

Abstract
Heaptocellular carcinoma (HCC) is still a great health problem around the world. Recently, the cetuximab has been implicated to have therapeutic values for HCC. However, cetuximab-resistance has also been synchronously reported pertaining to HCC treatment. This study aimed to evaluate the role of eIF5A2 in cetuximab-treated HCC cell proliferation, and whether eIF5A2 specific inhibitor GC7 has any effects on cetuximab-mediated proliferation inhibition in HCC cell lines. It was observed that GC7 significantly inhibited cell proliferation in HCC cell lines. GC7 synergized cetuximab to inhibit the proliferation in epithelial HCC cell lines HepG2, Huh7 and Hep3B, but not in mesenchymal cell lines SNU387 and SNU449. Knockdown of eIF5A-2 by specific siRNA exhibited the similar effects as GC7 did. In cetuximab-treated cells, cetuximab decreased the protein level of EGFR and phosphorylated STAT3 and unexpectedly up-regulated the expression level of eIF5A2, indicating the activation of eIF5A2 pathway. In turn, cetuximab also synergized GC7 to inhibit cell proliferation in epithelial cell lines. GC7 also suppressed hypoxia-induced cell proliferation in epithelial cell lines. These data suggest that eIF5A2 is an alternative pathway for cell proliferation in epithelial HCC cells escaping from the cytotoxicity of cetuximab. The eIF5A inhibitor GC7 might be a potent agent that promotes the cytotoxicity of cetuximab on epithelial HCC cells.
Keywords: Cetuximab, GC7, eIF5A, hepatocellular carcinoma
Introduction
Liver tumor is still a great threat for human health around the world. Among the primary liver tumors, hepatocellular carcinoma (HCC) is widely recognized as the most frequent form [1,2]. Hence, novel therapeutic strategies for HCC treatment are urgently required. Tumor resection and liver transplantation are currently most common surgical procedures for HCC. However, they are inapplicable for metastatic liver cancers [3]. Sorafenib is to date the only treatment proven to extend the survival of HCC patients in advanced stage and is widely implicated in clinical practices as a first-line treatment [4,5]. However, this does not mean that the need for an effective second-line treatment of advanced liver cancer has already been met. In fact, lots of novel agents have been under development [6], of which cetuximab has been shown to have great efficacy pertaining to the treatment of human malignancies.
Cetuximab is an effective inhibitor of epidermal growth factor receptor (EGFR). Cetuximab exerts its anti-cancer effects by suppressing EGFR in EGFR-overexpressing tumors, such as non-small cell lung cancer [7]. It was observed that EGFR activation contributed to the primary resistance of HCC cells to sorafenib, while inhibition of EGFR by cetuximab promoted sorafenib-mediated therapeutic effects in HCC [8]. Despite its potential, cetuximab has been indicated to be only modestly effective for treating HCC in phase II clinical trials [9]. In addition, a recent publication showed that cetuximab co-operated with the STAT3 inhibitor NSC 74839 to inhibit the proliferation of HCC cells via an EGFR-STAT3 pathway [10]. Combination of cetuximab with rapacymin [11,12] or microRNA-146a mimic [13] has also been shown to enhance the therapeutic efficacy of cetuximab on HCC. These publications altogether suggest that HCC cells may be potentially resistant to cetuximab. A single use of cetuximab should not have high therapeutic efficacy in HCCs. Combined therapy (cetuximab and other agents) may potently enhance the cytotoxicity of cetuximab in HCCs.
In the last decades, the eukaryotic translation initiation factor 5A (eIF5A) has been shown to be critically involved in oncogenic activities, including tumor growth and metastasis. Inhibition of eIF5A impairs melanoma growth [14], while overexpression of eIF5A promotes cell motility and metastasis in HCC [15]. In fact, eIF5A is an independent indicator for cell proliferation [16]. The prognostic significance and therapeutic potential of eIF5A in HCC has also been revealed [17]. eIF5A has two isoforms, namely eIF5A-1 and eIF5A-2. The function of eIF5A depends on a specific and unique post-translational modification, termed hypusination (a lysine residue is converted into hypusine). Hypusination is completed by two steps: (1) a 4-butylamine moiety of spermidine is transferred to the e-amino group of a specific lysine residue in the eIF5A molecule (Lys50 in human eIF5A), by the action of deoxyhypusine synthase (DHS), giving rise to the deoxyhypusil residue; (2) the deoxyhypusil residue carbon 2 is hydroxylated by desoxyhypusil hydroxylase (DHH) to form the hypusine residue [N-e-(4amino-2 hydroxybutyl) lysine] [14,18]. Previously, inhibitors of DHH (step 2) have been tested as anti-neoplastic agents, but unfortunately result in uncontrolled and unpredictable side effects. Hence, the N1-guanyl-1,7-diaminoheptane, known as GC7, has now been widely tested its property of inhibiting eIF5A hypusination [19]. GC7 is known as a DHS inhibitor (step 1) of high affinity and selectivity [19,20]. The anti-proliferative effects of this compound via inhibiting eIF5A have been observed in various cell lines such as HUVEC, NIH-3T3, CHO-K1, H9 and HeLa [20,21]. Therefore, inhibition of eIF5A hypusination by GC7 has been considered as a promising strategy to suppress tumor growth.
In this study, we aimed to explore whether eIF5A has any connection with the cetuximab-inhibited EGFR-STAT3 pathway in HCC. The toxic effects of GC7 on various HCC cell lines were hence investigated. In particular, the combined effects of GC7 and cetuximab on HCC cell proliferation were assessed.
Materials and methods
Cell lines and reagents
The human HCC cell lines, including epithelial HepG2, Hep3B, Huh7 cells and mesenchymal cells SNU-387 and SNU-449, were obtained from the Shanghai Institute of Biological Science, Shanghai, China. All cells were cultured in Dulbecco’s modified eagle medium (DMEM) (Gibco, Los Angeles, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin. Cells were maintained in a humidified incubator at 37°C under 5% CO2. For induction of hypoxia, cells were treated with 100 μM of deferoxamine (Sigma, St. Louis, MO, USA) for 4 h. For knockdown of specific genes, cells were transfected with specific siRNAs (GenePharma, Shanghai, China) using Lipofectamine 2000 (Invitrogen, Shanghai, China) based on the manufacturer’s instructions. Culture medium was refreshed every two days. For all the agents, stock solutions were prepared with dimethyl sulfoxide (DMSO). Working solutions were made in fresh medium when needed. To prevent toxicity, the working concentration of DMSO did not exceed 0.5% in any experiment. For other reagents, the Cell count kit-8 (cck8) was purchased from Sigma-Aldrich (St. Louis, MO, USA). all antibodies were from Santa Cruz Biotech (Santa Cruz, CA, USA).
Cell viability assay
Cells were suspended into single cells and seeded into 96-well plates (3×106 cells/well) with 100 μl DMEM media supplied with 10% FBS medium. After cell growth overnight, cells were treated with corresponding concentrations of GC7, with or without cetuximab for 48 h. After that, 10 μL CCK-8 solution was added into each well and cells were allowed to grow for additional 2 h under the same humidified incubator. Air bubbles were strictly avoided during the whole process. The absorbance of each well was measured at 450 nm using a synergy 2 multi-mode microplate reader (Bio Tek Instruments, Winooski, VT, USA). CCK-8 Kit was commercially from Sigma-Aldrich (St. Louis, MO, USA). Relative cell viability was calculated as a percentage of untreated controls. The half-maximal inhibitory concentration (IC50) was determined as previously described [11].
Western blot
Total cellular proteins were extracted from cultured cells with the RIPA buffer (Thermo Scientific, IL, USA) supplied with protease inhibitor (PI, Beyotime Biotechnology, Nanjing, China). Total proteins were quantified using the Bradford method (Bio-Rad Laboratories, CA, USA) with bovine serum albumin (BSA) as a standard. Briefly, an equal amount of 50 µg protein/sample were loaded into each lane on a 10% SDS-PAGE gel and later transferred into polyvinylidene fluoride membranes (Millipore, Massachusetts, USA). Membranes were subsequently blocked with 5% skim milk for 1 h at room temperature and incubated with primary antibodies overnight at 4°C. After PBS washes, corresponding secondary antibodies were incubated with the membranes for additional 1 h at room temperature. The immunoreactivity was developed by using an ECL reagent (Thermo Scientific, USA).
Assessment of eIF5A activity
HCC cells were incubated in the presence of [1,8-3H]-spermidine (5 μCi/mL, Perkin-Elmer/NEN) with GC7 or vehicle for 48 h. Cells were harvested and precipitated with 10% trichloroacetic acid (TCA) containing 1 mM spermidine and spermine, and then washed three times to remove the free [3H]-spermidine. A portion of the TCA precipitate was re-suspended in 1 N NaOH. The radioactivity of HCC cells were measured by liquid scintillation analyzer (Tri-Carb 2900TR, Perkin-Elmer). Another portion of TCA precipitate was dissolved in SDS buffer and used for fluorographic detection of 3H-labelled hypusinated-eIF5A isoforms after SDS-PAGE.
EdU incorporation assay
EdU incorporation assay was performed to visualize the proliferative activity of cells under distinct treatments. Briefly, cells were exposed to EdU (5-ethynyl-2’-deoxyuridine) (Invitrogen) for 3 h at 37°C prior to tests. The cells were then fixed with 4% formaldehyde for another 15min and treated with 0.5% Triton X-100 for additional 20 min at 37°C. After a consecutive of three times washes with PBS, the cells in each well were then reacted with 100 μL of 1× Apollo reaction cocktail for 30 min. Subsequently, the cells of each well were stained with 100 μL of Hoechst 33342 (5 μg/mL) for 30 min and the proliferative activity of cells were visualized under a fluorescent microscope (Leica Microsystems).
Statistical analysis
Data were obtained from three independent experiments. All data were expressed as mean ± standard deviation (SD). The inhibitory effects of different treatments were compared using the two-way ANOVA for repeated measurements. All statistics were done using Prism 5 (GraphPad Software, CA, USA). A p value less than 0.05 was considered of statistical significance.
Results
Inhibitive effects of cell proliferation by GC7 in HCC cell lines
To investigate the role of GC7 in HCC cell proliferation, we cultured five cell lines, including epithelial HepG2, Huh7, Hep3B cells and mesenchymal SNU387 and SNU449 cells. All these cells were treated with a various concentrations of GC7. It was observed that GC with its concentration less than 10 μM barely had any effects on cell viability in these cell lines. However, cell proliferative inhibition was observed with GC7 doses over 10 μM. And this inhibition effects were dose-dependent in all the five cell lines (Figure 1). These data suggest the proliferation inhibitive effects by GC7 in the five HCC cell lines.
Figure 1.

Inhibitive effects of cell proliferation by GC7 in HCC cell lines. A-E. Five HCC cell lines, including three epithelial cells HepG2, Huh7 and Hep3B, and two mesenchymal cells SNU387 and SNU449 were cultured and treated with a various concentrations of GC7. The cell viability under GC7 treatments were assessed by CCK-8 assay (*, P<0.05).
GC7 synergizes with cetuximab to promote cell proliferation in epithelial HCC cell lines
Next, we examined the effects of GC7 (10 μM) on cetuximab-mediated proliferation inhibition in the above five cell lines. The efficacy of GC7 in inhibiting the expression of eIF5A1 and eIF5A2 was initially assessed and confirmed (Figure 2A). It was observed that cetuximab exhibited dose-dependently inhibition of cell viability in all of the cell lines. In the epithelial cell lines, combination of GC7 and cetuximab caused further inhibition of cell viability (Figure 2B-D). However, in the mesenchymal SNU387 cells and SNU449 cells, combination of GC7 and cetuximab barely changed the cell viability as compared with a single treatment of cetuximab (Figure 2E, 2F). Calculation of IC50 also confirmed that the IC50 values in the epithelial HCC cells, instead of mesenchymal HCC cells, were significantly lower in combined treatments of GC7 and cetuximab than in single treatment of cetuximab (Table 1). EdU incorporation assay is an ideal technique to reveal the proliferating cells. Hence, the five cell lines were treated with single cetuximab (IC50 concentration), or combined with GC7. Treatment of cells with cetuximab alone did not change the ratio of EdU-positive cells. However, combined treatments of cetuximab and GC7 caused significant decreases of EdU-positive cells only in epithelial HepG2, Huh7 and Hep3B cells (Figure 2G-K). These data suggest that GC7 enhances the cytotoxicity of cetuximab in epithelial HCC cells.
Figure 2.
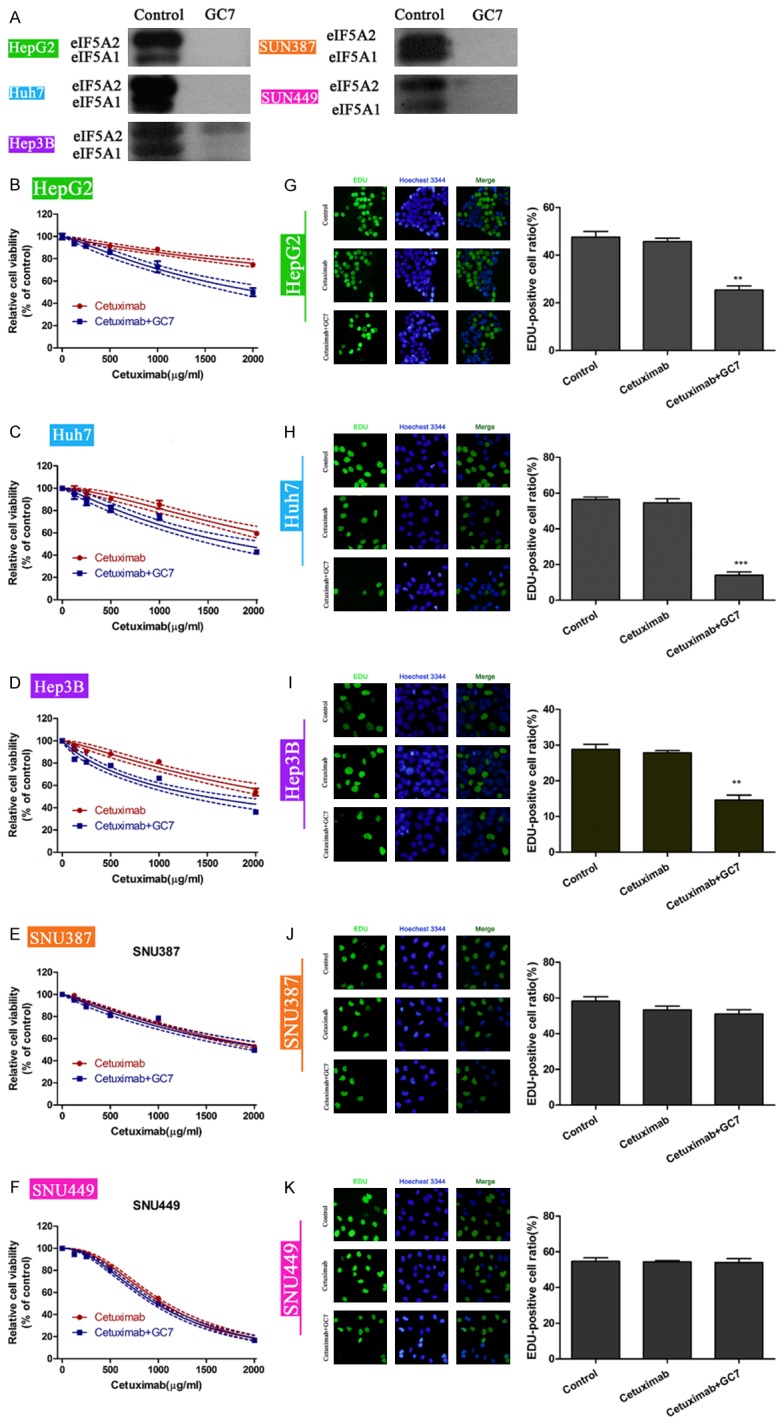
GC7 synergizes with cetuximab to promote cell proliferation in epithelial HCC cell lines. A. The efficacy of GC7 in inhibiting eIF5A was confirmed by western blotting. B-F. The five HCC cell lines were treated with cetuximab, or combination of cetuximab with GC7. Cell viability was assessed by CCK-8 assays. G-K. EdU incorporation assay was performed to assess the number of cells in proliferation. EdU-positive cells represent proliferating cells. The concentrations of cetuximab were the IC50 values in B-F panels. GC7 were treated for a fixed concentration of 10 μM (**, P<0.01).
Table 1.
IC50 values for single treatment of cetuximab or combined treatment of cetuximab with GC7 in the five HCC cell lines
| HCC cell lines | IC50 (μg/ml) | |
|---|---|---|
|
| ||
| Cetuximab | Cetuximab+GC7 | |
| HepG2 | 6881 | 2091 |
| Huh7 | 2648 | 1800 |
| Hep3B | 2490 | 1481 |
| SNU387 | 2294 | 2252 |
| SNU449 | 1049 | 974.2 |
eIF5A is activated in cetuximab-treated epithelial HCC cell lines
To further confirm the GC7-mediated enhancement of cetuximab cytotoxicity, we employed specific siRNA against eIF5As to knock down the expression of eIF5A2 in the five cell lines. The knockdown efficiency of specific siRNA was tested and confirmed by western blot (Figure 3A). Based on this siRNA, the five cell lines were depleted from eIF5A2 and treated with single cetuximab, or a combination of cetuximab and GC7. Interestingly, each cell line displayed similar cell viability between single cetuximab treatment group and combined treatment group, despite epithelial and mesenchymal cell lines (Figure 3B). This observation indicated that eIF5A2 is essential for GC7-mediated enhancement of cetuximab cytotoxicity. Furthermore, when eIF5A2 was depleted, cetuximab-mediated proliferation inhibition was even significant as compared to the group without eIF5A2 depletion. However, this phenomenon was only visible in the epithelial HepG2, Huh7 and Hep3B cells. These data suggest that eIF5A2 is another alternative pathway for cell proliferation in cetuximab-treated epithelial cancer cells.
Figure 3.

eIF5A2 is another alternative pathway for cell proliferation in cetuximab-treated epithelial cancer cells. A. Knockdown efficiency of specific siRNA against eIF5A2 was assessed in the five HCC cell lines. B. The five cell lines were firstly depleted from expressing eIF5A2 by specific siRNA. These cells were then subject to treatment of single cetuximab, or combined treatment of cetuximab and GC7 (10 μM). Cell viability in each cell line was assessed by CCK-8 assay under distinct conditions. C. All cells that were treated with cetuximab were transfected with or without specific siRNA against eIF5A2. Cell viability was accordingly assessed.
eIF5A is activated by cetuximab treatment in the epithelial HCC cell lines
Further, we performed western blot to detect the expression of eIF5A in response to treatment of cetuximab, with or without GC7 in the three epithelial HCC cell lines. As shown in Figure 4A, treatment of cetuximab decreased the expression of EGFR and the phosphorylated level of STAT3 without affecting the total STAT3 level. Interestingly, the protein level of eIF5A2 was also up-regulated by single treatment of cetuximab relative to no agent treatment. Combination of GC7 and cetuximab down-regulated the protein level of eIF5A2 as compared with single cetuximab treatment. Moreover, cetuximab treatment also increased the activities of both eIF5A1 and eIF5A2 (Figure 4B), further verifying that eIF5A2 is activated by cetuximab in the epithelial HCC cells.
Figure 4.
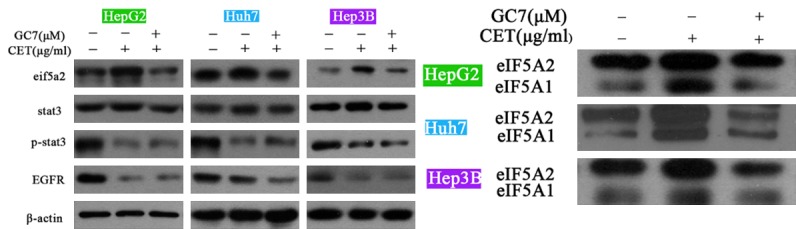
eIF5A is activated by cetuximab treatment in the epithelial HCC cell lines. The protein levels of eIF5A2, total STAT3, phosphorylated STAT3 and EGFR were assessed upon treatments of vehicle, single cetuximab, or combination of cetuximab and GC7 (A). The biological activities of eIF5A1 and eIF5A2 were also detected in distinct treatments (B). It was shown that cetuximab up-regulated the protein level of eIF5A2, and also increased the biological activities of both eIF5A1 and eIF5A2 in the three epithelial HCC cell lines. CET, cetuximab.
Cetuximab synergizes GC7 to promote its prolifereation inhibition in the epithelial HCC cell lines
In turn, we assessed whether cetuximab had any effects on GC7-mediated proliferation inhibition. It was shown that while GC7 inhibited the cell proliferation in a dose-dependent manner in HepG2, Hep3B and Huh7 cells, addition of cetuximab caused further inhibition of cell proliferation (Figure 5A-C). In the EdU incorporation assay, 10 μM of GC7 barely affected the proliferation ability, which was also evidenced by the data in Figures 1 and 5A-C. However, combination of GC7 and cetuximab significantly decreased the proliferation abilities in the epithelial HCC cells (Figure 5D-F). The IC50 values also verified that cetuximab enhanced the proliferation inhibitive effects by GC7 in the epithelial HCC cell lines (Table 2). Since cetuximab exerted its inhibition of proliferation mainly by EGFR-STAT3 pathway, we depleted STAT3 by specific siRNA (Figure 5J). Epithelial HCC cells that were transfected with or without specific siRNA again STAT3 were treated with GC7 with distinct doses. It was interestingly observed that knockdown of STAT3 could still further enhanced the GC7-mediated proliferation inhibition in the three cell lines. However, these enhanced effects of proliferation inhibition were less remarkable as compared with that in the Figure 5A-C (Figure 5G-I). These data suggest cetuximab also enhances the cytotoxicity of GC7 in the epithelial HCC cells. The enhancement of GC7 cytotoxicity by cetuximab depends on the expression of STAT3.
Figure 5.

Cetuximab synergizes GC7 to promote its prolifereation inhibition in the epithelial HCC cell lines. A-C. Three epithelial HCC cell lines HepG2, Huh7 and Hep3B were treated with various doses of GC7, with or without additional cetuximab. D-F. EdU incorporation assay was performed to assess the proliferating abilities in GC7 (10 μM) - treated cells with or without cetuximab (IC50 concentration in Table 2). G-I. Specific siRNA against STAT3 were employed to knock down the expression of STAT3 in the three cells HepG2, Huh7 and Hep3B. Cells were then treated with various doses of GC7 and were assessed for their viability under each condition. J. The knockdown efficacy of specific siRNA against STAT3 in HepG2, Huh7 and Hep3B cells (***, P<0.001).
Table 2.
IC50 values for single treatment of GC7 or combined treatment of GC7 with cetuximab in the three epithelial HCC cell lines
| HCC cell lines | IC50 (μM) | |
|---|---|---|
|
| ||
| GC7 | Cetuximab+GC7 | |
| HepG2 | 142.3 | 58.95 |
| Huh7 | 169.7 | 99.99 |
| Hep3B | 143.8 | 42.80 |
Effects of GC7 on the EGFR-STAT3 pathway
Next, it was observed that treatment of GC7 up-regulated the phoshorylated levels of STAT3 without affecting the protein level of EGFR in HepG2, Huh7 and Hep3B cells. More importantly, combination of cetuximab and GC7 decreased the expression of p-STAT3 and EGFR to levels lower than the control group (vehicle treatment) (Figure 6), indicating that combined therapy may exhibit much more potent therapeutic effects.
Figure 6.
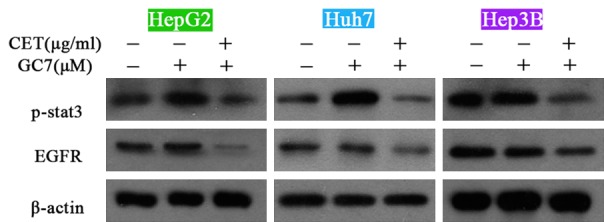
Effects of GC7 on the EGFR-STAT3 pathway. Treatment of GC7 up-regulated the phoshorylated levels of STAT3 without affecting the protein level of EGFR in HepG2, Huh7 and Hep3B cells. In addition, combination of cetuximab and GC7 decreased the expression of p-STAT3 and EGFR to levels lower than the control group (vehicle treatment). CET, cetuximab.
GC7 inhibits the hypoxia-induced cell proliferation in epithelial HCC cell lines
Lastly, a hypoxia model was used to induce cell proliferation. It was observed that compared to normoxia, hypoxia rescued cells from cetuximab-induced proliferation inhibition in HepG2, Huh7 and Hep3B cells. However, treatment of cells with GC7 significantly blunted the above hypoxia-induced effects. Instead, cell viability was further inhibited by addition of GC7 to cells in hypoxia environment (Figure 7A). In addition, we detected the biological activities of eIF5A1 and eIF5A2 in hypoxia-incubated cells. It was shown that hypoxia increased the activities of both eIF5A1 and eIF5A2 significantly as compared with control cells (Figure 7B). These data suggest that eIF5A is also involved in hypoxia-induced cell proliferation in epithelial HCC cell lines.
Figure 7.

GC7 inhibits the hypoxia-induced cell proliferation in epithelial HCC cell lines. A. A hypoxia model was used to induce cell proliferation in HCC cell lines. Compared to normoxia, hypoxia rescued cells from cetuximab-induced proliferation inhibition in HepG2, Huh7 and Hep3B cells. However, treatment of cells with GC7 significantly blunted the above hypoxia-induced effects. B. Hypoxia increased the biological activities of eIF5A in HepG2, Huh7 and Hep3B cells.
Discussion
The heavy burden of hepatocellular carcinoma (HCC), which is characterized by its high incidence and mortality rates, urgently requires novel developments of effective treatments for this malignancy [22]. Prognosis of unresectable or metastatic HCC still remains poor, possibly due to diagnosis at a late stage, underlying liver cirrhosis or resistance to chemotherapy [10]. Sorafenib is currently the first-line treatment for HCC. However, resistance to sorafenib is also reported. It has been revealed that EGFR activation is a potential determinant of primary resistance of HCC cells to sorafenib, while inhibition of EGFR by its specific inhibitor cetuximab promotes the efficacy of sorafenib [8]. Furthermore, a p-STAT3 inhibitor (NSC 74859) enhances the anti-proliferative activity of cetuximab in HCC [10]. These data suggest that EGFR-STAT3 pathway plays critical roles in HCC cell resistance to sorafenib. For the last years, the effects of cetuximab in treating HCC have been emerging. However, single use of cetuximab is only modestly effective in clinical trials [10], indicating that a cetuximab-resistance mechanism may exist, and other agents are urgently required to promote the cytotoxicity of cetuximab in the treatment of HCC.
This study investigated (1) whether eIF5A2 had any connection with cetuximab-treated HCC cells, and if any, (2) whether the specific eIF5A inhibitor GC7 could promote the efficacy of cetuximab in HCC. We found that treatment of GC7 to epithelial HCC cells significantly inhibited cell proliferation. Combination of GC7 and cetuximab enhanced either GC7- or cetuximab-mediated proliferation inhibition, indicating a synergistic effect between these two agents. Moreover, in cetuximab-treated cells (resistant predisposition), it was detected that eIF5A was elevated. Knockdown of eIF5A by siRNA or its inhibitor GC7 significantly blunted cell viability in cetuximab-treated cells. These data suggest that eIF5A2 is an alternative pathway for cell proliferation after HCC cell resistance to cetuximab. The single use of cetuximab to HCC cells may activate the eIF5A2 pathway which blunts the cetuximab-mediated proliferation inhibition in HCC cells. However, combination of cetuximab with GC7 significantly enhances the cytotoxicity of cetuximab, therefore illustrating that co-treatment of cetuximab and GC7 may be a potent therapeutic strategy against HCC.
Of note, the synergistic effect of GC7 with cetuximab was only observed in epithelial HCC cell lines HepG2, Huh7 and Hep 3B. This observation is the first report to reveal that HCC cells with distinct genotypes differ in response to GC7 and/or cetuximab. Moreover, this could be explained by the fact that epithelial cell lines had lower levels of p-STAT3 [10]. Activation of STAT3 was shown to play a role in tumor resistance to anticancer drugs, and down-regulation of pSTAT3 increased the sensitivity of human cancer cells to agents [23-25]. Hence, epithelial HCC cell lines that have lower levels of pSTAT3 predispose to anticancer drug sensitivity. Our findings may suggest that only epithelial HCC cell lines are sensitive to STAT3-related agents such as GC7 and cetuximab.
Interestingly, treatment of epithelial HCC cells with GC7 up-regulated the expression of pSTAT3, while it barely caused changes of EGFR expression. The elevation of p-STAT3 may help decrease cancer cell resistance to GC7 as aforementioned. There is also another possibility that GC7 itself may have some potential connection to the activation of STAT3. It would be interesting that further work is performed on the possible communication between GC7 and the activation of STAT3.
In all, our study illustrate that eIF5A2 is an alternative pathway for cell proliferation in cetuximab-treated HCC cells. Inhibition of eIF5A2 by GC7 may serve as a promising agent to promote the cytotoxicity of cetuximab in treating HCC. These effects are only remarkable in epithelial HCC cells. For mesenchymal HCC cells, other agents may be required.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81273260), Henan provincial key scientific and technological project (No. 132102310088).
Disclosure of conflict of interest
None.
Authors’ contribution
Conceived and designed the experiments: Hongwei Zhang; Performed the experiments: Fei Xue, Yanhui Liu, Haoyuan Chu; Analyzed the data: Qiang Tang, Erhui Xiao, Dongyi Zhang; Contributed reagents/materials/analysis tools: Yu Wen, Lei Yan; Wrote the paper: Fei Xue, Yanhui Liu, Haoyuan Chu.
References
- 1.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 2.Villanueva A, Minguez B, Forner A, Reig M, Llovet JM. Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu Rev Med. 2010;61:317–328. doi: 10.1146/annurev.med.080608.100623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruix J, Sherman M American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022. doi: 10.1002/hep.24199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 6.Rimassa L, Santoro A. The present and the future landscape of treatment of advanced hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S273–280. doi: 10.1016/S1590-8658(10)60516-6. [DOI] [PubMed] [Google Scholar]
- 7.Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, de Marinis F, Eberhardt WE, Paz-Ares L, Storkel S, Schumacher KM, von Heydebreck A, Celik I, O’Byrne KJ. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012;13:33–42. doi: 10.1016/S1470-2045(11)70318-7. [DOI] [PubMed] [Google Scholar]
- 8.Ezzoukhry Z, Louandre C, Trecherel E, Godin C, Chauffert B, Dupont S, Diouf M, Barbare JC, Maziere JC, Galmiche A. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer. 2012;131:2961–2969. doi: 10.1002/ijc.27604. [DOI] [PubMed] [Google Scholar]
- 9.Zhu AX, Stuart K, Blaszkowsky LS, Muzikansky A, Reitberg DP, Clark JW, Enzinger PC, Bhargava P, Meyerhardt JA, Horgan K, Fuchs CS, Ryan DP. Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer. 2007;110:581–589. doi: 10.1002/cncr.22829. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Shen X, Xia X, Xu G, Ma T, Bai X, Liang T. NSC 74859-mediated inhibition of STAT3 enhances the anti-proliferative activity of cetuximab in hepatocellular carcinoma. Liver Int. 2012;32:70–77. doi: 10.1111/j.1478-3231.2011.02631.x. [DOI] [PubMed] [Google Scholar]
- 11.Chen W, Hu QD, Xia XF, Liang C, Liu H, Zhang Q, Ma T, Liang F, Liang TB. Rapamycin enhances cetuximab cytotoxicity by inhibiting mTOR-mediated drug resistance in mesenchymal hepatoma cells. Cancer Biol Ther. 2014;15:992–999. doi: 10.4161/cbt.29113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng J, Li X, Lang X, Qiao C, Hu M, Yang J, Feng J, Lv M. Combination of cetuximab and rapamycin enhances the therapeutic efficacy in hepatocellular carcinoma. Technol Cancer Res Treat. 2014;13:377–385. doi: 10.7785/tcrt.2012.500389. [DOI] [PubMed] [Google Scholar]
- 13.Huang S, He R, Rong M, Dang Y, Chen G. Synergistic effect of MiR-146a mimic and cetuximab on hepatocellular carcinoma cells. Biomed Res Int. 2014;2014:384121. doi: 10.1155/2014/384121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jasiulionis MG, Luchessi AD, Moreira AG, Souza PP, Suenaga AP, Correa M, Costa CA, Curi R, Costa-Neto CM. Inhibition of eukaryotic translation initiation factor 5A (eIF5A) hypusination impairs melanoma growth. Cell Biochem Funct. 2007;25:109–114. doi: 10.1002/cbf.1351. [DOI] [PubMed] [Google Scholar]
- 15.Tang DJ, Dong SS, Ma NF, Xie D, Chen L, Fu L, Lau SH, Li Y, Li Y, Guan XY. Overexpression of eukaryotic initiation factor 5A2 enhances cell motility and promotes tumor metastasis in hepatocellular carcinoma. Hepatology. 2010;51:1255–1263. doi: 10.1002/hep.23451. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura K, Murozumi K, Shirahata A, Park MH, Kashiwagi K, Igarashi K. Independent roles of eIF5A and polyamines in cell proliferation. Biochem J. 2005;385:779–785. doi: 10.1042/BJ20041477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee NP, Tsang FH, Shek FH, Mao M, Dai H, Zhang C, Dong S, Guan XY, Poon RT, Luk JM. Prognostic significance and therapeutic potential of eukaryotic translation initiation factor 5A (eIF5A) in hepatocellular carcinoma. Int J Cancer. 2010;127:968–976. doi: 10.1002/ijc.25100. [DOI] [PubMed] [Google Scholar]
- 18.Chen KY, Liu AY. Biochemistry and function of hypusine formation on eukaryotic initiation factor 5A. Biol Signals. 1997;6:105–109. doi: 10.1159/000109115. [DOI] [PubMed] [Google Scholar]
- 19.Lee Y, Kim HK, Park HE, Park MH, Joe YA. Effect of N1-guanyl-1,7-diaminoheptane, an inhibitor of deoxyhypusine synthase, on endothelial cell growth, differentiation and apoptosis. Mol Cell Biochem. 2002;237:69–76. doi: 10.1023/a:1016535217038. [DOI] [PubMed] [Google Scholar]
- 20.Jakus J, Wolff EC, Park MH, Folk JE. Features of the spermidine-binding site of deoxyhypusine synthase as derived from inhibition studies. Effective inhibition by bis- and mono-guanylated diamines and polyamines. J Biol Chem. 1993;268:13151–13159. [PubMed] [Google Scholar]
- 21.Shi XP, Yin KC, Ahern J, Davis LJ, Stern AM, Waxman L. Effects of N1-guanyl-1,7-diaminoheptane, an inhibitor of deoxyhypusine synthase, on the growth of tumorigenic cell lines in culture. Biochim Biophys Acta. 1996;1310:119–126. doi: 10.1016/0167-4889(95)00165-4. [DOI] [PubMed] [Google Scholar]
- 22.Soerjomataram I, Lortet-Tieulent J, Parkin DM, Ferlay J, Mathers C, Forman D, Bray F. Global burden of cancer in 2008: a systematic analysis of disability-adjusted life-years in 12 world regions. Lancet. 2012;380:1840–1850. doi: 10.1016/S0140-6736(12)60919-2. [DOI] [PubMed] [Google Scholar]
- 23.Gariboldi MB, Ravizza R, Molteni R, Osella D, Gabano E, Monti E. Inhibition of Stat3 increases doxorubicin sensitivity in a human metastatic breast cancer cell line. Cancer Lett. 2007;258:181–188. doi: 10.1016/j.canlet.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Chiu HC, Chou DL, Huang CT, Lin WH, Lien TW, Yen KJ, Hsu JT. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem Pharmacol. 2011;81:1263–1270. doi: 10.1016/j.bcp.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 25.Chen CC, Chen WC, Lu CH, Wang WH, Lin PY, Lee KD, Chen MF. Significance of interleukin-6 signaling in the resistance of pharyngeal cancer to irradiation and the epidermal growth factor receptor inhibitor. Int J Radiat Oncol Biol Phys. 2010;76:1214–1224. doi: 10.1016/j.ijrobp.2009.09.059. [DOI] [PubMed] [Google Scholar]