

Abstract
Diabetic patients are at high risk of developing delayed cutaneous wound healing. Proper keratinocyte proliferation and migration are crucial steps during re-epithelialization. Melatonin (Mel) accelerates wound repair in full-thickness incisional wounds; however, its role in diabetic wound healing is unknown. This study explored the role of Mel in diabetic wound healing in vitro by using high glucose (HG)-cultured keratinocytes. Mel reduced the HG-induced mRNA expression and release of pro-inflammatory cytokines, including tumor necrosis factor-α, interleukin (IL)-1β, IL-6, and IL-8, in keratinocytes. Mel inhibited oxidative stress, as evidenced by reduced production of reactive oxygen species and malondialdehyde and increased activity of superoxide dismutase in HG-stimulated keratinocytes. Mel also inhibited HG-induced nucleotide binding oligomerization domain-like receptor family pyrin domain-containing 3 inflammasome activation in keratinocytes. HG-induced reduced migration and proliferation and increased apoptosis of keratinocytes were counteracted by Mel treatment. The pro-proliferative, pro-migratory, and anti-apoptotic effects of Mel on HG-treated keratinocytes were mediated by extracellular signal-regulated kinase signaling pathway. Results collectively suggested that Mel is an alternative therapeutic strategy to ameliorate poor condition for diabetic wound healing by regulating keratinocyte activity.
Keywords: Diabetic wound healing, high glucose, chronic inflammation, oxidative stress, melatonin, keratinocyte
Introduction
Diabetes mellitus (DM) is a chronic inflammatory disease. Diabetic foot ulcer (DFU) is a common and tiring complication of DM and the leading cause of disability and mortality among diabetics [1]. Up to 15% of diabetic patients develop chronic DFU, and 84% of these cases lead to amputation [1,2]. Although therapy for DFU was reinforced in the past few years, improvement remains limited. Thus, novel effective therapeutic treatments for this chronic diabetes complication are urgently needed.
Keratinocyte is critical for wound closure because of its role in re-epithelialization, which is mediated by proliferation and migration of keratinocytes [3]. Diabetic wound healing differs from the normal healing process [4]. Normal wound healing is a carefully orchestrated process involving proper induction of inflammatory cytokines. However, persistence of inflammation is associated with impaired re-epithelialization among diabetic patients [5]. Dysregulation of glucose homeostasis and elevated glucose levels are the central etiologies of diabetes [6]. Hyperglycemia increases the levels of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6, and increases oxidative stress, and these factors mainly cause impaired wound healing [7,8]. Excessive production of reactive oxygen species (ROS), resulting in damages to cellular membranes, lipids, proteins, and DNA, becomes deleterious to wound healing [5,9]. Studies have demonstrated that nucleotide binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is activated in DM patients [10]. Sustained activation of NLRP3 inflammasome in macrophages impairs wound healing in humans and mice with type 2 diabetes [11]. A recent study has shown that high glucose (HG) primes NLRP3 inflammasome in mesangial cells via a ROS-mediated pathway [12]. In addition, HG stimulation promotes ROS generation as well as inhibits the proliferation and migration and reduces the apoptosis of keratinocytes [13,14]. The role of extracellular signal-regulated kinase (ERK) signaling in specific aspects of wound healing, including cell proliferation, migration, and apoptosis, has begun to be investigated [15]. ERK phosphorylation is significantly suppressed in keratinocytes under HG conditions [13]. Moreover, the proliferative and migratory activities of adiponectin-stimulated keratinocytes were inhibited by ERK inhibitors [16]. However, it remains unknown whether ERK signaling is involved in proliferation, migration, and apoptosis of keratinocytes under HG conditions.
Melatonin (Mel), an indoleamine synthesized from tryptophan, is produced by the pineal gland and other organs, including retina, gastrointestinal tract, and skin [17]. Mel possesses widespread neuroprotective, antioxidant, and anti-inflammatory properties [18,19]. Studies have demonstrated that Mel can prevent several complications of diabetes, such as pancreatic, renal, liver, neural, and corneal injuries [20-24]. Mel notably accelerates wound repair in full-thickness incisional wounds [25]. Although a range of protective effects of Mel have been reported in diabetes-related diseases, the underlying molecular mechanisms of its action on diabetic wound healing have not yet been fully evaluated.
By using a cultured keratinocyte cell model, we found that Mel reduced HG-induced pro-inflammatory cytokine levels and oxidative stress. Mel inhibited HG-induced NLRP3 inflammasome activation in keratinocytes. Mel promoted the proliferation and migration and reduced the apoptosis of HG-cultured keratinocytes. The pro-proliferative, pro-migratory, and anti-apoptotic effects of Mel on HG-cultured keratinocytes were dependent on ERK signaling. Overall, Mel accelerated diabetic wound healing in vitro by regulating keratinocyte activity.
Materials and methods
Keratinocyte isolation and culture
Primary keratinocytes were derived from neonatal SD rats (postnatal day 1) as previously described [13]. The rats were successively washed with water, 5% polyvinylpyrrolidone, and 70% ethanol and then rinsed with phosphate-buffered saline (PBS) at least thrice. Full-thickness skins were cultivated from rat trunk under aseptic conditions and soaked in penicillin and streptomycin (all from Sigma-Aldrich, St. Louis, MO, USA) for 15 min. The fat and membranous materials were subsequently removed, and the tissues were cut into 1 cm × 1 cm pieces and incubated overnight in 2.5 mg/mL dispase (Invitrogen, Carlsbad, CA, USA) at 4°C. The epidermis was mechanically separated from the underlying dermis, washed, minced, digested with 0.25% trypsin-EDTA at 37°C for 30 min, and then cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 15% fetal bovine serum (FBS; Gibco). The mixture was filtered through a sterile 100 μm filter to remove the undigested fragments. The acquired suspension was subsequently centrifuged, suspended in DermaLife K Calcium-Free Medium and supplements (Lifeline Cell Technology, Walkersville, MD, USA), and plated onto collagen I-coated culture dishes (Becton Dickinson, Bedford, MA, USA).
Keratinocyte treatment
The keratinocyte treatments included the following: (1) Control group: cells were cultured in normal glucose (NG; 6 mM) medium; (2) HG group: cells were cultured in 26 mM glucose medium; (3) HG + Mel group: the cells were incubated with 1 mM Mel for 24 h prior to HG treatment. At the indicated time points after NG or HG incubation, the cells and supernatants were collected for subsequent experiments. In blocking experiments, keratinocytes were pretreated with 10 μM ERK inhibitor (U0126; Cell Signaling Technology, Danvers, MA, USA) for 30 min, washed, and incubated with different treatments for the indicated time points.
Quantitative real-time PCR (qPCR)
Total RNA from keratinocytes was extracted using the TRIzol method (Invitrogen) and processed according to the manufacturer’s instructions. Using a reverse transcription kit (Promega, Madison, WI, USA), approximately 5 mg of RNA was reverse transcribed into cDNA, which served as template for PCR. Amplification and detection were performed on an ABI Prism 7500 sequence detection system (Applied Biosystems, Hammonton, NJ, USA). The fold changes in mRNA level were calculated by 2-ΔΔCt. Glycerinaldehyd-3-phosphate dehydrogenase (GAPDH) was used as endogenous control. The primer sequences used are listed: for TNF-α, 5’-TTG ACCTCAGCGCTGAGTTG-3’ (forward), and 5’-CCTGTAGCCCACGTCGTAGC-3’ (reverse); for IL-1β, 5’-CAGGATGAGGACATGAGCACC-3’ (forward), and 5’-CTC TGCAGACTCAAACTCCAC-3’ (reverse); for IL-6, 5’-GTACTCCAGAAGACCAG AGG-3’ (forward), and 5’-TGCTGGTGACAACCACGGCC-3’ (reverse); for IL-8, 5’-TCAACGGGCAGAATCAAAGAG-3’ (forward), and 5’-CTCAGACAGCGAGGC ACATC-3’ (reverse), and for GAPDH, 5’-AGGTCGGTGTGAACGGATTTG-3’ (forward), and 5’-TGTAGACCATGTAGTTGAGGTCA-3’ (forward).
Enzyme-linked immunosorbent assay (ELISA)
Supernatants derived from keratinocytes cultivated for 72 h after NG or HG treatment were collected and stored at -20°C. TNF-α, IL-1β, IL-6, and IL-8 productions were determined using commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Absorbance was read on a microplate reader at 490 nm (BioTek Instruments Inc., Winooski, VT, USA).
Measurement of intracellular ROS
Measurement of intracellular ROS was based on ROS-mediated conversion of the non-fluorescent 2’,7’-dichlorofluorescein diacetate (DCFH-DA) into fluorescent 2’,7’-dichlorofluorescein (DCFH). Briefly, keratinocytes were seeded into a 6-well plate at a density of 1 × 104 cells/well. After performing different treatments, DCFH-DA was added into each well at a final concentration of 10 μM, and the plates were incubated at 37°C for 30 min in the dark. The cells were washed twice with PBS, and DCFH fluorescence from each well was excited at 485 nm; fluorescence emission was measured at 520 nm by using a microplate reader (BioTek).
Measurement of superoxide dismutase (SOD) and malondialdehyde (MDA) levels
SOD and MDA levels were determined using commercially available kits (Nanjing Jiancheng Biological Engineering Institute, Nanjing, China). Briefly, following different treatments, keratinocytes were lysed and centrifuged at 12,000 g for 10 min at 4°C, and then the supernatants were collected. SOD and MDA levels in the supernatant were measured according to the protocols and then analyzed on a microplate reader (BioTek). The absorbance was read at 450 (SOD) and 530 nm (MDA). The levels were calculated using a standard calibration curve and expressed in U/mL (SOD) and μmol/mg protein (MDA). Data were expressed relative to those of the control.
Cell viability assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, the keratinocytes were seeded on 96-well plates at a density of 1 × 104 cells/well and incubated with different treatments for 72 h. MTT (Sigma; 0.5 mg/mL) was subsequently added into each well and then incubated at 37°C for 4 h. The formazan product was dissolved in 100 μL of dimethyl sulfoxide at 37°C for 30 min, and absorbance at 570 nm was measured with a microplate reader (BioTek).
BrdU incorporation assay
Keratinocytes were seeded into 96-well plates (1 × 104 cells/well) and cultured for 72 h under different treatments. The cells were subsequently incubated with a medium containing BrdU (Boster Biological Engineering, Wuhan, China) for 2 h. BrdU incorporation was measured using a cell-proliferating ELISA kit (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s instructions.
Clonogenic assay
Keratinocytes were seeded on 6-well plates in triplicate at a density of 1000 cells/well in 1.5 mL of DMEM containing 10% FBS under different treatments. The cells were kept under humidified atmosphere of 95% air and 5% CO2 at 37°C and allowed to grow for 14 days. The culture medium was replaced every 3 days. The cell colonies were fixed in 4% paraformaldehyde and stained with 0.5% crystal violet (Sigma) followed by rinsing with PBS thrice. Colonies consisting of more than 50 cells were counted under a microscope (Olympus, Tokyo, Japan). All colonies were photographed for subsequent analysis, and area is measured using the Image J program (National Institutes of Health, USA).
Wound healing assay
Keratinocytes were seeded on 6-well plates at a density of 1 × 104 cells/well and cultured for 72 h under different treatments. After reaching subconfluency, wounds were created at the center of each well by scraping, and culture debris was removed by washing with PBS. At 0 and 24 h post-injury, keratinocyte migration was observed under a phase-contrast microscope (Olympus). Wound closure was determined by identifying the cell migration front and by calculating the ratio of the migration area to the area at time 0 h.
Transwell migration assay
Keratinocyte migration was quantitatively assayed on a Millicell chamber (8 μm pores; Transwell, Millipore, Billerica, MA). Subconfluent keratinocytes were added into the upper wells (1 × 104 cells/well), and a DMEM-containing hepatocyte growth factor was added to the lower chamber for chemotaxis. The chamber was incubated for 72 h at 37°C in a humidified air atmosphere with 5% CO2. The cells on the upper surface of the membrane were mechanically removed by scraping with a rubber blade, and the migrated cells on the lower surface of the membrane were fixed and stained with 0.5% crystal violet (Sigma). The total number of migrated cells on the lower surface of the membrane was counted under a microscope (Olympus).
Flow cytometry
Keratinocyte apoptosis was measured via flow cytometry by using an Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. Briefly, variously treated keratinocytes were harvested through trypsinization, washed twice with PBS, and resuspended in binding buffer. Annexin V-FITC and PI solution were subsequently added to stain the cells before analysis via FACScan flow cytometry (BD Biosciences) by using FlowJo software v7.6 (Tree Star Inc.). Apoptosis ratio was defined as the percentage of Annexin V-FITC-positive cells.
Measurement of mitochondrial membrane potential (MMP)
MMP of keratinocytes was measured using the lipophilic cationic probe 5,5’,6, ’tetrachloro-1,1’,3,3’-tetraethylbenzimidazol-carbocyanine iodide (JC-1; Molecular Probes, Carlsbad, CA, USA). JC-1 selectively enters into a mitochondrion, where it aggregates when the membrane potential exceeds 80-100 mV, causing a shift in fluorescence from 530 nm (green) to 590 nm (red). Briefly, following different treatments, keratinocytes were incubated with 5 mM JC-1 at 37°C for 20 min. The cells were subsequently washed with PBS thrice and immediately analyzed with a fluorescence microscope (Olympus). The ratio of red to green fluorescence intensity was measured, and data were expressed relative to those of the control group.
Caspase-3 and caspase-1 activity assay
Activities of caspase-3 and caspase-1 in keratinocytes were measured using colorimetric assay kits (BioVision, Palo Alto, CA, USA) according to the manufacturers’ protocols. Briefly, following different treatments, keratinocytes (1 × 106) were lysed using Cell Lysis Buffer and centrifuged at 10,000 g for 10 min at 4°C, and the supernatants were collected. For caspase-3 and caspase-1 activity assays, 50 μL of 2 × Reaction Buffer and 5 μL of caspase-3 substrate (DEVD-pNA, 4 mM) or caspase-1 substrate (YVAD-AFC, 1 mM) were added into 50 μL of cell lysate. The reaction mixtures were incubated at 37°C for 2 h, and absorbance was read at 405 nm by a microplate reader (BioTek). The activity of caspases was expressed in micromole of pNA released per minute per milliliter of cell lysate and compared with that of the control.
Western blot analysis
Proteins from keratinocytes were extracted through radioimmunoprecipitation assay buffer (Beyotime Biotechnology, Haimen, China). The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). After blocking with 5% nonfat milk in TBS containing 0.1% Tween 20 for 2 h at 37°C, the membranes were incubated overnight with primary antibodies against NLRP3, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), caspase-1-p20 (all obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA), phosphorylated (p)-ERKs, total ERK, p-p38 MAPK, p38 MAPK, p-JNK, JNK, and β-actin (all obtained from Cell Signaling, Beverly, MA) at 4°C. The membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) for 1 h at 37°C. The proteins were visualized using a chemiluminescence detection system (Pierce, Rockford, IL, USA). The blots were analyzed using a FluorChem FC system (Alpha Innotech, San Jose, California, USA).
Statistical analysis
All data were expressed as means ± standard deviation (SD). Statistical analysis was performed using one-way ANOVA followed by Dunnett’s post-hoc test. SPSS 16.0 software (Chicago, IL, USA) was used for statistical analysis. Significance was accepted at P < 0.05.
Results
Mel counteracts the increase in mRNA and protein expression of pro-inflammatory cytokines in HG-stimulated keratinocytes
DFU is accompanied by chronic inflammation. To analyze the anti-inflammatory effects of Mel on HG-stimulated keratinocytes, we measured the mRNA expression and release of pro-inflammatory cytokines by using qPCR and ELISAs. mRNA expression of TNF-α (Figure 1A), IL-1β (Figure 1B), IL-6 (Figure 1C), and IL-8 (Figure 1D) in cultured keratinocytes under HG condition for 72 h was significantly increased compared with that in the NG-treated keratinocytes. The release of TNF-α (Figure 1E), IL-1β (Figure 1F), IL-6 (Figure 1G), and IL-8 (Figure 1H) in the supernatants derived from the HG-treated keratinocytes was considerably higher than that in NG cultures. However, Mel treatment markedly reduced the increase in mRNA expression and production of pro-inflammatory cytokines in HG-challenged keratinocytes (Figure 1A-H). These results suggest that Mel reduced HG-induced mRNA expression and production of pro-inflammatory cytokines in keratinocytes.
Figure 1.
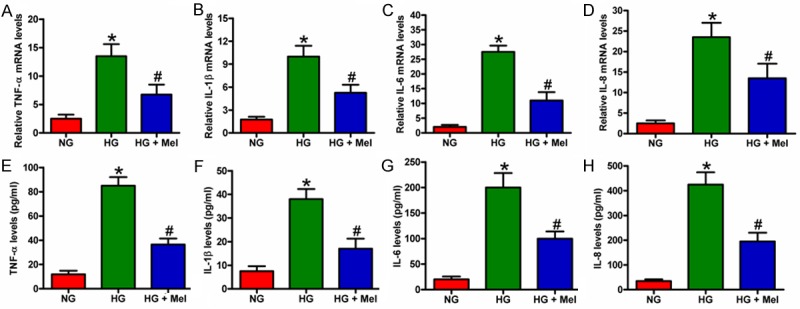
Mel reduced the mRNA expression and release of pro-inflammatory cytokines in HG-cultured keratinocytes. Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. (A-D) mRNA expression of (A) TNF-α, (B) IL-1β, (C) IL-6, and (D) IL-8 was measured by qPCR assays. GAPDH was used as endogenous control. (E-H) Release of (E) TNF-α, (F) IL-1β, (G) IL-6, and (H) IL-8 in the supernatants of the cells were detected by ELISAs. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group. NG: normal glucose; HG: high glucose; Mel: melatonin.
Mel alleviates oxidative stress in keratinocytes under HG condition
Increased oxidative stress is associated with DFU. We used DCFH-DA fluorescent staining to determine whether Mel can inhibit HG-induced ROS generation in keratinocytes. As shown in Figure 2A, intracellular ROS generation markedly increased in HG-treated keratinocytes. In addition, the level of MDA, a surrogate marker for estimation of damage induced by ROS, was significantly increased in HG-cultivated keratinocytes compared with that in the control (Figure 2B). However, Mel inhibited ROS and MDA production in HG-stimulated keratinocytes. By contrast, the activity of SOD, an anti-oxidative enzyme, was significantly reduced by HG challenge and attenuated by Mel treatment (Figure 2C). These results imply that Mel relieved oxidative stress in HG-stimulated keratinocytes.
Figure 2.
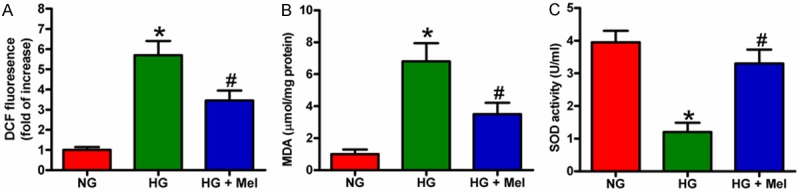
Mel inhibited ROS and MDA generation and enhanced SOD activity in HG-stimulated keratinocytes. Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. (A) After treatment, the cells were labeled with DCFH-DA to detect ROS generation. (B and C) (B) MDA level and (C) SOD activity were measured by specific commercial kits. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group. NG: normal glucose; HG: high glucose; Mel: melatonin.
Mel inhibits activation of NLRP3 inflammasome in HG-cultivated keratinocytes
To determine whether the activation of NLRP3 inflammasome was inhibited by Mel in keratinocytes under HG environment, we assessed the expression of NLRP3, ASC, and caspase-1-p20 through Western blot analysis. The expression of NLRP3, ASC, and caspase-1-p20 significantly increased in the HG-treated keratinocytes, although this trend was counteracted by Mel treatment (Figure 3A and 3B). In addition, Mel treatment reduced the increase in caspase-1 activity in HG-challenged keratinocytes (Figure 3C). These results indicate that Mel inhibited HG-induced NLRP3 inflammasome activation in keratinocytes.
Figure 3.
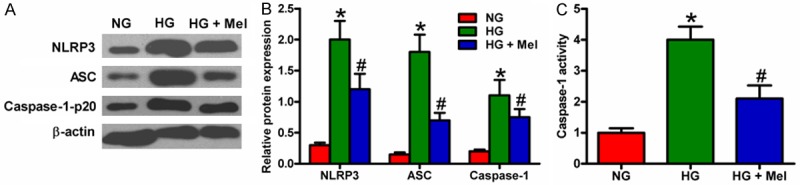
Mel inhibited HG-induced activation of NLRP3 inflammasome in keratinocytes. Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. (A) Western blot analyses of NLRP3, ASC, and caspase-1-p20 in keratinocytes. β-actin was used as endogenous control. (B) Quantification of NLRP3, ASC, and caspase-1-p20 in (A) was normalized to that of β-actin. (C) Caspase-1 activity in keratinocytes was measured by a colorimetric assay kit. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group.
Mel enhances the proliferation of HG-treated keratinocytes
We investigated the effect of Mel on keratinocyte proliferation using MTT, BrdU, and colony formation assays. MTT assay showed that Mel increased the viability of HG-treated keratinocytes compared with that of the HG group (Figure 4A). BrdU incorporation assay showed that HG significantly inhibited keratinocyte proliferation, which was attenuated by Mel treatment (Figure 4B). In addition, the number of colonies was significantly reduced in HG-challenged keratinocytes compared with that in the NG-treated cells. However, Mel treatment increased the number of colonies (Figure 4C and 4D). These data demonstrate that Mel promoted cell growth of HG-cultured keratinocytes.
Figure 4.
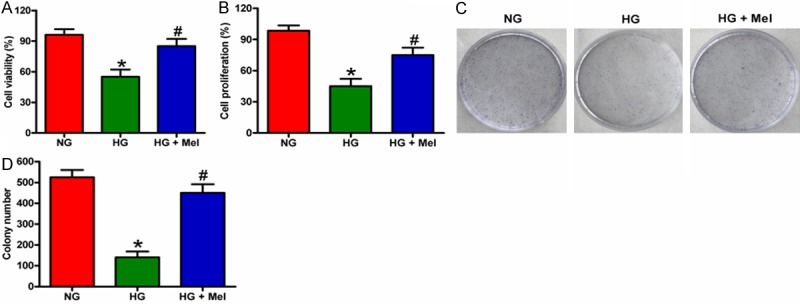
Mel increased the proliferation of HG-treated keratinocytes. (A and B) Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. (A) Cell viability was assessed by MTT assay. (B) Cell proliferation was measured by BrdU incorporation assay. (C and D) Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM). The cells were subsequently cultured for 14 days, and the medium was replaced every 3 days. The cell colonies were stained and photographed. (D) The number of colonies in (C) was counted. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group. NG: normal glucose; HG: high glucose; Mel: melatonin.
Mel inhibits HG-induced apoptosis of keratinocytes
To evaluate the role of Mel in the apoptosis of HG-cultured keratinocyte, we performed flow cytometry, JC-1 staining, and caspase-3 activity assays. As shown in Figure 5A, apoptosis increased in HG-stimulated keratinocytes, whereas Mel treatment negatively modulated apoptosis. The inhibitory effect of Mel on keratinocyte apoptosis was also confirmed by JC-1 staining (Figure 5B). In addition, Mel treatment markedly reduced the activity of caspase-3 in HG-challenged keratinocytes (Figure 5C). These results collectively suggest that HG promoted keratinocyte apoptosis, and this effect was attenuated by Mel treatment.
Figure 5.
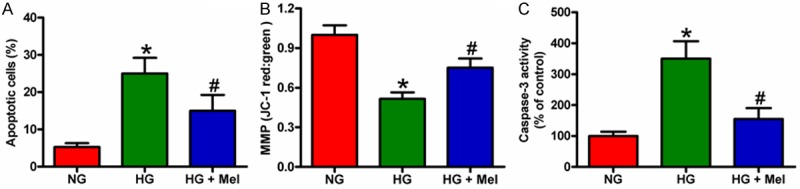
Mel reduced the apoptosis of HG-treated keratinocytes. Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. A. Apoptosis of keratinocytes was analyzed by flow cytometry and the apoptotic rate was calculated. B. JC-1 staining was performed to evaluate the cell apoptosis. C. Quantification of caspase-3 activity in keratinocytes. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group. NG: normal glucose; HG: high glucose; Mel: melatonin.
Mel induces migration of HG-treated keratinocytes
Keratinocyte migration is critical in wound re-epithelialization. We thus investigated the effect of Mel on keratinocyte migration by using two types of in vitro assay systems. Transwell migration assay showed that keratinocyte migration was significantly inhibited in HG medium, whereas Mel treatment significantly accelerated cell migration (Figure 6A). Wound healing assay also showed that Mel promoted the mobility of HG-treated keratinocytes (Figure 6B). These results demonstrate that Mel could reverse the impaired migration of keratinocytes caused by HG stimulation.
Figure 6.
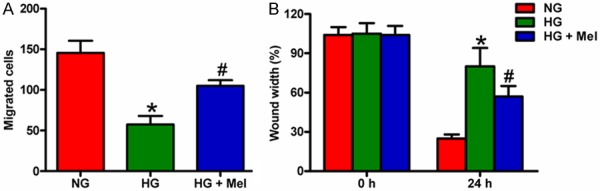
Mel promoted the migration of HG-treated keratinocytes. Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. A. Keratinocyte migration was assayed quantitatively using the Transwell migration assay. After adding keratinocytes into the upper wells and incubating overnight, the migrated cells were counted. B. Scratch wound healing assay was performed to evaluate the effect of Mel on keratinocyte migration. The percentage of wound closures at the indicated time points was measured and calculated. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group. NG: normal glucose; HG: high glucose; Mel: melatonin.
ERK signaling is involved in Mel-promoted survival and migration of HG-treated keratinocytes
MAPKs, including ERK1/2, JNK, and p38 MAPK, enhance the proliferation and migration and suppress apoptosis of keratinocytes [26]. Therefore, we investigated whether Mel could induce MAPK activation in keratinocytes. HG markedly inhibited the phosphorylation of ERK1/2 in keratinocytes, whereas Mel induced significant phosphorylation of ERK1/2. However, Mel exerted no effect on phosphorylation of p38 MAPK and JNK (Figure 7A). To confirm the role of ERK signaling in keratinocyte proliferation, we incubated keratinocytes with U0126 (10 μM) for 30 min prior to HG treatment. As shown in Figure 7B, Mel-enhanced proliferation of HG-challenged keratinocytes was attenuated by U0126 pretreatment. We subsequently investigated whether ERK signaling is involved in anti-apoptotic effect of Mel in HG-treated keratinocytes. We found that preincubation with U0126 significantly counteracted the Mel-induced inhibition of apoptosis of HG-treated keratinocytes (Figure 7C). In addition, the pro-migratory effect of Mel on HG-treated keratinocyte was weakened by U0126 administration (Figure 7D). These results indicate that ERK signaling mediated Mel-induced keratinocyte proliferation, apoptosis resistance, and migration in HG-treated keratinocytes.
Figure 7.
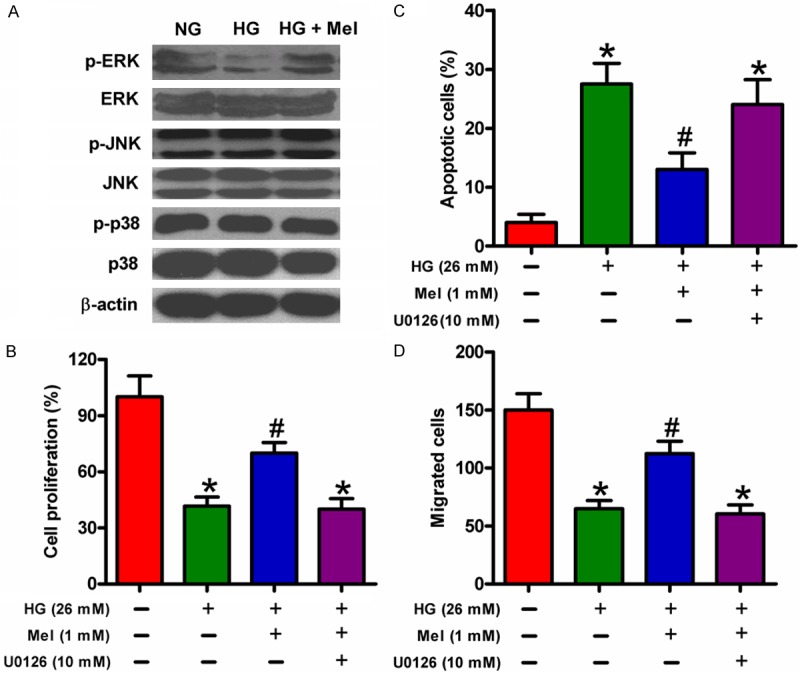
Pro-proliferative, pro-migratory, and anti-apoptotic effects of Mel on HG-treated keratinocytes were mediated by ERK signaling. A. Keratinocytes were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. Representative Western blot results of ERK1/2, p-ERK1/2, JNK, p-JNK, p38 MAPK, and p-p38 MAPK are shown. β-actin was used as endogenous control. B-D. Keratinocytes were preincubated with or without U0126 (10 μM) for 30 min. The cells were treated with or without 1 mM Mel 24 h prior to treatment with NG (6 mM) or HG (26 mM) for 72 h. B. Keratinocyte proliferation was analyzed using the BrdU assay. C. Flow cytometry was performed to measure apoptotic cells. D. Migration of keratinocyte was measured by Transwell migration assay. Data are means ± SD of three independent experiments. *P < 0.05 vs. NG group; #P < 0.05 vs. HG group. HG: high glucose; Mel: melatonin.
Discussion
The current study demonstrated the essential role of Mel in diabetic wound healing. Our key findings were as follows: First, Mel reduced the mRNA expression and release of pro-inflammatory cytokine in keratinocytes under HG condition. Second, Mel reduced HG-induced oxidative stress in keratinocytes. Third, HG-induced activation of NLRP3 inflammasome was inhibited by Mel treatment. Last, Mel-induced enhanced proliferation and migration as well as reduced apoptosis of keratinocytes were mediated by ERK signaling. Thus, all data confirmed that Mel contributes to important events in diabetic wound healing, including inflammation suppression, oxidative stress attenuation, proliferation and migration promotion, and apoptosis inhibition in keratinocytes under HG condition.
Diabetic wounds are generally chronic wounds characterized by sustained inflammatory phase. Prolonged inflammation may lead to permanent residence of neutrophils and macrophages in wound microenvironment, thereby abrogating normal healing and transforming wounds into non-healing chronic ulcers [27,28]. Wound bed of a diabetic wound is overpopulated by several pro-inflammatory cells and cytokines [29]. Pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, are primarily released by immune cells early in wound healing, and they act as effectors for keratinocytes and fibroblasts, stimulating tissue deposition and epithelialization [30,31]. Diabetic individuals are more susceptible to both wound infection and hyperinflammation because of elevated levels of pro-inflammatory cytokines, including TNF-α and IL-6 [32]. High levels of these cytokines are indeed found in diabetic ulcers [33]. Prolonged inflammation may be attributed to hyperglycemia and endogenous ligands [34]. Recent reports suggest that activation of NLRP3 inflammasome is a key contributor to delayed healing of diabetic wounds [10,35]. NLRP3 inflammasome has been proposed to sense and mediate downstream inflammatory events of “glucotoxicity” during pathogenesis of type 2 diabetes and thus is responsible for a constant pro-inflammatory status [36]. Mirza et al. [11] demonstrated that sustained activation of NLRP3 inflammasome in macrophages impairs wound healing in type 2 diabetic humans and mice. Mel alleviates acute lung injury by inhibiting activation of NLRP3 inflammasome [37]. The present study found that Mel inhibited pro-inflammatory cytokine levels and NLRP3 inflammasome activation in keratinocytes under HG condition, suggesting that Mel exerts anti-inflammatory effect during diabetic wound healing.
Oxidative stress is prevalent in diabetes. Excessive ROS causes damage to proteins, lipids, and DNA in different cells, ultimately leading to cell death and tissue dysfunction [5,9]. Hyperglycemia leads to oxidative stress when ROS production exceeds antioxidant capacity [38]. ROS accumulation participates in the development of complications of diabetes and propagates excessive inflammatory cascade, which is deleterious to wound healing and impedes formation of new tissues [5,9,39,40]. Consistent with these reports, our results demonstrate that keratinocytes cultivated in HG environment showed increased levels of ROS and MDA as well as reduced SOD activity. However, Mel treatment reversed the alterations of ROS and MDA production and SOD activity, suggesting that Mel exerted an anti-oxidative effect on HG-cultured keratinocytes. ROS can function as a secondary signaling molecule in cancer cells, playing a key role in cell proliferation, apoptosis, differentiation, migration, and invasion [41]. Furthermore, ROS is overproduced in keratinocytes after HG stimulation, and incubation of the ROS scavenger N-acetyl-L-cysteine enhances the proliferation and migration of keratinocytes, suggesting that ROS induced by HG environment may impair wound healing by suppressing the function of keratinocytes [13,14]. The present data demonstrate that Mel enhanced the proliferation and migration and inhibited the apoptosis of keratinocytes under HG condition, suggesting that Mel promotes diabetic wound closure by regulating keratinocyte activity.
Multiple signaling pathways are involved in proliferation and migration of keratinocytes, and ERK signaling pathway has been extensively investigated. Li et al. [13] showed that HG caused oxidative stress, thereby inhibiting ERK signaling pathways in keratinocytes. A recent study has demonstrated that adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signaling pathway [16]. We conjectured that ERK signaling pathway is involved in Mel-mediated enhancement of proliferation and migration and in suppression of apoptosis in HG-stimulated keratinocytes. ERK phosphorylation was significantly inhibited by HG treatment but rescued by Mel treatment. The increase in proliferation and migration and reduction in apoptosis of keratinocytes were specifically abolished by pretreatment with U0126, a selective inhibitor of ERK. These results demonstrated that increased migration and proliferation and reduced apoptosis of HG-stimulated keratinocytes caused by Mel were dependent on ERK signaling pathway.
In summary, Mel promotes diabetic wound healing in vitro by restricting the mRNA expression and release of pro-inflammatory cytokine, by reducing oxidative stress, by inhibiting NLRP3 inflammasome activation, by enhancing proliferation and migration, and by reducing apoptosis in HG-stimulated keratinocytes. The pro-proliferative, pro-migratory, and anti-apoptotic effects of Mel in HG-treated keratinocytes were mediated by ERK signaling pathway. Therefore, Mel is a potential therapeutic agent for DFU by regulating keratinocyte activity.
Disclosure of conflict of interest
None.
References
- 1.Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden of diabetic foot disease. Lancet. 2005;366:1719–1724. doi: 10.1016/S0140-6736(05)67698-2. [DOI] [PubMed] [Google Scholar]
- 2.Bartus CL, Margolis DJ. Reducing the incidence of foot ulceration and amputation in diabetes. Curr Diab Rep. 2004;4:413–418. doi: 10.1007/s11892-004-0049-x. [DOI] [PubMed] [Google Scholar]
- 3.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 4.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 5.Pierce GF. Inflammation in nonhealing diabetic wounds: the space-time continuum does matter. Am J Pathol. 2001;159:399–403. doi: 10.1016/S0002-9440(10)61709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wertheimer E, Spravchikov N, Trebicz M, Gartsbein M, Accili D, Avinoah I, Nofeh-Moses S, Sizyakov G, Tennenbaum T. The regulation of skin proliferation and differentiation in the IR null mouse: implications for skin complications of diabetes. Endocrinology. 2001;142:1234–1241. doi: 10.1210/endo.142.3.7988. [DOI] [PubMed] [Google Scholar]
- 7.Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- 8.Kant V, Gopal A, Pathak NN, Kumar P, Tandan SK, Kumar D. Antioxidant and anti-inflammatory potential of curcumin accelerated the cutaneous wound healing in streptozotocin-induced diabetic rats. Int Immunopharmacol. 2014;20:322–330. doi: 10.1016/j.intimp.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 9.auf dem Keller U, Kumin A, Braun S, Werner S. Reactive oxygen species and their detoxification in healing skin wounds. J Investig Dermatol Symp Proc. 2006;11:106–11. doi: 10.1038/sj.jidsymp.5650001. [DOI] [PubMed] [Google Scholar]
- 10.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes. 2014;63:1103–1114. doi: 10.2337/db13-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng H, Gu J, Gou F, Huang W, Gao C, Chen G, Long Y, Zhou X, Yang M, Liu S, Lu S, Luo Q, Xu Y. High Glucose and Lipopolysaccharide Prime NLRP3 Inflammasome via ROS/TXNIP Pathway in Mesangial Cells. J Diabetes Res. 2016;2016:6973175. doi: 10.1155/2016/6973175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Zhao Y, Hao H, Dai H, Han Q, Tong C, Liu J, Han W, Fu X. Mesenchymal stem cell-conditioned medium improves the proliferation and migration of keratinocytes in a diabetes-like microenvironment. Int J Low Extrem Wounds. 2015;14:73–86. doi: 10.1177/1534734615569053. [DOI] [PubMed] [Google Scholar]
- 14.Long M, Rojo de la Vega M, Wen Q, Bharara M, Jiang T, Zhang R, Zhou S, Wong PK, Wondrak GT, Zheng H, Zhang DD. An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes. 2016;65:780–793. doi: 10.2337/db15-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeigler ME, Chi Y, Schmidt T, Varani J. Role of ERK and JNK pathways in regulating cell motility and matrix metalloproteinase 9 production in growth factor-stimulated human epidermal keratinocytes. J Cell Physiol. 1999;180:271–284. doi: 10.1002/(SICI)1097-4652(199908)180:2<271::AID-JCP15>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 16.Shibata S, Tada Y, Asano Y, Hau CS, Kato T, Saeki H, Yamauchi T, Kubota N, Kadowaki T, Sato S. Adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signaling pathway. J Immunol. 2012;189:3231–3241. doi: 10.4049/jimmunol.1101739. [DOI] [PubMed] [Google Scholar]
- 17.Acuna-Castroviejo D, Escames G, Venegas C, Diaz-Casado ME, Lima-Cabello E, Lopez LC, Rosales-Corral S, Tan DX, Reiter RJ. Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci. 2014;71:2997–3025. doi: 10.1007/s00018-014-1579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paredes SD, Terron MP, Marchena AM, Barriga C, Pariente JA, Reiter RJ, Rodriguez AB. Effect of exogenous melatonin on viability, ingestion capacity, and free-radical scavenging in heterophils from young and old ringdoves (Streptopelia risoria) Mol Cell Biochem. 2007;304:305–314. doi: 10.1007/s11010-007-9513-7. [DOI] [PubMed] [Google Scholar]
- 19.Park K, Lee Y, Park S, Lee S, Hong Y, Kil Lee S, Hong Y. Synergistic effect of melatonin on exercise-induced neuronal reconstruction and functional recovery in a spinal cord injury animal model. J Pineal Res. 2010;48:270–281. doi: 10.1111/j.1600-079X.2010.00751.x. [DOI] [PubMed] [Google Scholar]
- 20.Negi G, Kumar A, Sharma SS. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-kappaB and Nrf2 cascades. J Pineal Res. 2011;50:124–131. doi: 10.1111/j.1600-079X.2010.00821.x. [DOI] [PubMed] [Google Scholar]
- 21.Kanter M, Uysal H, Karaca T, Sagmanligil HO. Depression of glucose levels and partial restoration of pancreatic beta-cell damage by melatonin in streptozotocin-induced diabetic rats. Arch Toxicol. 2006;80:362–369. doi: 10.1007/s00204-005-0055-z. [DOI] [PubMed] [Google Scholar]
- 22.Derlacz RA, Poplawski P, Napierala M, Jagielski AK, Bryla J. Melatonin-induced modulation of glucose metabolism in primary cultures of rabbit kidney-cortex tubules. J Pineal Res. 2005;38:164–169. doi: 10.1111/j.1600-079X.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 23.Guven A, Yavuz O, Cam M, Ercan F, Bukan N, Comunoglu C, Gokce F. Effects of melatonin on streptozotocin-induced diabetic liver injury in rats. Acta Histochem. 2006;108:85–93. doi: 10.1016/j.acthis.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Gul M, Emre S, Esrefoglu M, Vard N. Protective effects of melatonin and aminoguanidine on the cornea in streptozotocin-induced diabetic rats. Cornea. 2008;27:795–801. doi: 10.1097/ICO.0b013e318169d67c. [DOI] [PubMed] [Google Scholar]
- 25.Pugazhenthi K, Kapoor M, Clarkson AN, Hall I, Appleton I. Melatonin accelerates the process of wound repair in full-thickness incisional wounds. J Pineal Res. 2008;44:387–396. doi: 10.1111/j.1600-079X.2007.00541.x. [DOI] [PubMed] [Google Scholar]
- 26.Tang L, Wu JJ, Ma Q, Cui T, Andreopoulos FM, Gil J, Valdes J, Davis SC, Li J. Human lactoferrin stimulates skin keratinocyte function and wound re-epithelialization. Br J Dermatol. 2010;163:38–47. doi: 10.1111/j.1365-2133.2010.09748.x. [DOI] [PubMed] [Google Scholar]
- 27.Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol. 2000;115:245–253. doi: 10.1046/j.1523-1747.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 28.Dinh T, Tecilazich F, Kafanas A, Doupis J, Gnardellis C, Leal E, Tellechea A, Pradhan L, Lyons TE, Giurini JM, Veves A. Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes. 2012;61:2937–2947. doi: 10.2337/db12-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirza RE, Koh TJ. Contributions of cell subsets to cytokine production during normal and impaired wound healing. Cytokine. 2015;71:409–412. doi: 10.1016/j.cyto.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 31.Park HJ, Lee SH, Son DJ, Oh KW, Kim KH, Song HS, Kim GJ, Oh GT, Yoon DY, Hong JT. Antiarthritic effect of bee venom: inhibition of inflammation mediator generation by suppression of NF-kappaB through interaction with the p50 subunit. Arthritis Rheum. 2004;50:3504–3515. doi: 10.1002/art.20626. [DOI] [PubMed] [Google Scholar]
- 32.Borst SE. The role of TNF-alpha in insulin resistance. Endocrine. 2004;23:177–182. doi: 10.1385/ENDO:23:2-3:177. [DOI] [PubMed] [Google Scholar]
- 33.Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N, Behl Y, Alikhani M, Yang J, Braasch C, Graves DT. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1) Diabetologia. 2010;53:378–388. doi: 10.1007/s00125-009-1529-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, Nowygrod S, Wolf BM, Caliste X, Yan SF, Stern DM, Schmidt AM. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bitto A, Altavilla D, Pizzino G, Irrera N, Pallio G, Colonna MR, Squadrito F. Inhibition of inflammasome activation improves the impaired pattern of healing in genetically diabetic mice. Br J Pharmacol. 2014;171:2300–2307. doi: 10.1111/bph.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Li X, Grailer JJ, Wang N, Wang M, Yao J, Zhong R, Gao GF, Ward PA, Tan DX, Li X. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J Pineal Res. 2016;60:405–414. doi: 10.1111/jpi.12322. [DOI] [PubMed] [Google Scholar]
- 38.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev. 2004;25:612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 39.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 40.Vermeij WP, Backendorf C. Skin cornification proteins provide global link between ROS detoxification and cell migration during wound healing. PLoS One. 2010;5:e11957. doi: 10.1371/journal.pone.0011957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang P, Zeng Y, Liu T, Zhang C, Yu PW, Hao YX, Luo HX, Liu G. Chloride intracellular channel 1 regulates colon cancer cell migration and invasion through ROS/ERK pathway. World J Gastroenterol. 2014;20:2071–2078. doi: 10.3748/wjg.v20.i8.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]