

Abstract
Background: GBM represents the most aggressive type of glioma which is featured by extremely aggressive invasion and destructive malignancy with a high proliferation rate. The aim of this study was to investigate the in vitro anti-tumor effect of icaritin in human GBM cell line U87. Methods: The effect of icaritin on In vitro cell viability was determined by MTT assay and colony formation assay. The inducing effect of icaritin on cell cycle arrest, mitochondrial membrane potential loss, apoptosis, autophagy and intracellular ROS generation was assessed by flow cytometry. The apoptotic cell death was also confirmed by TUNEL assay. The expression levels of target or marker molecules were examined by western blot. The activity of caspase-3, -8 and -9 was detected with ELISA kit. Results: Our results showed that icaritin significantly induced both caspase-dependent apoptosis and autophagy in human GBM cell line U87. Additionally, our findings revealed that icaritin exerted anti-tumor effect by modulating Stat3 through generating ROS and subsequent activation of AMPK and inhibition of mTOR. Further investigation also showed that icaritin-induced autophagy served as a pro-death function and possibly contributed to icaritin-induced apoptosis. Conclusion: Icaritin potently inhibit the cell growth of human GBM cell line U87 through inducing both caspase-dependent apoptosis and autophagy. Base on our findings, icaritin can be considered as a promising candidate therapeutic agent for treatment of GBM, though further studies are needed.
Keywords: Icaritin, GBM, apoptosis, autophagy, Stat3
Introduction
Gliomas include astrocytoma, oligodendroglioma, ependymoma, mixed glioma [1].
The most common primary malignant tumors in the central nervous system are malignant gliomas, which include anaplastic astrocytoma (AA, WHO grade III), anaplastic oligodendroglioma (AO, WHO grade III) and glioblastoma multiforme (GBM, WHO grade IV astrocytoma). GBM, accounting for the majority of malignant gliomas, represents the most aggressive type of glioma which is featured by extremely aggressive invasion and destructive malignancy with a high proliferation rate [2,3]. Despite great advances in surgical techniques, radiotherapy strategies and chemotherapy modalities recently, the median survival is only 12 to 15 months for patients with GBM [4,5]. These dismal outcomes render GBM urgent subjects of cancer research and more aggressive therapeutic strategy should be developed to improve prognosis.
Cell death can occur through different ways including apoptosis, autophagy, necrosis, cornification and tentative definitions of atypical cell death modalities [6]. Compared with apoptosis, which is categorized as the type-I programmed cell death (PCD) and serves as the main mechanisms of action for most chemotherapeutic agents against human malignancies [7], autophagy that represents caspase-independent cell death pathway is regarded as type-II PCD [8]. Autophagy (also referred to as macroautophagy) is a fundamental catabolic process by which cellular materials including cellular proteins, cytoplasm, organelles and even the damaged cell in its entirety are encapsulated within vesicles and degraded by lysosomes for for energy production or stress elimination [9]. In cancer cells, autophagy can serves as a cell survival pathway, because cancer cells can utilize autophagy pathway to maintain homeostasis in starvation and stress through suppressing the accumulation of harmful proteins and chromosomal instability [10]. Moreover, cancer cells present with deficient autophagy and acquired tolerance to cell death, which results in cytokine production, inflammatory response and genome damage, ultimately leading to tumorigenesis [11]. However, autophagy can also produce tumor-suppressive effect by inducing autophagic cell death, either in collaboration with apoptosis or as a back-up mechanism in case of the former is defective [12,13]. A lot of efforts studies have made to reveal the role of autophagy in tumor development and progression [14,15].
Icaritin, a hydrolytic product of icariin, is one of the main active ingredients of a traditional Chinese herbal medicine Epimedium Genus. A large body of evidence have revealed the pharmacological and biological activities of icaritin, including induction of differentiation of embryonic stem cells into cardiomyocytes [16], immunomodulatory effect against systemic lupus erythematosus [17], cardiopreotective effect against myocardial ischemia and reperfusion injury [18], anti-atherosclerotic effect [19], hepatoprotective effect [20], neuroprotective effect against β-amyloid-induced neurotoxicity [21], and osteoprotective effect [22]. In addition, icaritin has been proved to be a potent anti-cancer agent in both solid tumor and hematological malignancies [23-25]. Therefore, it is reasonable to postulate that icaritin can exert inhibitory effect on the growth of GBM cells. More importantly, it has been found that icaritin can pass the blood brain barrier, which makes it an ideal candidate drug for GBM treatment [26]. In this study, our results revealed that icaritin induces apoptosis and autophagy in GBM cell line U87. Moreover, this present study provided experimental evidence that icaritin exerted anti-proliferative effect by modulating Sp1 though generating ROS and regulating AMPK/mTOR signaling.
Materials and methods
Cell culture
The GBM cell line U87 was obtained from Cell Bank of Shanghai Institute of Biotechnology and Cell Biology (Shanghai, China) and maintained at a density of 1×105 cells in DMEM medium (Gibco, Grand Island, NY) containing 10% FBS (Hyclone, Logan, UT), 100 IU/mL penicillin, and 100 mg/mL streptomycin (Sigma-Aldrich, St. Lious, MO). The cells were kept in a CO2 incubator at 37°C and the culture condition was maintained as 90% humidity and 5% CO2.
Cell viability assay
U87 cells were plated at a density of 5.0×103 cells/well in 96-well culture plates for 24 hours before challenged by different concentrations of icaritin for 24 and 48 hours. The cell viability s was determined by using a CellTiter96® Aqueous One Cell Proliferation Assay Kit (Promega, Madison, WI) according to the instructions of the manufacturer. The relative cell viability was presented after normalized to untreated cells.
Cell apoptosis assay
Cell apoptosis was assessed using FITC Annexin V Apoptosis Detection Kit (BD Biosciences Pharmingen, San Diego, CA) according to manufacturer’s instructions. Briefly, U87 cells were treated with various concentrations of icaritin for 48 hours, collected and washed with ice-cold PBS, and then suspended in 500 μl of annexin V binding buffer. 100 μl aliquot was taken, 2 μl of annexin V-FITC and 2 μl of PI were added, and the mixture was incubated for 5 minutes at room temperature in the dark. After the addition of 400 μl of binding buffer, 1×104 cells were analyzed on a FACSCAN flow cytometer (Beckman Coulter Inc, Fullerton, CA) by using CellQuest software. Annexin V-FITC positive cells were considered to be undergoing apoptosis and those negative for FITC were considered to be alive.
Caspase-3, caspase-8 and caspase-9 activity assay
After the U87 cells were treated with various concentration of icaritin for 48 hours, the cytosolic proteins were extracted in hypotoniccell lysis buffer and subjected to Caspase Activity Assay Kit (Beyotime, Shanghai, China) following the manufacture’s instructions. The activity was determined by measuring the absorbance at 405 nm using a microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
Mitochondrial membrane potential (MMP) assay
The changes in MMP were examined using fluorochrome dye JC-1 following a standard protocol. Briefly, U87 cells were challenged with the indicated dose of icaritin for 48 hours before the cells were harvested and incubated with JC-1. The cells were then gently rinsed with PBS to remove excessive dye before the fluorescence signal was quantitatively analyzed by flow cytometry (Beckman Coulter Inc, Fullerton, CA).
Quantification of autophagic cells by flow cytometry
The autophagic cells were quantified by acridine orange (AO) staining as previously described [27]. Briefly, U87 cells were stained with AO (1 μg/ml) for 15 minutes following treatment with various concentration of icaritin. Then the florescence signal was detected by FACSCAN flow cytometer (Beckman Coulter Inc, Fullerton, CA). In flow cytometric analysis of the AO stained cells, cytoplasm and nucleolus in non-autophagic cells showed green fluorescence (500-550 nm, FL-1 channel) whereas acidic vesicular organelles (AVO) in autophagic cells (quadrant A1) showed bright red fluorescence (650 nm, FL-3 channel). Since the red fluorescence intensity presents the number of AVO in autophagic cells, autophagic cells can be quantified on the basis of the intensity of red fluorescence.
Knockdown of Stat3 using siRNA
The siRNA oligos for Stat3 gene knockdown were purchased from Santa Cruz (Santa Cruz, CA) and a scramble sequence (Ribio, Guangzhou, China) was used as control. The U87 cells in logarithmic growth phase were seeded in 6-well plates at a density of 3×105 cells per well and incubated overnight and then transfected with siRNA or scramble siRNA respectively using Lipofectamine 2000 (Invitrogen, Grand Island, NY) according to the manufacturer’s protocol. Transfected cells were incubated for another 48 hours and the knockdown were verified by western blot analysis.
Western blots
Western blot was performed following standard protocol. Briefly, following incubation with various concentration of icaritin for 48 hours, U87 cells were harvested from flasks, and lysed with ice-cold lysis buffer (Beyotime, Shanghai, China) for 30 minutes on ice. Then collected proteins were subjected to SDS-PAGE gels and transferred onto PVDF membranes (Millipore, MA). Proteins were probed with specific primary antibodies and a rabbit polyclonal antibody to β-actin used as a gel loading control. Specific primary antibodies against cytochrome C, β-actin, PARP, caspase-3, COX IV, AMPK, phosphor-AMPK, beclin 1 and LC3B were purchased from Beyotime (Shanghai, China), antibodies against Fas, mTOR, phospho-mTOR, Stat3 and phospho-Stat3 were purchased from Abcam (Shanghai, China). After another washing with TBST, secondary antibody (HRP-conjugated secondary antibody, Boster, Wuhan, China) was added for blots detection. Signals were detected using chemiluminescent substrate (KPL., Guildford, UK) and the blot intensity were quantified using BandScan software (Glyko, Novato, CA).
Stat3 overexpression in U87 cells
The transfection of Stat3 overexpressing plasmid was performed as previously described [28]. Briefly, U87 cells were seeded in 96-well culture plate in culture medium and grew to 70% confluence before transfection. The Stat3 overexpression plasmid was constructed by inserting a full-length cDNA fragment obtained with reverse transcription and PCR with the specific primer for Stat3 primers into the pCMV vector (Beyotime, Shanghai, China). Then, the resulting pCMV- Stat3 vector was cloned into U87 cells to induce exogenous Sp1 expression. U87 cells transfected with an empty pCMV vector was taken as a control. Stable Stat3-overexpressing clones were selected at 48 hours after transfection.
Statistical analysis
The data are presented as mean ± SD (Standard Deviation) and represent the results of three separate experiments each conducted in quadruplicate unless otherwise stated. All statistical analysis was performed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). Values were presented as the mean ± SD. Statistical comparisons were performed by one-way ANOVA followed by Dunnett’s t-test. The difference with a P value less than 0.05 was defined as statistically significant.
Results
Icaritin suppresses cell growth of U87 cells
First, we examined whether icaritin could exert anti-proliferative effect in U87cells by treating U87 cells with different concentrations of icaritin for 24, 48 and 72 hours. As shown in Figure 1A and 1B, MTT results showed that icaritin could inhibit the proliferation of U87 cells in a dose- and time-dependent manner.
Figure 1.
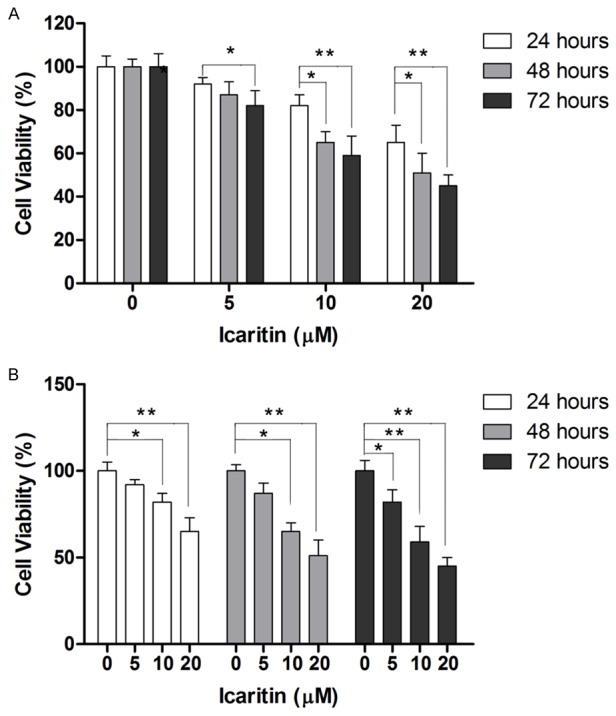
Icaritin inhibits cell proliferation in human GBM cell line U87. A: Icaritin reduces cell viability in time-dependent manner. B: Icaritin reduces cell viability in dose-dependent manner. *P<0.05, **P<0.01.
Icaritin induces caspase-dependent apoptosis
Next, we examined whether icaritin could induce apoptosis in U87 cells. As shown in Figure 3A, apoptotic cell proportion increased along with the increase of icaritin concentration. When U87 cells were incubated with 10 μM icaritin for 48 hours, apoptotic cell death occurred in 23% of cells (Figure 2A). The apoptosis-inducing effect of Icaritin was also supported by the findings that levels of activated caspase-3 and cleaved PARP were significantly elevated (Figure 2B and 2C). Since apoptosis in cells can occur via caspase-dependent and caspase-independent pathways, which are mediated by different factors, a caspase inhibitor z-VAD-fmk was used to demonstrate the mechanism by which Icaritin induced apoptosis in U87 cells. As shown in Figure 2D, z-VAD-fmk almost completely abolished Icaritin-induced apoptosis in U87 cells, suggesting that Icaritin induced apoptosis in U87 cells in a caspase-dependent manner. Caspase-dependent apoptosis can be triggered by extrinsic stimuli through cell surface death receptor or intrinsic stimuli through mitochondrial signaling, featured by activation of caspase-8 and caspase-9, respectively. Therefore, the activity of caspase-8 and caspase-9 was examined to further investigate the apoptosis-inducing mechanism of icaritin. As shown in Figure 3A, the activity of caspase-8 and caspase-9 was dose-dependently increased by icaritin treatment. The activation of intrinsic apoptotic pathway was also confirmed by decrease in mitochondrial membrane potential (MMP), and release of cytochrome C from mitochondria to cytosol in icaritin-treated U87 cells (Figure 3B and 3C). On the other hand, activation of extrinsic pathway was also supported by the increased expression of Fas in icaritin-treated U87 cells (Figure 3D). Taken together, our results suggested that ICARITIN induced apoptosis by activating both intrinsic and extrinsic caspase-dependent pathway.
Figure 3.

Icaritin induces apoptosis via both intrinsic and extrinsic pathway in human GBM cell line U87. Assays are performed following treatment with icaritin at indicated concentrations for 48 hours. A: Icaritin activates caspase-8 and caspase-9 as determined by ELISA kit. B: Icaritin leads to loss of MMP as measured by flow cytometry following florescent staining. C: Icaritin causes release of cytochrome C from mitochondria to cytosol as assessed by western blot. D: Icaritin alters the expression of markers Fas for extrinsic apoptosis pathway as assessed by western blot. *P<0.05, **P<0.01.
Figure 2.

Icaritin induces caspase-dependent apoptosis in human GBM cell line U87. Assays are performed following treatment with Icaritin at indicated concentrations for 48 hours. A: Icaritin induces apoptotic cell death as measured by flow cytometry. B: Icaritin increases caspase-3 activity as determined by ELISA assay. C: Icaritin activates PARP and caspase-3c as assessed by western blot. D: Caspase inhibitor z-VAD-fmk abolishes icaritin-induced apoptosis as measured by flow cytometry.
Icaritin induces autophagy in U87 cells
Next, we investigated whether icaritin induced autophagy in U87 cells. Acridine orange (AO) staining was performed to detect acidic vesicular organelles (AVO) in autophagic cells. As shown in Figure 4A, a significant increase in number of AVO was observed compared with control cells. In addition, flow cytometric analysis showed that icaritin induced autophagy in a dose-dependent manner in U87 cells (Figure 4A). To further confirm the autophagy-inducing effect of icaritin, we conducted western blot analysis to examine the level of LC3 and beclin 1 protein, the prominent autophagy markers. As shown in Figure 4B, icaritin at both 10 and 20 μM significantly increased the level of LC3B-II and Beclin 1, evidencing that icaritin induced autophagy in U87 cells.
Figure 4.

Icairtin induces autophagy in human human GBM cell line U87. Assays are performed following treatment with Icairtin at indicated concentrations for 48 hours. A: Icairtin causes autophagy was detected following AO staining. B: Icaritin elevates the expression level of LC3B-II and beclin 1 as assessed by western blot. *P<0.05, **P<0.01.
Interplay between icaritin-induced apoptosis and autophagy
In the following experiments, autophagy inhibitor as well as apoptosis was used to investigate the role of autophagy in the anti-tumor effect of icaritin. As shown in Figure 5A, inhibition of autophagy with 3-MA (autophagy inhibitor) significantly attenuated icaritin-induced decrease in cell viability (3-MA itself has no marked effect on cell viability), indicating that icaritin-induced autophagy contributed to its anti-proliferative effect and served as a form of cell death. Mounting evidence have elucidated that complex crosstalk presents between autophagy and apoptosis [29,30]. In this study, our results showed that z-VAD-fmk (caspase inhibitor) itself slightly increased autophagic cell death (not significant) whereas combination of icaritin and z-VAD-fmk significantly increased the proportion of autophagic cells relative to icaritin as single agent, as demonstrated by flow cytometric analysis as well the expression level of LC3B-II and beclin 1, indicating that apoptotic cell death was partially transformed into autophagic cell death in the case of apoptosis inhibition (Figure 5B). Then the effect of autophagy inhibition on icaritin-induced apoptosis was explored. As demonstrated by the flow cytometric results and the change in caspase-3 activity, autophagy inhibition significantly enhanced icaritin-induced apoptosis in U87 cells, suggesting that autophagy acts as pro-apoptototic factor here (Figure 5C).
Figure 5.

Icaritin-induces autophagy contributes to apoptosis. Assays are performed following treatment with icaritin at 20 μM for 48 hours in the presence or absence of 3-MA or z-VAD-fmk. A: Inhibition of autophagy impairs the anti-proliferative effect of icaritin as determined by MTT assay. B: Inhibition of apoptosis enhances autophagy as determined by flow cytometry. C: Inhibition of apoptosis increases the expression of LC3B-II and beclin 1 as assessed by western blot. D: Inhibition of autophagy decreases apoptosis as determined by flow cytometry. E: Inhibition of autophagy decreases caspase-3 activation as determined by ELISA kit. *P<0.05.
Inhibition of Stat3 signaling is involved in icaritin-induced apoptosis and autophagy
The role of Stat3 in apoptosis has been well documented in a number of cancer cells including GBM cells [31]. Interestingly, a recent study by Zou et al found that suppressed Stat3 activation was associated with induction of autophagy in human malignant glioma cells [32]. Therefore, we investigated whether icaritin induced apoptosis and autophagy in U87 cells by regulating Stat3 signaling. Our results revealed that icaritin repressed the phosphorylation of Stat3 in a dose-dependent manner (Figure 6A). To verify the role of Stat3 in apoptosis and autophagy regulation in U87 cells, Stat3 was knockdown using siRNA. As shown in Figure 6B and 6C, knockdown Stat3 in U87 cells resulted in increase in both apoptotic and autophagic cell population, suggesting that suppression of Stat3 signaling in cells would promote both apoptosis and autophagy. Moreover, our results showed that treatment of icaritin plus knockdown of Stat3 for 48 hours promoted both apoptosis and autophagy, indicating that icaritin promoted apoptosis and autophagy, at least partially, by modulating Stat3 signaling.
Figure 6.

Icaritin induces apoptosis and autophagy via inhibiting Stat3 signaling. Assays are performed following treatment with icaritin at indicated concentrations for 48 hours (the concentration of icaritin is 20 μM unless stated otherwise). A: Icaritin suppresses the activation of Stat3 as assessed by western blot. B: Overexpression of Stat3 significantly abolishes the apoptosis-inducing effect of icaritin as determined by flow cytometry. C: Overexpression of Stat3 significantly abolishes the autophagy-inducing effect of icaritin as determined by flow cytometry. **P<0.01.
Icaritin modulates Stat3 activation through ROS/AMPK/mTOR signaling
Recently, a number of studies have highlighted the role of mTOR as an upstream factor for Stat3 signaling [33]. In this study, we examined whether icaritin modulated the activation of Stat3 through regulating mTOR. As shown in Figure 7A, dose-dependent inhibition mTOR phosphorylation was observed when U87 cells were treated with icairtin for 48 hours. Then mTOR atcivation was artificially manipulated to demonstrate the role of mTOR signaling in Stat3 regulation. Our results showed that mTOR inhibition by rapamycin led to significantly suppression in Stat3 signaling (Figure 7B). More importantly, our results showed that the suppressing effect of icaritin on Stat3 activation was significantly attenuated by mTOR activator (phosphatidic acid, PA) in U87 cells (Figure 7B), suggesting that icaritin regulated the Stat3 signaling by regulating mTOR.
Figure 7.

Icaritin modulates Stat3 signaling through activating ROS/AMPK. Assays are performed following treatment with icaritin at indicated concentrations for 48 hours (the concentration of icaritin is 20 μM unless stated otherwise). A: Icaritin induces mTOR inhibition as well as AMPK activation. B: Icaritin modulates Stat3 through mTOR. C: Icaritin induces ROS generation in U87 cells in a dose-dependent manner. D: Icaritin modulates mTOR and Stat3 through ROS/AMPK signaling. *P<0.05, **P<0.01.
Next, we investigated the upstream signaling responsible for the regulatory effect of icaritin on mTOR. A number of signaling pathways have been found to be act as upstream modulating factors for mTOR, including AMPK [34]. Given the ability of icaritin to induce ROS generation and modulating cellular metabolic functions in cancer cell [35,36], we postulated that icaritin might modulate mTOR and hence Stat3 through activating ROS/AMPK. As shown in Figure 7C, icaritin treatment led to a dose-dependent increase in intracellular ROS level. In addition, western blot analysis showed that icaritin increased phosphorylation of AMPK without causing changes in total AMPK level (Figure 7A). Next, we examined the involvement of ROS/AMPK signaling in the modulatory effect of mTOR and Stat3 activation. Similar to the effect of icaritin, both ROS activator (CoCl2) and AMPK activator (AICAR) caused inhibition on mTOR and Stat3 (Figure 7D). On the other hand, ROS inhibitor (NAC) and AMPK inhibitor (compound C) were able to significantly abolish the modulatory effect of icaritin on mTOR and Stat3 (Figure 7D). Collectively, our results showed that icaritin regulated the Stat3 activation by inhibiting mTOR through ROS/AMPK signaling.
Discussions
Icaritin, a prenylflavonoid derivative from medicinal plant of Epimedium Genus, has been used in folk medicine for ages. A number of studies have made efforts to elucidate the role of icaritin in protection of neuron, the promotion of cardiac differentiation, and the prevention of steroid-associated osteonecrosis [21,37,38]. Interestingly, the anti-cancer activities of icaritin, first discovered in human breast cancer MCF-7 cells [39], have been widely investigated in a variety of human cancers. Icaritin can exert anti-cancer effect by inducing apoptosis [40], suppressing initiation and malignant growth of cancer [23], activating necrosis pathway [25], reversing multidrug resistance [41], and blocking cell cycle progression [35]. In the case of GBM, icaritin has been found to be able to sensitize GBM cells to TRAIL-induced apoptosis [42] as well as inhibit invasion and epithelial-to-mesenchymal transition [43]. The results presented here clearly demonstrated that icaritin is effective in inducing both apoptotic and autophagic cell death in GBM cell line U87. Moreover, our findings also suggest that icaritin induce apoptosis and autophagy in GBM cells by targeting Stat3, which is mediated by the ROS/AMPK/mTOR signaling.
Accumulating evidence has proved that inducing apoptosis is the major route for chemotherapeutic agents to eradicate cancer cells [44]. Apoptotic cell death can occur through two distinct pathways, caspase-independent and caspase-dependent pathways. It has well documented that AIF and Endo mediates caspase-independent apoptosis through promoting chromatin condensation and fragmentation of the nucleus after translocation from the intermembrane space of mitochondria to the nucleus [45] and directly digesting nuclear DNA following entering the nucleus [46], respectively. In the present study, our results showed that caspase inhibitor almost completely abolished the icaritin-induced apoptosis in U87 cells, excluding the participation of caspase-independent pathway in apoptosis-inducing effect of icaritin in U87 cells, which is consistent with a previous study by Sun et al showing icaritin inducing caspase-dependent apoptosis in hepatocarcinoma cells [47]. Moreover, given that caspase-dependent can occur via intrinsic mitochondrial pathway and extrinsic death receptor pathway [48], we then examine the mechanism basis of icaritin-induced caspase-dependent apoptosis. Our results showed that icaritin caused mitochondrial dysfunction in U87 cells. In addition, our results also revealed that icaritin can increase the intracellular level of Fas ligand, which can activate caspase-8 by interacting with Fas receptor and hence inducing apoptosis [49]. These results suggested that icaritin induced apoptosis in U87 cells via both intrinsic and extrinsic pathway, which agrees with previous in vitro studies in human hepatocellular carcinoma cell line and burkitt lymphoma cell line [47,50]. However, another study by Wang et al revealed that icaritin triggered apoptosis in human osteosarcoma cells and prostate carcinoma cells in vitro only through mitochondrial pathway [51,52]. These combined results showed that the mechanism by which icaritin induces apoptosis might be cell specific and relevant to activation of distinct signaling elicited by icaritin.
Autophagy in normal cells is a fundamental process for providing homeostatic and housekeeping functions through lysosome degradation of cytoplasmic organelles or cytosolic components [53]. In the context of cancer cells, autophagy has been documented in responses to various metabolic stress and anticancer agents, but the role of autophagy in cancer remains controversial [54]. It has been found that autophagy can serve as survival pathway in cancer cells [55]. On the contrary, autophagic cell death, defined as Type II PCD and presumed to result from excess autophagy, acts as a route for cancer cell elimination [56]. Here, we found that icaritin-induced reduction in cell viability was partially abolished by an autophagy inhibitor, 3-MA, indicating that icaritin-induced autophagy here acts as contributing factor to cell death. However, a very recent in vitro study reported that icaritin acts synergistically with epirubicin to suppress bladder cancer growth through inhibition of autophagy [57]. These conflicting results highlight the complex role of icaritin in the autophagy in tumor cells, which needs to be clarified with further studies.
Mounting evidences have showed that autophagy and apoptosis are important cellular processes which have crosstalk with each other [30]. For example, autophagy can promote apoptosis by degrading inhibitor of apoptosis proteins (IAPs) or Fas-associated phosphates 1 (Fap 1) [58]. On the other hand, the cross inhibition between autophagy and apoptosis has also been noted [59]. It has been found that blockage of autophagy by volatile oil of acori graminei Rhizoma potentiated the proapoptotic effect in GBM cell line, indicating a protective role of autophagy in VOA-induced cell death [34]. Another recent study by Cheng et al has also showed that inhibition of autophagy by 3-MA enhanced Hono-Mag-induced apoptosis in LN229 cells [60]. However, our results showed that inhibition of apoptosis enhanced icaritin-induced autophagy while inhibition of autophagy impaired icaritin-induced apoptosis, suggesting that icaritin-induced acts as pro-apoptotic factor and U87 cells tends to die via autophagy in the case of apoptosis inhibition in response to icaritin challenge. These mixed results suggest that the complex interplay between autophagy and apoptosis might occur in a stimulus-specific manner even in same tumor cells, which requires further investigation.
mTOR, a serine/threonine kinase activated by signaling pathway originating from growth factors and nutrient availability but inhibited in response to starvation, plays a crucial role in cell proliferation, growth, survival and suppression of mTOR is required for induction of autophagy by limiting the inhibitory effect on the ULK1 kinase complex [61]. A number of various signaling pathways has been proposed to regulate mTOR signaling, including positive regulation of mTOR (PI3K/Akt and p38 MAPK signaling) suppressing autophagy and negative regulation of mTOR (AMPK and p53 signaling) promoting autophagy [62]. In present study, our results also demonstrate that icaritin induces apoptotic and autophagic cell death via suppressing mTOR, further supporting the role of mTOR as a target for GBM therapy.
STAT3 is a latent transcription factor that mediates extracellular signals such as cytokines and growth factors through interaction with polypeptide receptors at the cell surface [63]. Following activation mainly through primarily by tyrosine phosphorylation, phosphorylated STAT3 dimerizes, translocates to the nucleus, and binds to sequence-specific DNA elements for consequent transcription of target genes [63]. STAT3 has been found to be constitutively activated or is required to maintain the transformed phenotype in human malignancies including solid tumor and hematological malignancy [64] and involved in a variety of biological activities of cancer cells, including apoptosis, autophagy and cell cycle progression. In our study, the data here showed that icaritin induces apoptosis and autophagy by inhibiting Stat3 signaling as a consequence of suppressing mTOR signaling. Besides its role in apoptosis, autophagy and cell cycle regulation, Stat3 also plays a role in glioma metastasis and angiogenesis [65]. Therefore, by inhibiting Stat3, icaritin could possibly suppress metastasis and angiogenesis of GBM in addition to inhibiting tumor growth.
In conclusion, our study firstly report that icaritin can exert anti-cancer effect in GBM in vitro through inducing apoptosis and autophagy. Moreover, our results showed that icaritin promotes both apoptotic and autophagic cell death by modulating Stat3 through activating ROS/AMPK signaling and subsequent inhibiting mTOR. The compelling results of this study warrant further study to evaluate the therapeutic value of icaritin as a potential anti-neoplastic agent.
Disclosure of conflict of interest
None.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Tonn JC, Brada M, Pentheroudakis G Group EGW. High-grade malignant glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v190–193. doi: 10.1093/annonc/mdq187. [DOI] [PubMed] [Google Scholar]
- 4.Noda SE, El-Jawahri A, Patel D, Lautenschlaeger T, Siedow M, Chakravarti A. Molecular advances of brain tumors in radiation oncology. Semin Radiat Oncol. 2009;19:171–178. doi: 10.1016/j.semradonc.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Desjardins A, Rich JN, Quinn JA, Vredenburgh J, Gururangan S, Sathornsumetee S, Reardon DA, Friedman AH, Bigner DD, Friedman HS. Chemotherapy and novel therapeutic approaches in malignant glioma. Front Biosci. 2005;10:2645–2668. doi: 10.2741/1727. [DOI] [PubMed] [Google Scholar]
- 6.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nuñez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92:57–70. doi: 10.1016/s0163-7258(01)00159-0. [DOI] [PubMed] [Google Scholar]
- 8.de Bruin EC, Medema JP. Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat Rev. 2008;34:737–749. doi: 10.1016/j.ctrv.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res. 2010;70:3431–3434. doi: 10.1158/0008-5472.CAN-09-4027. [DOI] [PubMed] [Google Scholar]
- 13.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 14.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu DY, Lou YJ. Inducible effects of icariin, icaritin, and desmethylicaritin on directional differentiation of embryonic stem cells into cardiomyocytes in vitro. Acta Pharmacol Sin. 2005;26:477–485. doi: 10.1111/j.1745-7254.2005.00076.x. [DOI] [PubMed] [Google Scholar]
- 17.Liao J, Liu Y, Wu H, Zhao M, Tan Y, Li D, Long H, Dai Y, Yung S, Chan TM, Lu Q. The role of icaritin in regulating Foxp3/IL17a balance in systemic lupus erythematosus and its effects on the treatment of MRL/lpr mice. Clin Immunol. 2016;162:74–83. doi: 10.1016/j.clim.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Xing B, Yang L, Shi J, Zhou X. Icaritin Attenuates Myocardial Ischemia and Reperfusion Injury Via Anti-Inflammatory and Anti-Oxidative Stress Effects in Rats. Am J Chin Med. 2015;43:1083–1097. doi: 10.1142/S0192415X15500627. [DOI] [PubMed] [Google Scholar]
- 19.Zhang ZK, Li J, Yan DX, Leung WN, Zhang BT. Icaritin Inhibits Collagen Degradation-Related Factors and Facilitates Collagen Accumulation in Atherosclerotic Lesions: A Potential Action for Plaque Stabilization. Int J Mol Sci. 2016:17. doi: 10.3390/ijms17020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu P, Jin X, Lv H, Li J, Xu W, Qian HH, Yin Z. Icaritin ameliorates carbon tetrachloride-induced acute liver injury mainly because of the antioxidative function through estrogen-like effects. In Vitro Cell Dev Biol Anim. 2014;50:899–908. doi: 10.1007/s11626-014-9792-8. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Zhang X, Wang H, Qi L, Lou Y. Neuroprotective effects of icaritin against beta amyloid-induced neurotoxicity in primary cultured rat neuronal cells via estrogen-dependent pathway. Neuroscience. 2007;145:911–922. doi: 10.1016/j.neuroscience.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 22.Qin L, Yao D, Zheng L, Liu WC, Liu Z, Lei M, Huang L, Xie X, Wang X, Chen Y, Yao X, Peng J, Gong H, Griffith JF, Huang Y, Zheng Y, Feng JQ, Liu Y, Chen S, Xiao D, Wang D, Xiong J, Pei D, Zhang P, Pan X, Wang X, Lee KM, Cheng CY. Phytomolecule icaritin incorporated PLGA/TCP scaffold for steroid-associated osteonecrosis: Proof-of-concept for prevention of hip joint collapse in bipedal emus and mechanistic study in quadrupedal rabbits. Biomaterials. 2015;59:125–143. doi: 10.1016/j.biomaterials.2015.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Guo Y, Li S, Han R, Ying J, Zhu H, Wang Y, Yin L, Han Y, Sun L, Wang Z, Lin Q, Bi X, Jiao Y, Jia H, Zhao J, Huang Z, Li Z, Zhou J, Song W, Meng K, Cai J. A novel anti-cancer agent Icaritin suppresses hepatocellular carcinoma initiation and malignant growth through the IL-6/Jak2/Stat3 pathway. Oncotarget. 2015;6:31927–31943. doi: 10.18632/oncotarget.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu T, Wang S, Wu J, Lin Z, Sui X, Xu X, Shimizu N, Chen B, Wang X. Icaritin induces lytic cytotoxicity in extranodal NK/T-cell lymphoma. J Exp Clin Cancer Res. 2015;34:17. doi: 10.1186/s13046-015-0133-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou C, Chen Z, Lu X, Wu H, Yang Q, Xu D. Icaritin activates JNK-dependent mPTP necrosis pathway in colorectal cancer cells. Tumour Biol. 2015;37:3135–44. doi: 10.1007/s13277-015-4134-3. [DOI] [PubMed] [Google Scholar]
- 26.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 27.Mohan N, Chakrabarti M, Banik NL, Ray SK. Combination of LC3 shRNA plasmid transfection and genistein treatment inhibited autophagy and increased apoptosis in malignant neuroblastoma in cell culture and animal models. PLoS One. 2013;8:e78958. doi: 10.1371/journal.pone.0078958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahu RP, Srivastava SK. The role of STAT-3 in the induction of apoptosis in pancreatic cancer cells by benzyl isothiocyanate. J Natl Cancer Inst. 2009;101:176–193. doi: 10.1093/jnci/djn470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie T, Zhang N, Ye ZM. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis. 2015;6:e1604. doi: 10.1038/cddis.2014.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Wang K, Lei Y, Li Q, Nice EC, Huang C. Redox signaling: Potential arbitrator of autophagy and apoptosis in therapeutic response. Free Radic Biol Med. 2015;89:452–465. doi: 10.1016/j.freeradbiomed.2015.08.030. [DOI] [PubMed] [Google Scholar]
- 31.Wu J, Feng X, Zhang B, Li J, Xu X, Liu J, Wang X, Wang J, Tong X. Blocking the bFGF/STAT3 interaction through specific signaling pathways induces apoptosis in glioblastoma cells. J Neurooncol. 2014;120:33–41. doi: 10.1007/s11060-014-1529-8. [DOI] [PubMed] [Google Scholar]
- 32.Zou M, Hu C, You Q, Zhang A, Wang X, Guo Q. Oroxylin A induces autophagy in human malignant glioma cells via the mTOR-STAT3-Notch signaling pathway. Mol Carcinog. 2015;54:1363–1375. doi: 10.1002/mc.22212. [DOI] [PubMed] [Google Scholar]
- 33.Ma J, Meng Y, Kwiatkowski DJ, Chen X, Peng H, Sun Q, Zha X, Wang F, Wang Y, Jing Y, Zhang S, Chen R, Wang L, Wu E, Cai G, Malinowska-Kolodziej I, Liao Q, Liu Y, Zhao Y, Sun Q, Xu K, Dai J, Han J, Wu L, Zhao RC, Shen H, Zhang H. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J Clin Invest. 2010;120:103–114. doi: 10.1172/JCI37964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Jiang Z, Ma H, Ning L, Chen H, Li L, Qi H. Volatile Oil of Acori Graminei Rhizoma-Induced Apoptosis and Autophagy are dependent on p53 Status in Human Glioma Cells. Sci Rep. 2016;6:21148. doi: 10.1038/srep21148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Q, Liu WW, Li B, Chen HJ, Zhu WS, Yang GX, Chen MJ, He GY. Anticancer effect of icaritin on human lung cancer cells through inducing S phase cell cycle arrest and apoptosis. J Huazhong Univ Sci Technolog Med Sci. 2014;34:497–503. doi: 10.1007/s11596-014-1305-1. [DOI] [PubMed] [Google Scholar]
- 36.Sun F, Indran IR, Zhang ZW, Tan MH, Li Y, Lim ZL, Hua R, Yang C, Soon FF, Li J, Xu HE, Cheung E, Yong EL. A novel prostate cancer therapeutic strategy using icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis. 2015;36:757–768. doi: 10.1093/carcin/bgv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wo YB, Zhu DY, Hu Y, Wang ZQ, Liu J, Lou YJ. Reactive oxygen species involved in prenylflavonoids, icariin and icaritin, initiating cardiac differentiation of mouse embryonic stem cells. J Cell Biochem. 2008;103:1536–1550. doi: 10.1002/jcb.21541. [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Qin L, Sheng H, Wang XL, Wang YX, Yeung DK, Griffith JF, Yao XS, Xie XH, Li ZR, Lee KM, Leung KS. A novel semisynthesized small molecule icaritin reduces incidence of steroid-associated osteonecrosis with inhibition of both thrombosis and lipid-deposition in a dose-dependent manner. Bone. 2009;44:345–356. doi: 10.1016/j.bone.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye HY, Lou YJ. Estrogenic effects of two derivatives of icariin on human breast cancer MCF-7 cells. Phytomedicine. 2005;12:735–741. doi: 10.1016/j.phymed.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Tong JS, Zhang QH, Huang X, Fu XQ, Qi ST, Wang YP, Hou Y, Sheng J, Sun QY. Icaritin causes sustained ERK1/2 activation and induces apoptosis in human endometrial cancer cells. PLoS One. 2011;6:e16781. doi: 10.1371/journal.pone.0016781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun L, Chen W, Qu L, Wu J, Si J. Icaritin reverses multidrug resistance of HepG2/ADR human hepatoma cells via downregulation of MDR1 and Pglycoprotein expression. Mol Med Rep. 2013;8:1883–1887. doi: 10.3892/mmr.2013.1742. [DOI] [PubMed] [Google Scholar]
- 42.Han H, Xu B, Hou P, Jiang C, Liu L, Tang M, Yang X, Zhang Y, Liu Y. Icaritin Sensitizes Human Glioblastoma Cells to TRAIL-Induced Apoptosis. Cell Biochem Biophys. 2015;72:533–42. doi: 10.1007/s12013-014-0499-y. [DOI] [PubMed] [Google Scholar]
- 43.Xu B, Jiang C, Han H, Liu H, Tang M, Liu L, Ji W, Lu X, Yang X, Zhang Y, Liu Y. Icaritin inhibits the invasion and epithelial-to-mesenchymal transition of glioblastoma cells by targeting EMMPRIN via PTEN/AKt/HIF-1alpha signalling. Clin Exp Pharmacol Physiol. 2015;42:1296–1307. doi: 10.1111/1440-1681.12488. [DOI] [PubMed] [Google Scholar]
- 44.Han YT, Chen XH, Gao H, Ye JL, Wang CB. Physcion inhibits the metastatic potential of human colorectal cancer SW620 cells in vitro by suppressing the transcription factor SOX2. Acta Pharmacol Sin. 2016;37:264–275. doi: 10.1038/aps.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang HS, Kim JY, Lee JH, Lee BW, Park KH, Shim KH, Lee MK, Seo KI. Celastrol isolated from Tripterygium regelii induces apoptosis through both caspase-dependent and -independent pathways in human breast cancer cells. Food Chem Toxicol. 2011;49:527–532. doi: 10.1016/j.fct.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 46.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 47.Sun L, Peng Q, Qu L, Gong L, Si J. Anticancer agent icaritin induces apoptosis through caspase-dependent pathways in human hepatocellular carcinoma cells. Mol Med Rep. 2015;11:3094–3100. doi: 10.3892/mmr.2014.3007. [DOI] [PubMed] [Google Scholar]
- 48.Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- 49.Abd El-Ghany RM, Sharaf NM, Kassem LA, Mahran LG, Heikal OA. Thymoquinone triggers anti-apoptotic signaling targeting death ligand and apoptotic regulators in a model of hepatic ischemia reperfusion injury. Drug Discov Ther. 2009;3:296–306. [PubMed] [Google Scholar]
- 50.Li ZJ, Yao C, Liu SF, Chen L, Xi YM, Zhang W, Zhang GS. Cytotoxic effect of icaritin and its mechanisms in inducing apoptosis in human burkitt lymphoma cell line. Biomed Res Int. 2014;2014:391512. doi: 10.1155/2014/391512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang XF, Wang J. Icaritin suppresses the proliferation of human osteosarcoma cells in vitro by increasing apoptosis and decreasing MMP expression. Acta Pharmacol Sin. 2014;35:531–539. doi: 10.1038/aps.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang X, Zhu D, Lou Y. A novel anticancer agent, icaritin, induced cell growth inhibition, G1 arrest and mitochondrial transmembrane potential drop in human prostate carcinoma PC-3 cells. Eur J Pharmacol. 2007;564:26–36. doi: 10.1016/j.ejphar.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 53.Meijer AJ, Dubbelhuis PF. Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun. 2004;313:397–403. doi: 10.1016/j.bbrc.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 54.Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res. 2012;22:43–61. doi: 10.1038/cr.2011.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem. 2009;284:21412–21424. doi: 10.1074/jbc.M109.026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan XW, Li L, Huang Y, Huang H, Xu DF, Gao Y, Chen L, Ren JZ, Cao JW, Hong Y, Cui XG. Icaritin acts synergistically with epirubicin to suppress bladder cancer growth through inhibition of autophagy. Oncol Rep. 2016;35:334–342. doi: 10.3892/or.2015.4335. [DOI] [PubMed] [Google Scholar]
- 58.Nezis IP, Shravage BV, Sagona AP, Johansen T, Baehrecke EH, Stenmark H. Autophagy as a trigger for cell death: autophagic degradation of inhibitor of apoptosis dBruce controls DNA fragmentation during late oogenesis in Drosophila. Autophagy. 2010;6:1214–1215. doi: 10.4161/auto.6.8.13694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng YC, Hueng DY, Huang HY, Chen JY, Chen Y. Magnolol and honokiol exert a synergistic anti-tumor effect through autophagy and apoptosis in human glioblastomas. Oncotarget. 2016;7:29116–30. doi: 10.18632/oncotarget.8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 63.Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 64.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding H, Shen J, Yang Y, Che Y. Saw Palmetto Extract Inhibits Metastasis and Antiangiogenesis through STAT3 Signal Pathway in Glioma Cell. Evid Based Complement Alternat Med. 2015;2015:926946. doi: 10.1155/2015/926946. [DOI] [PMC free article] [PubMed] [Google Scholar]