RIP3 (Figure 6F-H). These findings implicated MLKL as a key mediator of necrosis signaling downstream of the RIP1/RIP3 complex, which is consistent with previous report [17]. Collectively, these results proved that HGF promotes necroptosis in H9c2 cells under hypoxia.
Figure 6.
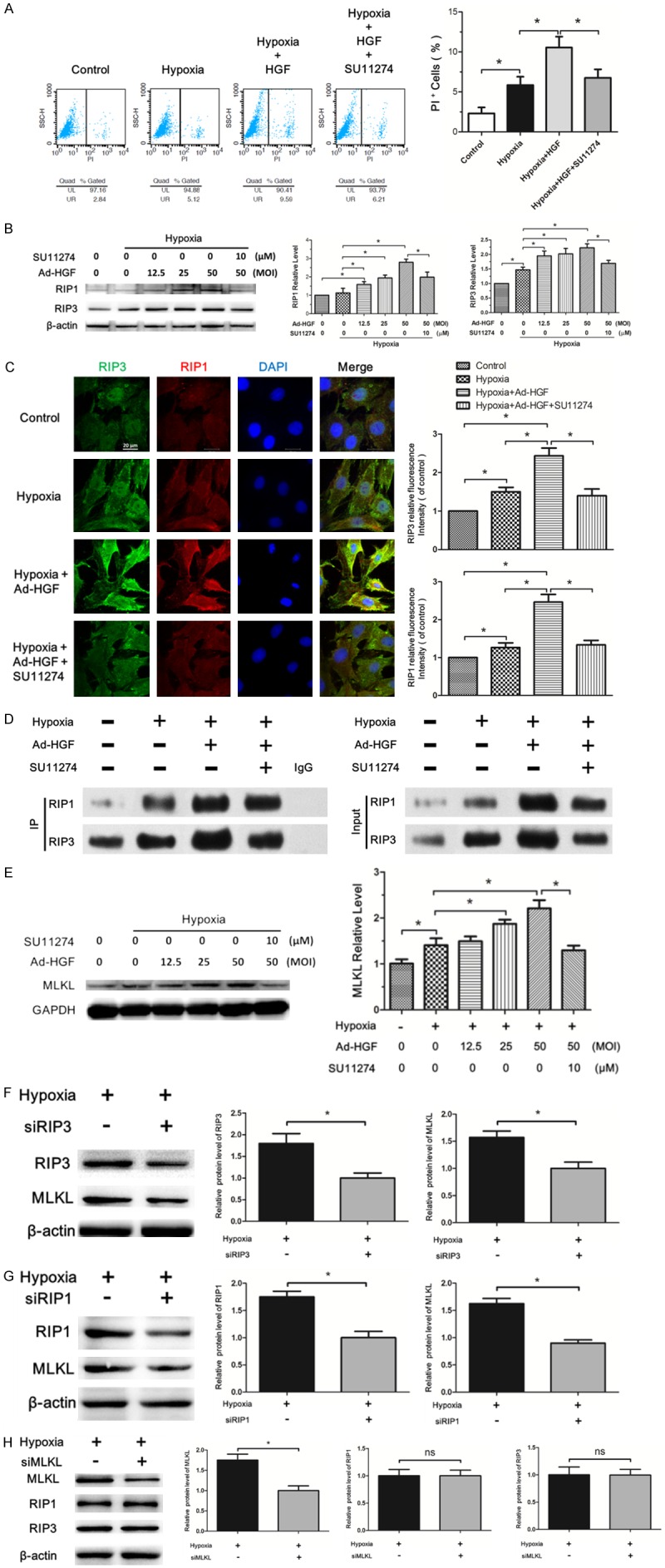
HGF promotes necroptosis in H9c2 cells under hypoxia. A. Flow cytometry analysis showing the percent of PI positive H9c2 cells under the indicated treatment. The statistical analysis of the result was described in the bar chat. B. Western blot analysis for the RIP1 and RIP3 protein level in H9c2 cells treated with Ad-HGF, SU11274, hypoxia or negative control as indicated. The bar graph displayed the statistical analysis of the above protein levels. C. Representative confocal images of RIP3 (green), RIP1 (red), and DAPI (blue) in H9c2 cells from the indicated groups. The pictures were captured using confocal fluorescence microscopy at uniform exposure. The bar chart described the relative fluorescence intensity of the staining proteins. D. The changes of RIP1-RIP3 complex in H9c2 cells after the indicated treatment, indicated by immunoprecipitation. E. Western blot for MLKL level in H9c2 cells under the indicated treatment. F-H. The lysates from H9c2 cells transfected with or without siRNA targeted RIP3, RIP1 or MLKL were analyzed by western blot with antibodies against RIP3, MLKL, RIP1 as indicated. The bar chart showed the statistical analysis of the according proteins. Data are expressed as mean ± SD, n=3, *P<0.05.
HGF regulated the interplay among autophagy, apoptosis and necroptosis
The above data confirmed HGF upregulated autophagy and necroptosis and inhibited apoptosis in H9c2 cells under hypoxia. Here, we further addressed how HGF regulated the interplay among autophagy, apoptosis and necroptosis. We first evaluated whether HGF affects interaction between Bcl-2 and Beclin1, or Bax using co-immunoprecipitation assays in H9c2 cells. Interestingly, Ad-HGF treatment significantly decreased binding of Bcl-2 to Beclin1 but enhanced Bcl-2 binding to Bax in H9c2 cells under hypoxia (Figure 7A and 7B).
Figure 7.
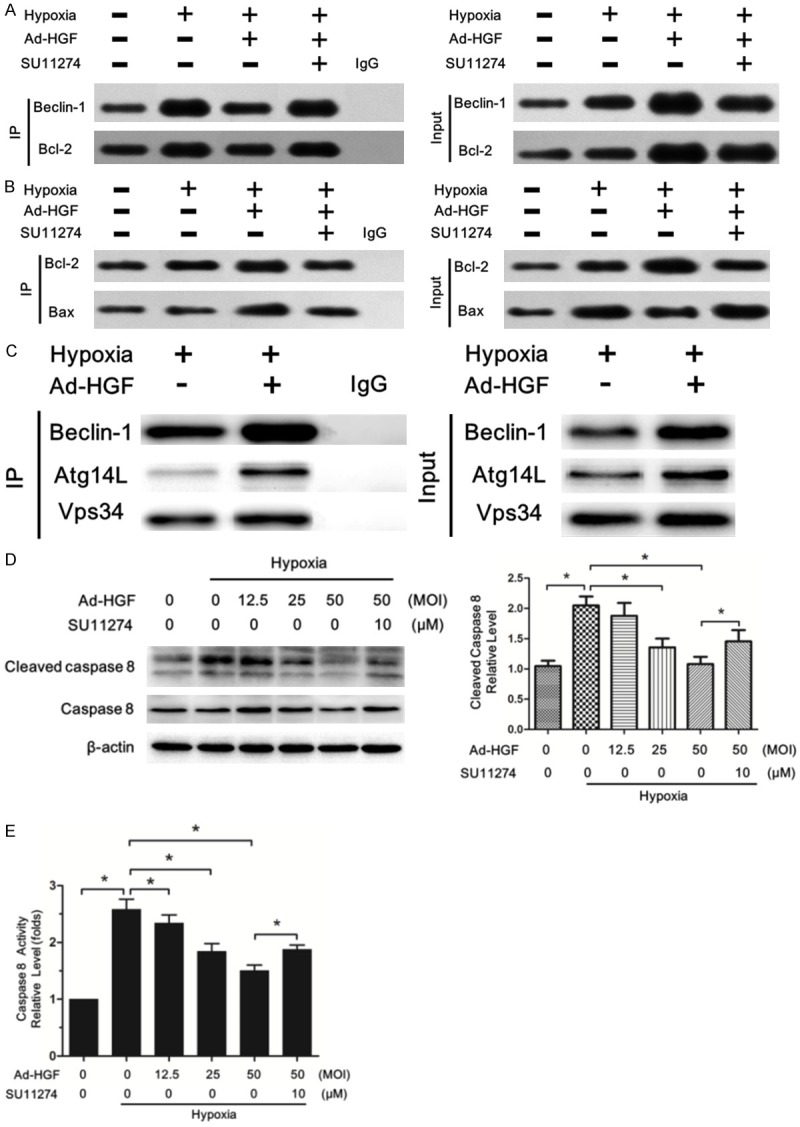
HGF regulated the interplay among autophagy, apoptosis and necroptosis. A. Cells were treated with or without SU11274 under hypoxia for six hours after thirty-six hours’ transfection with or without Ad-HGF. Lysates were extracted for immunoprecipitation to evaluate the interaction between Beclin-1 and Becl-2. Representative images are shown. B. The interaction between Bcl-2 and Bax in H9c2 cells after the indicated treatment was analysed using immunoprecipitation. The input whole-cell lysates were probed with Bcl-2 and Bax antibodies. C. Interaction among endogenous Beclin1 and Atg14L and exogenously expressed Flag-Vps34 in H9c2 cells after the indicated treatment was detected by immunoprecipitation. D. Western blot analysis of cleaved caspase 8 and caspase 8 level in H9c2 cells was shown. The bar chart manifested the statistical analysis of the protein levels. E. Ad-HGF treatment decreased the caspase 8 activity in H9c2 cells under hypoxia. Data are expressed as mean ± SD, n=3, *P<0.05.
These data suggested HGF treatment induced the dissociation of Beclin-1 from the Beclin-1/Bcl-2 complex, which may offer the chance for beclin-1 protein to be active in promoting autophagy. Furthermore, HGF-induced sequestration of Bax by Bcl-2 allows Bax to become inactive, thereby inhibiting apoptosis. The Beclin1-Vps34-Atg14L complex plays an important role in promoting the autophagosome formation [29]. We further used co-immunoprecipitation assay to examine whether Beclin1 dissociating from Beclin-1/Bcl-2 complex induced by HGF joined the formation of Beclin1-Vps34-Atg14L complex. As expected, Ad-HGF treatment significantly increased the formation of Beclin1-Vps34-Atg14L complex in H9c2 cells under hypoxia (Figure 7C), which accounted for promoting autophagy.
Active caspase-8 induces apoptosis and inhibits necroptosis by cleaving RIP1/RIP3 complex. When caspase-8 is inhibited, the amyloid RIP1/RIP3 necrosome is formed and necroptosis ensued [17,30]. We next analyzed whether HGF regulated caspase 8, thereby affecting apoptosis and necroptosis. Hypoxia markedly increased the cleaved caspase 8 protein level in H9c2 cells which was significantly decreased by Ad-HGF treatment in a dose-dependent fashion (Figure 7D). The protease activity test also demonstrated Ad-HGF reduced the caspase 8 activity in H9c2 cells under hypoxia (Figure 7E). Thus, Ad-HGF treatment weakened caspase 8 activity in H9c2 cells under hypoxia, on the one hand, inhibiting apoptosis and on the other hand, promoting necroptosis.
To elucidate the overall effect of Ad-HGF on H9c2 cells under hypoxia, we used CCK-8 assay to assess the cell activity after Ad-HGF treatment. Interestingly, Ad-HGF significantly improved cell vitality under the hypoxia injury (Supplementary Figure 3A), which may be due to the pro-survival properties of HGF on the whole.
Our study in vivo and in vitro has confirmed Ad-HGF promoted autophagy and necroptosis and inhibited apoptosis in cardiac tissues and H9c2 cells under hypoxia. But the effect of these results on the normal cells is still unknown. We therefore investigated the effect of the supernatant from H9c2 cell under hypoxia after Ad-HGF treatment on the vitality of H9c2 cells in normoxic culture. Surprisingly, we found the supernatant from H9c2 cells of Ad-HGF treatment under hypoxia dramatically increased the vitality of H9c2 cells in normoxic culture for 24, 48 and 72 hours. In addition, the supernatant from H9c2 cells under hypoxia alone also enhanced the vitality of H9c2 cells in normoxic culture for 24 hours than the control, but showed no significant difference between them in later 48 or 72 hours’ incubation (Supplementary Figure 3B-D). In summary, we confirmed supernatant from H9c2 cells of Ad-HGF treatment under hypoxia improved the cell vitality in normoxic culture, which may indirectly reflect the effect of Ad-HGF on the normal myocardial tissue around the infarct zones. And the beneficial substances existed in the supernatant from cells of Ad-HGF treatment under hypoxia warrants further investigation.
Discussion
We firstly showed that the distribution of autophagy, apoptosis and necroptosis differs in temporal and spatial context after MI. The main findings of our study were that: (1) Ad-HGF treatment improves the cardiac remodeling of SD rats following MI by upregulating autophagy and necroptosis and inhibiting apoptosis, (2) which is associated with the regulation of the interplay among autophagy, apoptosis and necroptosis.
Adverse remodeling after MI is a big challenge worldwide. Although substantial progress has been achieved in lowering acute mortality rates after AMI, much of the ongoing morbidity and mortality relates to chronic adverse remodeling that occur after infarction [2]. And the numbers of cell death in MI is the most critical determinant of subsequent left ventricular remodeling and heart failure [31]. For many years, apoptosis was considered as the only form of regulated cell death and studies investigating MI mainly focused on apoptosis [3,4]. Recent years, autophagy and necroptosis have been found to be another two regulated cell death styles existing in various diseases including MI [5-7]. Recent few studies have focused on autophagy or necroptosis in MI [7,32]; however, whether the whole of three cell death styles in MI affect subsequent cardiac remodeling process is still largely unknown.
In order to tackle these issues, we firstly investigated the distribution of autophagy, apoptosis and necroptosis in cardiac tissues after MI. Interestingly, we find autophagy, apoptosis and necroptosis differs in temporal and spatial context after MI, although all of them significantly increased after MI. In 7 days’ heart of MI, LC3-II staining was mainly concentrated in the infarcted area, which indicating more autophagy occurred in the infarct zones than the peri-infarct zones. Inversely, in 28 days’ heart autophagy in peri-infarct zones was markedly higher than the infarct zones, but both of which were lower than the 7 days’ heart with MI. However, apoptosis mainly appeared in the peri-infarct zones rather than the infarct zones in 7 days’ and 28 days’ heart after MI. When compared with the 7 days’ heart of MI, 28 days’ heart showed lower apoptosis level. No difference of apoptosis level was observed in the non-infarcted areas of the three investigated groups. Necroptosis was evaluated using PI labeling by intraperitoneal injection of PI one hour before termination [25]. Intriguingly, we found much more necroptosis appeared in the infarct zones than the other areas in both 7 days’ and 28 days’ hearts of MI. Furthermore, necroptosis in the infarct zones of the 28 days’ heart markedly declined as compared with the 7 days’ heart, but showed no difference in the non-infarcted areas of the three groups. These results offer us new evidence about the distribution of autophagy, apoptosis and necroptosis after MI, regulation of which affecting the cardiac remodeling need our further research.
As a secreted cytokine, HGF plays a protective role in MI [19]. But how HGF regulate autophagy, apoptosis and necroptosis and affect cardiac remodeling after MI has not been reported. HGF level enhances at the onset of MI, but declining after twenty four hours and returns to normal three days later [33]. In order to increase HGF expression level in the infarcted myocardium, meanwhile extending its expression time, we performed our study using adenovirus vector carrying HGF (Ad-HGF), which with CMV promoter could achieve summit expression in myocardial tissue on the first day post-injection and can be maintained for at least four weeks [21]. Our results confirmed Ad-HGF treatment significantly improving the cardiac remodeling by increasing the HGF and p-Met expression level in the cardiac peri-infarct zones of SD rat. Several observations were particularly noteworthy. Firstly, we found that Ad-HGF improved cardiac function including preserving LVEF and LVFS 28 days after MI. Secondly, cardiac fibrosis was attenuated with Ad-HGF treatment. Thirdly, the accumulation of aggresomes indicating damaged organelles and protein aggregates in chronic heart failure was markedly reduced after Ad-HGF intervention. Taken together, these data demonstrate that Ad-HGF improves cardiac remodeling after MI in rat, culminating in better preserved cardiac structure and function. To determine how Ad-HGF regulates autophagy, apoptosis and necroptosis after MI, the changes of accordingly representative proteins including autophagy markers, Beclin-1, LC3-II, P62, apoptotic markers, cleaved caspase 3, caspase 3, necroptotic markers, RIP1 and RIP3 were interrogated. The results confirmed Ad-HGF promoted autophagy and necroptosis and inhibited apoptosis after MI.
To validate the phenomenon observed in vivo and investigate the underlying mechanisms, we further perform the study in vitro. Model of ischemic hypoxemia was used to mimic in vivo myocardial infarction [34] and rat myocardial cell line, H9c2 cell was employed for later research. Using TEM method, we observed the typical cell morphological characteristics of autophagy, apoptosis and necroptosis in H9c2 cells under hypoxia, respectively. It’s worth noting that as early as 3 hours after hypoxia, autophagosomes were mainly observed, but apoptotic cell showing nuclear shrinkage and chromatin condensation was observed after hypoxia for 6 hours. Necroptotic cell showing swollen nucleus, increased cell volume and disrupted cell membrane integrity with translucent cytoplasm was observed in H9c2 cell after hypoxia for 9 hours. These results indicate the threshold value of hypoxia injury for the occurrence of autophagy, apoptosis or necroptosis differs. Autophagy is the most sensitive to hypoxia, but necroptosis the least. This may be partly responsible for their different distributions of autophagy, apoptosis and necroptosis in the heart after MI. The related proteins of autophagy, apoptosis or necroptosis have changed after hypoxia for 3 hours, the rational explanation of which is that signaling pathways go first, then the results.
Autophagy is generally defined as a cellular program that ensures survival under conditions of stress. The ability of autophagy to clear damaged or denatured subcellular constituents such as aggregated protein as well as to maintain mitochondrial homeostasis appears to play important roles in the cytoprotective and homeostatic functions of autophagy [35]. In study in vitro, we confirmed Ad-HGF dose-dependently promoted autophagy in H9c2 cells under hypoxia. Ad-HGF treatment not only increased the number of autolysosome and autophagosome, raised the pro-autophagic proteins including LC3-II, Beclin-1, but also decreased the accumulation of damaged proteins and organelles, in the form of aggresomes. SU11274, a specific inhibitor of HGF’s c-Met receptor, effectively block the impact of Ad-HGF on autophagy. These results are consistent with our findings in vivo. Upregulation of autophagy during myocardial stress is generally compensatory, alleviating energy loss and scavenging damaged mitochondria and protein aggregates [8], which is beneficial for the cardiac remodeling process after MI. One study reported agonist antibodies could activate the c-Met receptor and protect cardiomyocytes from cobalt chloride-induced autophagy [36]. In this study, cobalt chloride was employed to imitate cell chronic hypoxia, but we used serum-glucose free medium and anaerobic jar to mimic hypoxia for H9c2 cells. This may explain the conflict of the results.
Apoptosis, a parameter linked to fibrosis, occurs in nearly all models of cardiac remodeling [37]. Experimental models have substantiated the sufficiency of clinically relevant degrees of apoptosis to induce heart failure, as well as the necessity of myocyte apoptosis for the development of heart failure [38]. The anti-apoptotic effect of Ad-HGF on H9c2 cells under hypoxia was further demonstrated in our in vitro study, which was in accordance with our study in vivo and previous reports [39,40]. Ad-HGF treatment markedly decreased the percent of apoptotic cells under hypoxia insult. Further research revealed that Ad-HGF enhanced the anti-apoptotic protein levels including Bcl-2 and Bcl-xL, meanwhile reducing the pro-apoptotic Bax level. These results highlight the therapeutic effects of Ad-HGF on cardiac remodeling after MI.
Necroptosis, a regulated form of necrosis, plays a vital role in development, tissue homeostasis, and disease pathogenesis [41]. To date, few studies have reported the relationship between HGF and necroptosis in MI. Our study, for the first time, confirmed HGF intervention increased the percentage of necroptotic H9c2 cells under hypoxia. Previous studies have proved necroptosis is triggered by the interaction of RIP1 with RIP3, a serine/threonine kinase, resulting in the formation of necrosome [15,16]. Our further results of both western blot and immunofluorescence confirmed Ad-HGF increased the RIP1 and RIP3 protein levels in H9c2 cell under hypoxia injury. In addition, the immunofluorescence also revealed RIP1 and RIP3 proteins displaying diffuse distribution but colocalization in the cells, which was validated by the co-immunoprecipitation that Ad-HGF indeed promoted the interaction between RIP1 and RIP3 proteins by enhancing RIP1-RIP3 necrosome level. As the executor of necroptosis, MLKL activation results in its recruitment and oligomerization at phosphatidylinositol phosphates in the plasma membrane leading to plasma membrane rupture and cell death [42]. As expect, MLKL protein levels markedly increased in H9c2 cell after Ad-HGF treatment under hypoxia. Moreover, either knockdown of RIP1 or RIP3 by specific siRNA interference assay in H9c2 cells decreased the MLKL protein levels; however, knockdown of MLKL had no impact on RIP1 and RIP3 protein levels. These findings indicated MLKL as a key mediator of necroptosis signaling downstream of the kinase RIP1 and RIP3, which is consistent with previous report [17]. Collectively, we confirmed Ad-HGF promoted necroptosis in H9c2 cells under hypoxia, which is in accordance with what we observed in vivo.
Being different from apoptosis and autophagy, the occurrence of necroptosis involves release of the dying cell’s intracellular components, and the effects of this release on neighboring cells can yield inflammation [38,42]. Inflammation is a multicellular process that destroys pathogens and promotes recovery from injury through a variety of cooperating changes, including cell death [43]. However, Mark Luedde and colleagues reported that RIP3-dependent necroptosis modulates post-ischaemic adverse remodeling in a mouse model of MI. In their study, mice deficient for RIP3 showed a significantly better ejection fraction and less hypertrophy in magnetic resonance imaging studies thirty days after MI with LAD ligation method [7]. The rational explanation is that the protective effect of HGF on cardiac remodeling after MI may come from the whole impact of HGF on autophagy, apoptosis and necroptosis, not merely necroptosis. This explanation was further supported by our observation that supernatant from H9c2 cells of Ad-HGF treatment under hypoxia significantly improved the cell vitality in normoxic culture, which may indirectly reflect the effect of Ad-HGF on the normal myocardial tissue around the infarct zones. But the beneficial substances existed in the supernatant from cells of Ad-HGF treatment under hypoxia warrants further investigation. Furthermore, we also observed Ad-HGF treatment increased the HMGB1 protein level in myocardial tissue and serum in vivo (data not shown). As an indicator of necroptosis, HMGB1 was released from necroptotic cells, but not from autophagic or apoptotic cells [14]. Studies report HMGB1 induces myocardial regeneration after infarction via enhanced cardiac c-kit cell proliferation and differentiation [44] and improves cardiac remodeling after MI [45,46]. These reports suggest the protective aspect of necroptosis on post-MI cardiac remodeling. Further studies are needed to clarify it.
We have demonstrated Ad-HGF intervention promote autophagy and necroptosis and inhibit apoptosis in H9c2 cell under hypoxia. But how Ad-HGF regulates the interplay among autophagy, apoptosis and necroptosis need our further research. Our co-immunoprecipitation assays showed Ad-HGF treatment significantly decreased the binding of Bcl-2 to Beclin1 but enhanced Bcl-2 binding to Bax in H9c2 cells under hypoxia. These data indicate HGF induced the dissociation of Beclin-1 from the Beclin-1/Bcl-2 complex, which may offer the chance for Beclin-1 protein to be active in promoting autophagy. Moreover, HGF-induced sequestration of Bax by Bcl-2 allows Bax to become inactive, thereby inhibiting apoptosis. Studies reported the Beclin1-Vps34-Atg14L complex plays an important role in promoting autophagy by increasing the autophagosome formation [8,29]. We further used co-immunoprecipitation assay to examine whether Beclin1 dissociating from Beclin-1/Bcl-2 complex induced by HGF joined the formation of Beclin1-Vps34-Atg14L complex. As expected, Ad-HGF treatment significantly increased the formation of Beclin1-Vps34-Atg14L complex in H9c2 cells under hypoxia, which accounted for promoting autophagy. Caspase-8 not only activates downstream caspases to bring about apoptosis, but also cleaves RIP1 and RIP3 abrogating their ability to signal necroptosis [17]. Both the western blot and caspase 8 activity assays showed Ad-HGF significantly decreased the caspase 8 protein and activity levels in a dose-dependent fashion, which obligated the cell to undergo necroptosis under hypoxia and block apoptosis.
In conclusion, the current study shows the distribution of autophagy, apoptosis and necroptosis differs in temporal and spatial context after MI. Ad-HGF treatment improves the cardiac remodeling of SD rats following MI by upregulating autophagy and necroptosis and inhibiting apoptosis, which is associated with the regulation of the interplay among them. Thus, our findings offer new evidence and strategies for the treatment of MI and post-MI cardiac remodeling.
Acknowledgements
This work has been funded by the National Natural Science Foundation of China (No. 81441011 & No. 81170102/H0203), the Chinese Medical Association of the Sunlight Foundation (SCRFCMDA201217), the Fourth Period Project “333” of Jiangsu Province (BRA2012207), China, Supporting program of Science and Technology of Jiangsu (Social Development, BK2010021), Jiangsu provincial science and Technology Department basic research program (Natural Science Foundation of China BK20141020) and the Graduate Innovation Foundation of Jiangsu Province (JX22013341). We are indebted to Dr. Di Yang, Dr. Xiangjian Chen, Dr. Fang Wang and Dr. Huiwen Zhang (The First Affiliated Hospital of Nanjing Medical University) for help with skillful technical assistance.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Tao L, Shen S, Fu S, Fang H, Wang X, Das S, Sluijter JP, Rosenzweig A, Zhou Y, Kong X, Xiao J, Li X. Traditional Chinese Medication Qiliqiangxin attenuates cardiac remodeling after acute myocardial infarction in mice. Sci Rep. 2015;5:8374. doi: 10.1038/srep08374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottlieb RA. Mitochondrial signaling in apoptosis: mitochondrial daggers to the breaking heart. Basic Res Cardiol. 2003;98:242–249. doi: 10.1007/s00395-003-0404-0. [DOI] [PubMed] [Google Scholar]
- 4.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 5.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 7.Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, Hippe HJ, Linkermann A, Wolf MJ, Rose-John S, Lullmann-Rauch R, Adam D, Flogel U, Heikenwalder M, Luedde T, Frey N. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103:206–216. doi: 10.1093/cvr/cvu146. [DOI] [PubMed] [Google Scholar]
- 8.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 10.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 12.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol. 2011;57:9–17. doi: 10.1016/j.jacc.2010.08.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Wang X. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem Sci. 2014;39:587–593. doi: 10.1016/j.tibs.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 16.Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci U S A. 2010;107:21695–21700. doi: 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 18.Sala V, Crepaldi T. Novel therapy for myocardial infarction: can HGF/Met be beneficial? Cell Mol Life Sci. 2011;68:1703–1717. doi: 10.1007/s00018-011-0633-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deuse T, Peter C, Fedak PW, Doyle T, Reichenspurner H, Zimmermann WH, Eschenhagen T, Stein W, Wu JC, Robbins RC, Schrepfer S. Hepatocyte growth factor or vascular endothelial growth factor gene transfer maximizes mesenchymal stem cell-based myocardial salvage after acute myocardial infarction. Circulation. 2009;120:S247–254. doi: 10.1161/CIRCULATIONAHA.108.843680. [DOI] [PubMed] [Google Scholar]
- 20.Ellison GM, Torella D, Dellegrottaglie S, Perez-Martinez C, Perez de Prado A, Vicinanza C, Purushothaman S, Galuppo V, Iaconetti C, Waring CD, Smith A, Torella M, Cuellas Ramon C, Gonzalo-Orden JM, Agosti V, Indolfi C, Galinanes M, Fernandez-Vazquez F, Nadal-Ginard B. Endogenous cardiac stem cell activation by insulin-like growth factor-1/hepatocyte growth factor intracoronary injection fosters survival and regeneration of the infarcted pig heart. J Am Coll Cardiol. 2011;58:977–986. doi: 10.1016/j.jacc.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Xu Z, Tao Z, Yang Y, Wang H, Wang L, Wu Z, Tan Q, Zhou N, Zhang M, Chen P, Yang Z. Cardiac-specific expression of the hepatocyte growth factor (HGF) under the control of a TnIc promoter confers a heart protective effect after myocardial infarction (MI) Curr Gene Ther. 2014;14:63–73. doi: 10.2174/1566523214666140207104209. [DOI] [PubMed] [Google Scholar]
- 22.Chen B, Tao Z, Zhao Y, Chen H, Yong Y, Liu X, Wang H, Wu Z, Yang Z, Yuan L. Catheter-based intramyocardial delivery (NavX) of adenovirus achieves safe and accurate gene transfer in pigs. PLoS One. 2013;8:e53007. doi: 10.1371/journal.pone.0053007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou N, Fu Y, Wang Y, Chen P, Meng H, Guo S, Zhang M, Yang Z, Ge Y. p27 kip1 haplo-insufficiency improves cardiac function in early-stages of myocardial infarction by protecting myocardium and increasing angiogenesis by promoting IKK activation. Sci Rep. 2014;4:5978. doi: 10.1038/srep05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, Sluijter JP. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107:270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 26.Houtgraaf JH, de Jong R, Kazemi K, de Groot D, van der Spoel TI, Arslan F, Hoefer I, Pasterkamp G, Itescu S, Zijlstra F, Geleijnse ML, Serruys PW, Duckers HJ. Intracoronary infusion of allogeneic mesenchymal precursor cells directly after experimental acute myocardial infarction reduces infarct size, abrogates adverse remodeling, and improves cardiac function. Circ Res. 2013;113:153–166. doi: 10.1161/CIRCRESAHA.112.300730. [DOI] [PubMed] [Google Scholar]
- 27.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Ma B, Cao W, Li W, Gao C, Qi Z, Zhao Y, Du J, Xue H, Peng J, Wen J, Chen H, Ning Y, Huang L, Zhang H, Gao X, Yu L, Chen YG. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014;24:912–924. doi: 10.1038/cr.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takemura G, Fujiwara H. Role of apoptosis in remodeling after myocardial infarction. Pharmacol Ther. 2004;104:1–16. doi: 10.1016/j.pharmthera.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 32.Lin C, Liu Z, Lu Y, Yao Y, Zhang Y, Ma Z, Kuai M, Sun X, Sun S, Jing Y, Yu L, Li Y, Zhang Q, Bian H. Cardioprotective effect of Salvianolic acid B on acute myocardial infarction by promoting autophagy and neovascularization and inhibiting apoptosis. J Pharm Pharmacol. 2016;68:941–952. doi: 10.1111/jphp.12567. [DOI] [PubMed] [Google Scholar]
- 33.Konopka A, Janas J, Piotrowski W, Stepinska J. Hepatocyte growth factor--a new marker for prognosis in acute coronary syndrome. Growth Factors. 2010;28:75–81. doi: 10.3109/08977190903403984. [DOI] [PubMed] [Google Scholar]
- 34.Li R, Geng HH, Xiao J, Qin XT, Wang F, Xing JH, Xia YF, Mao Y, Liang JW, Ji XP. miR-7a/b attenuates post-myocardial infarction remodeling and protects H9c2 cardiomyoblast against hypoxia-induced apoptosis involving Sp1 and PARP-1. Sci Rep. 2016;6:29082. doi: 10.1038/srep29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol. 2014;2014:502676. doi: 10.1155/2014/502676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallo S, Gatti S, Sala V, Albano R, Costelli P, Casanova E, Comoglio PM, Crepaldi T. Agonist antibodies activating the Met receptor protect cardiomyoblasts from cobalt chloride-induced apoptosis and autophagy. Cell Death Dis. 2014;5:e1185. doi: 10.1038/cddis.2014.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 38.Moe GW, Marin-Garcia J. Role of cell death in the progression of heart failure. Heart Fail Rev. 2016;21:157–167. doi: 10.1007/s10741-016-9532-0. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Liu J, Tao Z, Wu P, Cheng W, Du Y, Zhou N, Ge Y, Yang Z. Exogenous HGF Prevents Cardiomyocytes from Apoptosis after Hypoxia via Up-Regulating Cell Autophagy. Cell Physiol Biochem. 2016;38:2401–2413. doi: 10.1159/000445592. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura T, Mizuno S, Matsumoto K, Sawa Y, Matsuda H. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J Clin Invest. 2000;106:1511–1519. doi: 10.1172/JCI10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, Chen Y, Doan J, Murray J, Molkentin JD, Liu Q. Transforming growth factor beta-activated kinase 1 signaling pathway critically regulates myocardial survival and remodeling. Circulation. 2014;130:2162–2172. doi: 10.1161/CIRCULATIONAHA.114.011195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJ, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016;26:721–32. doi: 10.1016/j.tcb.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol. 2014;14:51–59. doi: 10.1038/nri3561. [DOI] [PubMed] [Google Scholar]
- 44.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, Leoni O, Palumbo R, Battistini L, Rastaldo R, Muller S, Pompilio G, Anversa P, Bianchi ME, Capogrossi MC. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:e73–83. doi: 10.1161/01.RES.0000186276.06104.04. [DOI] [PubMed] [Google Scholar]
- 45.Kitahara T, Takeishi Y, Harada M, Niizeki T, Suzuki S, Sasaki T, Ishino M, Bilim O, Nakajima O, Kubota I. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008;80:40–46. doi: 10.1093/cvr/cvn163. [DOI] [PubMed] [Google Scholar]
- 46.Kohno T, Anzai T, Naito K, Miyasho T, Okamoto M, Yokota H, Yamada S, Maekawa Y, Takahashi T, Yoshikawa T, Ishizaka A, Ogawa S. Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovasc Res. 2009;81:565–573. doi: 10.1093/cvr/cvn291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.