

Abstract
To explore whether polycomb repressor Bmi1 plays an important role in dentin and mandible development homeostasis by maintaining redox balance, 3-week-old Bmi1 gene knockout (Bmi1-/-) mice were treated with the antioxidant N-acetylcysteine (NAC) for 2 weeks in their drinking water and phenotypes of the tooth and mandibles were compared with vehicle-treated Bmi1-/- mice and wild-type mice by radiograph, histochemistry and immunohistochemistry. Alterations of oxidative stress, DNA damage, cell proliferation and cell cycle-related parameters were also examined in mandibles. Results showed that the tooth volume and the dentin sialoprotein immunopositive areas, the cortical thickness, alveolar bone volume, osteoblast number and activity, and mRNA expression levels of Runx2, alkaline phosphatase and type I collagen were all reduced significantly in Bmi1-/- mice compared with their wild-type littermates, whereas these parameters were increased significantly in NAC-treated Bmi1-/- mice compared with vehicle-Bmi1-/- mice, although they were not normalized. The activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) were reduced, DNA damage markers including γ-H2AX and 8-oxoguanine levels were increased, the number of Ki67 positive cells was decreased, whereas protein expression levels of p16, p19, p21, p27 and p53 were up-regulated in mandibles from Bmi1-/- mice compared with those from wild-type mice; alterations of these antioxidative enzyme activities, DNA damage markers, cell proliferation and cell cycle-related parameters were all partially rescued by the treatment with antioxidant NAC in Bmi1 deficient mice. These results demonstrated that Bmi1 deficiency resulted in defects in dentin and alveolar bone formation, while the treatment with antioxidant could improve these defects obviously. Therefore, our results indicate that Bmi1 plays an important role in stimulating dentin formation and alveolar bone formation by maintaining redox homeostasis, preventing DNA damage and inhibiting cyclin-dependent kinase inhibitors.
Keywords: Antioxidant, Bmi1, DNA damage, dentin, mandible
Introduction
The development of teeth is characterized by a series of reiterative molecular interactions accompany with the growth of jawbones. The mouse has been used very successfully to identify many of the molecular signaling interactions during the early stage of development of mandibles. Numerous studies have been performed to investigate canonical signaling pathways of bone morphogenetic proteins (BMPs), Wnt signaling, Notch signaling, fibroblast growth factors (FGFs) and Sonic hedgehog (Shh), which can function synergistically or antagonistically during tooth development and intramembranous ossification in embryonic period [1-3]. However, the molecular basis of mandible growth is poorly understood in postnatal mice.
Bmi1 (B lymphoma Mo-MLV insertion region 1) is a member of the polycomb family of transcriptional repressors. It is reported that Bmi1 is expressed by incisor stem cells and that deletion of Bmi1 resulted in fewer stem cells, perturbed gene expression and defective enamel production [4]. Previous study showed that neonatal Bmi1 mice exhibited skeletal growth retardation, with reduced chondrocyte proliferation and increased apoptosis. Bmi1 maintains self-renewal of bone marrow mesenchymal stem cells (BM-MSCs) and alters the cell fate of BM-MSCs by enhancing osteoblast differentiation and inhibiting adipocyte differentiation [5]. Furthermore, Bmi1 is involved in mitochondrial function maintenance and DNA protection from damage. Increased and persistent high levels of ROS caused by impaired mitochondrial function are sufficient to induce organism senescence via DNA damage in Bmi1 null mice. The thymocyte and kidney maturation defect characteristic of Bmi1-deficient mice is largely rescued by treatment with antioxidants [6,7]. However, in view of the difference between dentin formation and enamel production, intramembranous ossification and cartilaginous ossification, it is unclear whether Bmi1 deficiency could lead to dentin and alveolar bone defects in development by disturbing redox homeostasis and inducing DNA damage in mandible.
To answer this question, Bmi1-/- mice were treated with the antioxidant N-acetylcysteine (NAC, 1 mg mL-1) in their drinking water. Their mandible phenotype was then compared with that of vehicle-treated Bmi1-/- and wild-type mice.
Materials and methods
Mice and genotyping
The Bmi1 heterozygote (Bmi1+/-) mice (129Ola/FVB/N hybrid background) provided in this study had been backcrossed 10-12 times in the C57BL/6J background and mated to generate Bmi1 homozygote (Bmi1-/-) and their wild-type (WT) littermates genotyped by PCR, as described previously [5,7,8]. This study was approved by the Institutional Animal Care and Use Committee. Age- and sex-matched Bmi1-/- and wild-type littermates were used in this study.
Administration of N-acetylcysteine
In vivo, 3-week-old Bmi1-/- mice were randomized to the drinking water with or without N-acetylcysteine (NAC, 1 mg ml-1) for 2 weeks, as previously described [6]. The phenotypes were analyzed at 5 weeks of age.
Radiography
Mandibles were removed and dissected free of soft tissue. Contact radiographs were taken using a Faxitron model 805 radiographic inspection system (Faxitron, München, Germany), at 22 kV voltage and with a 4-minute exposure time. X-Omat TL film (Eastman Kodak, Rochester, NY, USA) was used and processed routinely.
Micro-computed tomography (micro-CT)
Mandibles were fixed overnight in 70% ethanol and analyzed by micro-CT with a SkyScan 1072 scanner and associated analysis software (SkyScan, Antwerp, Belgium) as described [9]. Briefly, image acquisition was performed at 100 kV and 98 mA with a 0.98 degree rotation between frames. During scanning, the samples were enclosed in tightly fitting plastic wrap to prevent movement and dehydration. Thresholding was applied to the images to segment the bone from the background. 2D images were used to generate 3D renderings using the 3D Creator software supplied with the instrument. The resolution of the micro-CT images is 18.2 μm.
Histology
Mandibles were removed, fixed in PLP fixative (2% paraformaldehyde containing 0.075 M lysine and 0.01 M sodium periodate) overnight at 4°C and processed histologically as described [9]. Mandibles were decalcified in EDTA-glycerol solution for 5-7 days at 4°C. Decalcified right mandibles were dehydrated and embedded in paraffin, and 5 μm sections cut on a rotary microtome. The sections were stained with Hematoxylin and Eosin (HE), or histochemically for total collagen, alkaline phosphatase (ALP) activity, or immunohistochemically as described below.
Immunohistochemical staining
Immunohistochemical staining was carried out for biglycan, dentin sialoprotein (DSP), γ-H2AX, 8-hydroxydeoxyguanosine (8-OHdG) and 53Bp1 using the avidin-biotin-peroxidase complex technique with affinity-purified rabbit anti-mouse biglycan antibody (Abcam, Cambridge, UK), dentin sialoprotein (Santa Cruz, CA, USA), γ-H2AX (Cell Signaling Technology, MA, USA), 8-OHdG (Abcam, Cambridge, UK), and 53Bp1 (Novus Biological, USA) following previously described methods [9].
Quantitative real-time PCR
RNA isolated from mandible bodies via Trizol reagent (Invitrogen) was synthesized single stranded cDNA. Sample mRNA levels were semiquantified by RT-PCR or quantified by real-time RT-PCR as previously described [9].
Western blot analysis
Proteins were extracted from mandibular bones and quantitated using a protein assay kit (Bio-Rad, Mississauga, Ontario, Canada). Protein samples (30 μg) were fractionated by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was carried out as described [9] using antibodies against SOD1 (Abcam), Prdx4 (BD), γ-H2AX (Ser139) (Cell Signaling Technology), CHK2 (Novus Biological), p16 (Santa Cruz), p19 (Santa Cruz), p21 (Santa Cruz), p27 (Santa Cruz), p53 (Santa Cruz) and β-actin (Bioworld Technology).
Biochemical measurements
Mandible tissues from 5-week-old mice were homogenized in cold saline. Homogenate (10%) was centrifuged at 4000 rpm at 4°C for 10 min. Supernatant was used for measurements of total superoxide dismutase (T-SOD) (A001-1 SOD detection kit) and glutathione peroxidase (GSH-PX) (A005 GSH-PX detection kit). Detection kits were from Nanjing Jiancheng Bioengineering Institute in China. All examinations were performed according to the manufacturer’s instructions.
Computer-assisted image analysis
After H&E staining or histochemical or immunohistochemical staining of sections from five mice of each genotype, images of fields were photographed with a Sony digital camera. Images of micrographs from single sections were digitally recorded using a rectangular template, and recordings were processed and analyzed using Northern Eclipse image analysis software as described [9].
Statistical analysis
Five mice per group were averaged to provide a mean value. Data from image analysis are presented as mean ± s.e.m. Statistical comparisons were made using a two-way ANOVA, with P<0.05 considered significant.
Results
Bmi1 regulates development and redox balance of the teeth and mandibles
Bmi1-/- teeth and mandibles were smaller than wild-type teeth and mandibles. Radiolucency was increased in all teeth, including molars and incisors, and in the mandibles of Bmi1-/- mice compared with those of their wild-type littermates (Figure 1A). Histology showed that the root wall thickness of the first molars was reduced and dental alveolar bone volume was reduced at 5-week-old Bmi1-/- mice compared to their wild-type littermates (Figure 1B). These results demonstrate that Bmi1 deficiency led to tooth and mandible growth retardation.
Figure 1.
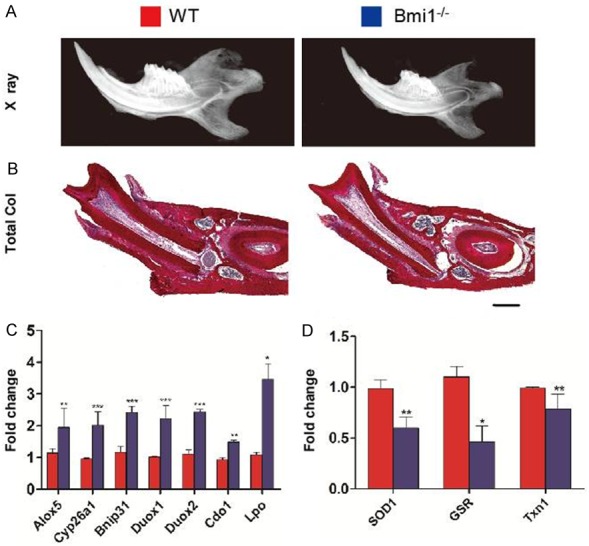
Bmi1 regulates development and redox balance of the teeth and mandibles. A: Radiographs of the mandibles from 5-week-old wild-type (WT) and Bmi1-/- mice. B: Representative micrographs of decalcified paraffin-embedded sections through the first molars from 5-week-old wild-type and Bmi1-/- mice stained with serious red for total collagen as described in Materials and Methods. Scale bars represent 400 μm. C: Quantitative RT-PCR expression analysis of gene products involved in redox homeostasis in either WT or Bmi1-/- teeth and mandibles. D: Quantitative RT-PCR expression analysis of antioxidative enzymes in WT and Bmi1-/- teeth and mandibles. Results are normalized to Gapdh expression. Each value is the mean ± s.e.m. of determinations in five animals of each group. *P<0.05; **P<0.01; ***P<0.001 relative to the wild-type mice.
In an effort to explain these observed defects of tooth and mandible development in Bmi1-/- mice, we made use of several previous gene expression studies that have identified a multitude of polycomb target genes [6]. Our results confirmed that Bmi1-/- mandible de-repressed a number of previously identified polycomb-regulated gene products that can regulate intracellular redox homeostasis (Figure 1C). However, gene expression levels of antioxidative enzymes including SOD1, glutathione reductase and thioredoxin1 in mandibles were down-regulated in Bmi1-/- mice demonstrated by real-time RTPCR (Figure 1D). These results therefore indicate that Bmi1 may stimulate tooth and mandible development by regulating redox homeostasis.
NAC supplementation improves mineralization defects in Bmi1-/- teeth and mandibles
Bone mineral density was lower in Bmi1-/- mice relative to wild-type mice (Figure 2A). Comparison between wild-type and Bmi1-/- littermates of micro-CT scanned sections through the incisor before the first molar, and through the first, second, and third molars showed that the mineralized tooth volume in incisor and molars and the mineralized cortical and alveolar bone volume in mandibles were decreased in Bmi1-/- mice (Figure 2B). These mineralization defects in teeth and alveolar bone were almost completely rescued by the NAC supplementation (Figure 2).
Figure 2.
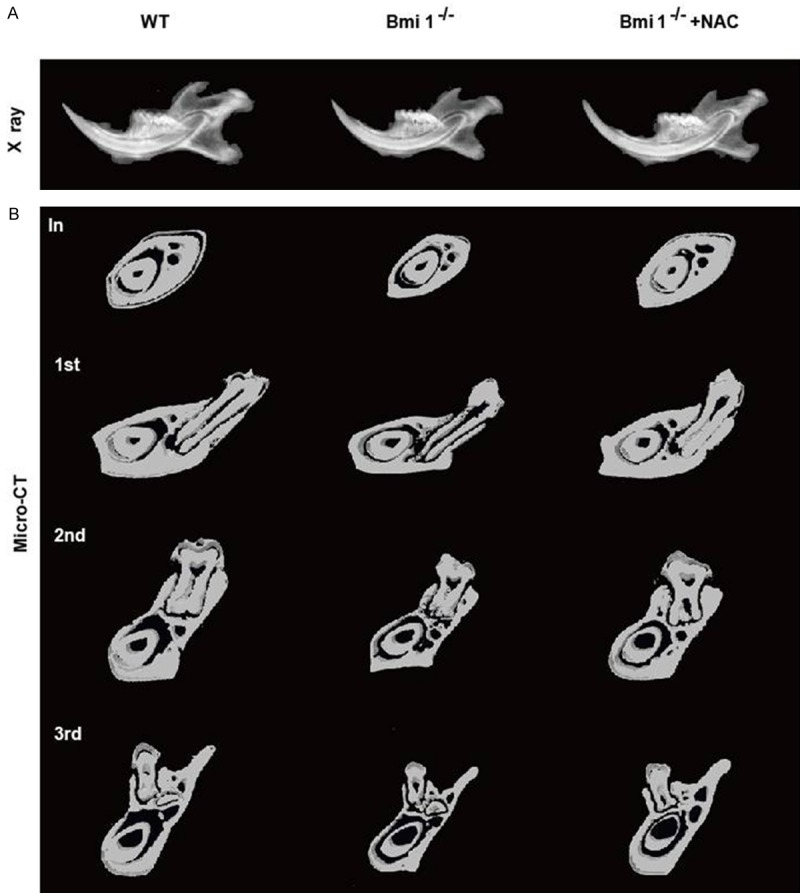
NAC supplementation improves mineralization defects in Bmi1-/- teeth and mandibles. A: Radiographs of the mandibles from 5-week-old vehicle-treated wild-type (WT) and Bmi1-/- mice and NAC-treated Bmi1-/- mice. B: Micro-CT scanned sections through the incisor before the first molar (In), and through the first (1st), second (2nd) and third (3rd) molars from vehicle-treated wild-type (WT) and Bmi1-/- mice and NAC-treated Bmi1-/- mice.
NAC supplementation improves decreased tooth volume and cortical and alveolar bone volume in Bmi1-/- mandibles
Total (mineralized and unmineralized) dentin thickness and the alveolar bone volumes were decreased in Bmi1-/- mice compared with their wild-type littermates (Figure 3A and 3B). Quantitative data showed that the dentin thickness of the first molars (Figure 3C), the alveolar bone volume (Figure 3D) and cortical thickness (Figure 3E) were all reduced significantly in Bmi1-/- mice compared with their wild-type littermates. Decreased dentin and cortical thickness and alveolar bone volume in Bmi1 deficient mandibles were largely rescued by the NAC supplementation (Figure 3).
Figure 3.
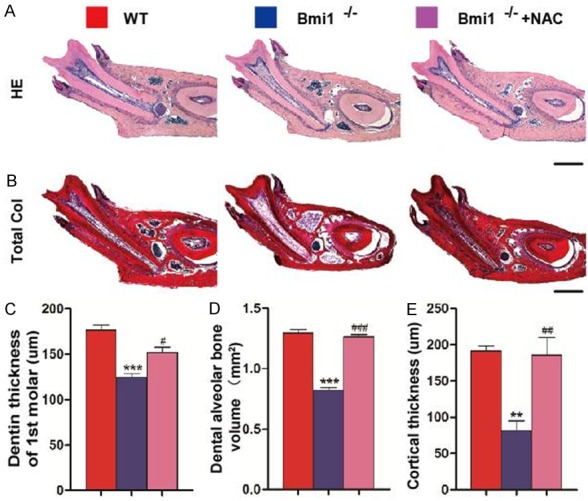
NAC supplementation improves decreased tooth volume and cortical and alveolar bone volume in Bmi1-/- mandibles. Representative micrographs of decalcified paraffin-embedded sections through the first molars from 5-week-old vehicle-treated wild-type (WT) and Bmi1-/- mice and NAC-treated Bmi1-/- mice stained with (A) HE, and with (B) serious red for total collagen as described in Materials and Methods. Scale bars represent 400 μm. (C) Dentin thickness of the first molars, (D) alveolar bone volume of the mandibles, (E) the cortical thickness was measured. Each value is the mean ± s.e.m. of determinations in five animals of each group. **P<0.01; ***P<0.001 relative to the wild-type mice. #P<0.05; ##P<0.01; ###P<0.001 compared with Bmi1-/- mice.
NAC supplementation improves abnormal predentin maturation and dentin formation in Bmi1-/- mice
As seen in sections stained with H&E, the ratio of the areas of predentin to dentin was increased in the first molars (Figure 4A and 4D) in Bmi1-/- mice compared with their wild-type littermates. Positive immunoreactivity for biglycan was detected in the region of the predentin (Figure 4B). The ratio of biglycan positive area to dentin was increased in the first molars (Figure 4E) in Bmi1-/- mice compared with their wild-type littermates. Positive immunoreactivity for DSP was detected in the predentin and dentin in the molars (Figure 4C). The DSP-positive area was decreased dramatically in the first molars in Bmi1-/- mice compared with their wild-type littermates (Figure 4F). Decreased dentin maturation and formation in Bmi1 deficient teeth were significantly rescued by the NAC supplementation (Figure 4).
Figure 4.
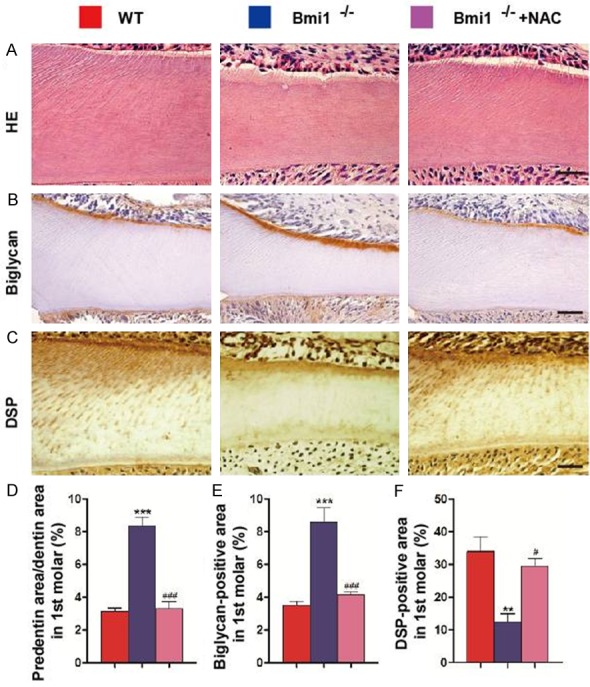
NAC supplementation improves abnormal predentin maturation and dentin formation in Bmi1-/- mice. Paraffin embedded sections through the first molars from 5-week-old vehicle-treated wild-type (WT) and Bmi1-/- mice and NAC-treated Bmi1-/- mice stained with (A) HE, immunohistochemically for (B) biglycan and (C) dentin sialoprotein (DSP) and photographed. Scale bars represent 50 μm. (A) Representative HE staining of micrographs of the root walls of the first molars. (D) Quantitative thickness of predentin in the root walls of the first molars. (B) Representative micrographs of the root wall of the first molars stained immunohistochemically for biglycan, and (C) the root wall of the first molars stained immunohistochemically for DSP. (E) Quantitative biglycan immunopositive areas in the first molars. (F) Quantitative DSP immunopositive areas in the first molars. Each value is the mean ± s.e.m. of determinations in 5 animals of each group. **P<0.01; ***P<0.001 relative to the wild-type mice. #P<0.05; ###P<0.001 compared with Bmi1-/- mice.
NAC supplementation improves impaired osteoblastic alveolar bone formation in Bmi1-/- mice
To clarify whether cortical and alveolar bone growth defect was associated with altered osteoblastic bone formation, paraffin-embedded sections were stained with H&E and histochemically for ALP. The osteoblast number and surface were decreased in the Bmi1-/- alveolar bone (Figure 5A, 5C and 5D). The ALP-positive area was also reduced in Bmi1-/- mice (Figure 5B and 5E). Meanwhile, we examined the expression of genes involved in bone formation. RNA was isolated from the mandibles and real-time RT-PCR was performed. Expression levels of the osteoblastic genes including Runx2, ALP and osteocalcin were down-regulated in Bmi1-/- mice compared to wild-type mice (Figure 5F-H). However, reduced osteoblast number and surface and osteoblastic gene expression levels in Bmi1 deficient mandibles were largely restored by NAC supplementation (Figure 5).
Figure 5.
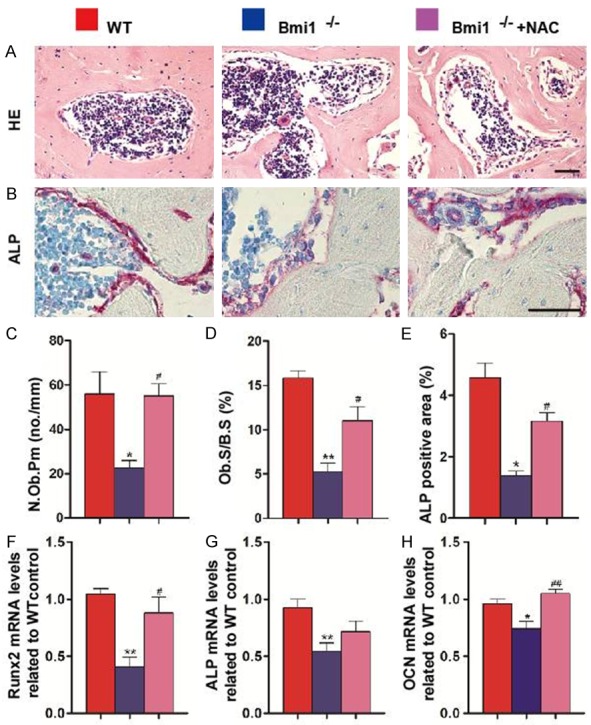
NAC supplementation improves impaired osteoblastic alveolar bone formation in Bmi1-/- mice. Representative micrographs of paraffin-embedded sections of mandibles from 5-week-old vehicle-treated wild-type (WT) and Bmi1-/- mice and NAC-treated Bmi1-/- mice stained with (A) H&E and histochemically for (B) ALP. Scale bars represent 50 μm. (C) Number of osteoblasts per mm bone parameter (N.Ob/B.Pm, #/mm) and (D) the surface of osteoblasts relative to the bone surface (Ob.S/B.S, %) were determined in the dental alveolar bone of H&E-stained mandibles respectively. (E) ALP-positive area as a percentage of the tissue area in the dental alveolar bone. Expression of (F) Runx2, (G) ALP and (H) OCN was assessed by real-time RT-PCR of mandibular extracts. mRNA expression normalized to Gapdh is shown relative to levels in wild-type mice as the mean ± s.e.m. of determinations in 5 animals of the same genotype. *P<0.05; **P<0.01 relative to the wild-type mice. #P<0.05; ##P<0.01 compared with Bmi1-/- mice.
NAC supplementation improves redox imbalance in Bmi1-/- mandibles
The activities of total superoxide dismutase (T-SOD) and glutathione peroxidase (GSH-PX) (Figure 6A and 6B), the mRNA levels of glutathione peroxidase (Gpx4), glutathione reductase (GSR) (Figure 6C), and the protein levels of SOD1 and peroxiredoxin4 (Prdx4) (Figure 6D) were reduced significantly in Bmi1-/- mice compared with wild-type mice, however, these parameters were increased significantly in NAC-treated Bmi1-/- mice compared with vehicle-treated Bmi1-/- mice.
Figure 6.
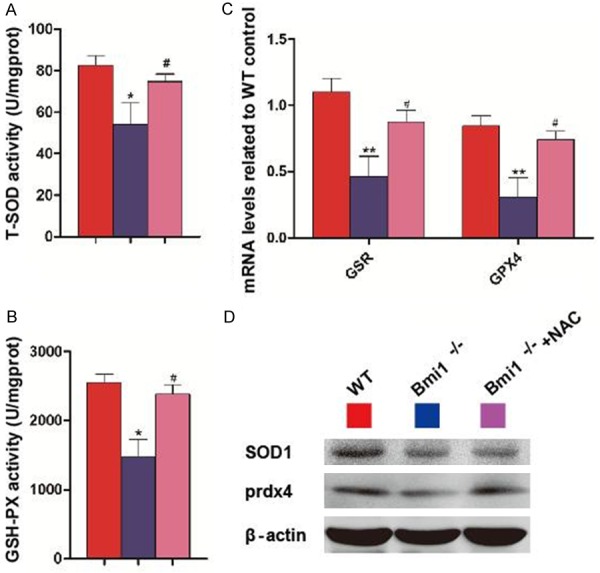
NAC supplementation improves redox imbalance in Bmi1-/- mandibles. Concentrations of (A) total superoxide dismutase (T-SOD), and (B) glutathione peroxidase (GSH-PX) determined by spectrophotometry. (C) Glutathione peroxidase (Gpx4) and glutathione reductase (GSR) mRNA relative levels in mandibles demonstrated by real-time RT-PCR. mRNA expression normalized to Gapdh is shown relative to levels in wild-type mice as the mean ± s.e.m. of determinations in 5 animals of the same genotype. (D) Western blots of mandible extracts for expression of SOD1 and peroxiredoxin4 (Prdx4). *P<0.05; **P<0.01 relative to the wild-type mice. #P<0.05 compared with Bmi1-/- mice.
NAC supplementation improves DNA damage in Bmi1-/- mandible
The percentages of γ-H2AX, 8-hydroxydeoxyguanosine (8-OHdG) and 53Bp1-positive cells, and protein expression levels of γ-H2AX, checkpoint kinase 2 (CHK2), p16, p19, p21, p27 and p53 in mandibles were up-regulated significantly in Bmi1-/- mice compared with wild-type mice, whereas these parameters were down-regulated dramatically in NAC-treated Bmi1-/- mice compared with vehicle-treated Bmi1-/- mice, although they were not normalized (Figure 7A-E).
Figure 7.
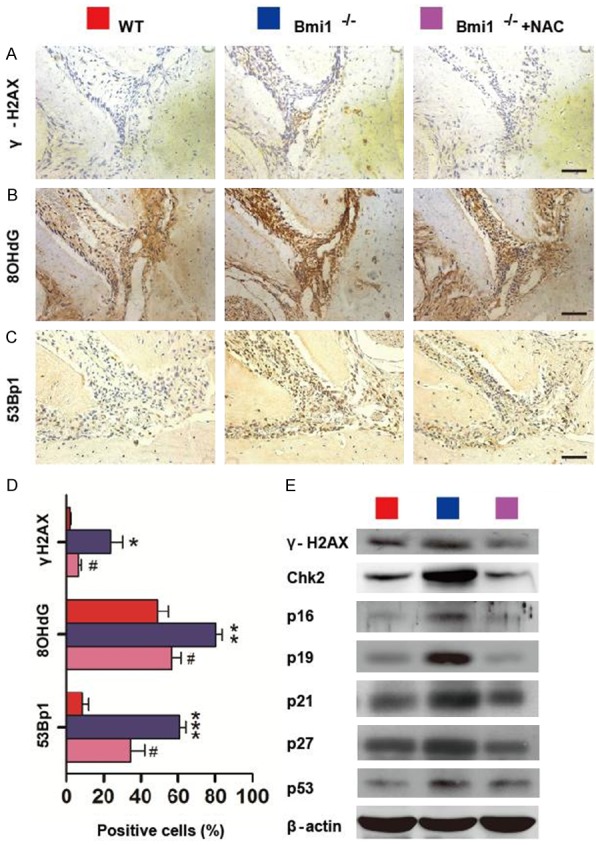
NAC supplementation improves DNA damage in Bmi1-/- mandible. Sections through the first molars and incisors from 5-week-old vehicle-treated wild-type (WT) and Bmi1-/- mice and NAC-treated Bmi1-/- mice stained immunohistochemically for (A) γ-H2AX, (B) 8-hydroxydeoxyguanosine (8-OHdG) and (C) 53Bp1-positive cells. Scale bars represent 50 μm. (D) Numbers of γ-H2AX-positive cells, 8-OHdG-positive cells and 53Bp1-positive cells in Hertwig’s epithelial root sheath (HERs) were determined by image analysis, and the percentages of immunopositive cells relative to total cells were presented as the mean ± s.e.m. of determinations in 5 animals of each group. (E) Western blots of mandibular extracts were carried out for expression of γ-H2AX, checkpoint kinase 2 (CHK2), p16, p19, p21, p27 and p53, with β-actin as a loading control. *P<0.05; **P<0.01; ***P<0.001 relative to the wild-type mice. #P<0.05 compared with Bmi1-/- mice.
Discussion
In this study, we demonstrated that Bmi1 deficiency resulted in dentin and mandible development defects with decreased dentin formation and osteoblastic alveolar bone formation. Our results also demonstrated that dentin and alveolar bone development defects caused by Bmi1 deficiency could be largely rescued by antioxidant NAC supplementation. These findings indicate that Bmi1 plays a critical role in the protection from dentin and alveolar bone development defects by maintaining redox balance.
Previous studies suggest that deletion of Bmi1 perturbed defective enamel production [4]. The enamel-producing ameloblasts are generated from epithelial cells adjacent to the dental papilla called the inner enamel epithelium (IEE), while dentin-producing odontoblasts differentiate from the outermost layer of the dental papilla and gradually migrate to the center of the dental papilla as they secrete dentin matrix [2]. In this study, we therefore examined whether deletion of Bmi1 perturbed dentin formation. Results from the present study revealed that the ratio of predentin to dentin and that of biglycan positive area to the molar area were increased severely in Bmi1-/- mice compared with their wild-type littermates. Dentin sialophosphoprotein (DSP) distribution in collagen matrix of the forming dentin suggests this protein plays an important role in the regulation of mineral deposition. Our results showed a decreased DSP immunopositive area in Bmi1-/- mice consistent with a reduced dentin thickness. Our results therefore demonstrated that Bmi1 deficiency impaired dentin formation and mineralization.
In previous study, the phenotype of long bones from Bmi1 deficient mice has been shown that the bone mineral density was reduced and trabecular bone volume and osteoblast number were decreased in Bmi1-/- mice [5]. In the current study, we analyzed the phenotype of mandibles from the Bmi1 deficient mice and found that the bone mineral density, the mineralized cortical and alveolar bone volume, osteoblast numbers, ALP activity in osteoblasts and expression of the osteoblastic genes in mandibles were all decreased. These results suggest that deletion of Bmi1 disturbs osteoblastic bone formation in both long bones and mandibles. The mandibular body develops by intramembranous bone formation. These results therefore suggest that Bmi1 plays an important role not only in endochondral ossification, but also in intramembranous ossification.
Previous studies suggest imbalances between the oxidant-antioxidant statuses have been affected in health status and the pathogenesis of oral tissues, including saliva, periodontitis, oral leukoplakia and oral cancer [10-13]. This study therefore examined whether perturbed dentin production and intramembranous bone formation caused by Bmi1 deficiency was associated with increased oxidative stress. It was shown from previous results that Bmi1 deficiency leads to an increased p53 accumulation at promoters and gene repression of antioxidant genes through recruitment of corepressors [14]. Our results also revealed that decreased levels of endogenous antioxidants, including significantly reducing activities of T-SOD and GSH-PX, down-regulating mRNA levels of Gpx4, and the protein levels of SOD1 and Prdx4. Consequently, increased oxidative stress induced by Bmi-1 deficiency may contribute to the defects of dentin and alveolar bone development.
It is reported that oxidative stress can trigger activation of the DNA damage response (DDR) pathway [15]. The current study further demonstrated the DDR pathway occurred in Bmi1-deficient dentin and mandibles, including significant increases in γ-H2AX, 8-OHdG and 53Bp1-positive cells, the up-regulation of protein expression levels of γ-H2AX, CHK2, p16, p19, p21, p27 and p53. Our results were similar to those induced by redox imbalance [6,16], suggesting that increased oxidative stress caused by Bmi1 deficiency can activate DDR in dentin and mandibles.
NAC, an essential precursor to many endogenous antioxidants involved in the decomposition of peroxides, attenuates oxidative stress by replenishing intracellular glutathione stores [17]. NAC treatment reduced conjunctival epithelial cell dysfunction by inhibit reactive oxygen species [18]. NAC treatment ameliorates the skeletal phenotype of diastrophic dysplasia and kidney injury [19,20]. In clinical trials, NAC improves lung function in patients with chronic obstructive pulmonary disease, highlighting the potential benefit of ROS-directed therapy [21,22]. The current study demonstrated that dentin and alveolar bone development defects caused by Bmi1 deficiency could be largely rescued by antioxidant supplementation by reducing oxidative stress and DNA damage. Consequently, our results support the hypothesis that the action of Bmi1 in maintaining redox balance is critical for the protection from abnormal tooth and mandible development.
Bmi1 is not only involved in redox maintenance and DNA protection from damage, it is also involved in cell cycle regulation by inhibiting p16INK4a/Rb and p19AFR/p53 pathways [5]. Our results demonstrated that the expression levels of p16, p19, p53, p21 and p27 proteins were up-regulated significantly in Bmi1 deficient tooth and mandibles. We also found that these protein expression levels were down-regulated significantly by the antioxidant NAC supplementation. These results suggest that p16 and p19 signal pathways are also regulated by antioxidants. However, the exact regulating mechanism of antioxidants on these molecules remains to be investigated.
In conclusion, this study demonstrated that dentin and alveolar bone development defects caused by Bmi1 deficiency was associated with increased oxidative stress and DNA damage, whereas these alterations were largely rescued by antioxidant NAC supplementation. Results from this study indicate that Bmi1 plays an important role in stimulating dentin formation and alveolar bone formation by maintaining redox homeostasis, preventing DNA damage and inhibiting cyclin-dependent kinase inhibitors.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (2012CB966902 to D.M. and N.C. 2014CB942900 to D.M.), from the Basic Research Program of Chongqing (CSTC2013jcyjC00009 to D.M.) and the National Natural Science Foundation of China (81271109 to N.C.).
Disclosure of conflict of interest
None.
References
- 1.Olley R. Corrigendum: Expression analysis of candidate genes regulating successional tooth formation in the human embryo. Front Physiol. 2015;6:41. doi: 10.3389/fphys.2015.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jheon AH, Seidel K, Biehs B, Klein OD. From molecules to mastication: the development and evolution of teeth. Wiley Interdiscip Rev Dev Biol. 2013;2:165–182. doi: 10.1002/wdev.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franz-Odendaal TA. Induction and patterning of intramembranous bone. Front Biosci (Landmark Ed) 2011;16:2734–2746. doi: 10.2741/3882. [DOI] [PubMed] [Google Scholar]
- 4.Biehs B, Hu JK, Strauli NB, Sangiorgi E, Jung H, Heber RP, Ho S, Goodwin AF, Dasen JS, Capecchi MR, Klein OD. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat Cell Biol. 2013;15:846–852. doi: 10.1038/ncb2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang HW, Ding J, Jin JL, Guo J, Liu JN, Karaplis A, Goltzman D, Miao D. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res. 2010;25:640–652. doi: 10.1359/jbmr.090812. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira II, Xu XL, van Lohuizen M, Motoyama N, Deng CX, Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin J, Lv X, Chen L, Zhang W, Li J, Wang Q, Wang R, Lu X, Miao D. Bmi-1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance. Aging Cell. 2014;13:797–809. doi: 10.1111/acel.12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao G, Gu M, Zhu M, Gao J, Yin Y, Marshall C, Xiao M, Ding J, Miao D. Bmi-1 absence causes premature brain degeneration. PLoS One. 2012;7:e32015. doi: 10.1371/journal.pone.0032015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin Y, Wang Q, Sun W, Wang Y, Chen N, Miao D. p27(kip1) deficiency accelerates dentin and alveolar bone formation. Clin Exp Pharmacol Physiol. 2014;41:807–816. doi: 10.1111/1440-1681.12276. [DOI] [PubMed] [Google Scholar]
- 10.Gumus P, Emingil G, Ozturk VO, Belibasakis GN, Bostanci N. Oxidative stress markers in saliva and periodontal disease status: modulation during pregnancy and postpartum. BMC Infect Dis. 2015;15:261. doi: 10.1186/s12879-015-1003-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava KC, Austin RD, Shrivastava D, Pranavadhyani G. Oxidant-antioxidant status in tissue samples of oral leukoplakia. Dent Res J (Isfahan) 2014;11:180–186. [PMC free article] [PubMed] [Google Scholar]
- 12.Golz L, Memmert S, Rath-Deschner B, Jager A, Appel T, Baumgarten G, Gotz W, Frede S. LPS from P. gingivalis and hypoxia increases oxidative stress in periodontal ligament fibroblasts and contributes to periodontitis. Mediators Inflamm. 2014;2014:986264. doi: 10.1155/2014/986264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choudhari SK, Chaudhary M, Gadbail AR, Sharma A, Tekade S. Oxidative and antioxidative mechanisms in oral cancer and precancer: a review. Oral Oncol. 2014;50:10–18. doi: 10.1016/j.oraloncology.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Chatoo W, Abdouh M, David J, Champagne MP, Ferreira J, Rodier F, Bernier G. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529–542. doi: 10.1523/JNEUROSCI.5303-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 16.Shao L, Li H, Pazhanisamy SK, Meng A, Wang Y, Zhou D. Reactive oxygen species and hematopoietic stem cell senescence. Int J Hematol. 2011;94:24–32. doi: 10.1007/s12185-011-0872-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 2012;17:311–321. doi: 10.1111/j.1440-1797.2012.01572.x. [DOI] [PubMed] [Google Scholar]
- 18.Park JH, Kang SS, Kim JY, Tchah H. The Antioxidant N-Acetylcysteine Inhibits Inflammatory and Apoptotic Processes in Human Conjunctival Epithelial Cells in a High-Glucose Environment. Invest Ophthalmol Vis Sci. 2015;56:5614–5621. doi: 10.1167/iovs.15-16909. [DOI] [PubMed] [Google Scholar]
- 19.Monti L, Paganini C, Lecci S, De Leonardis F, Hay E, Cohen-Solal M, Villani S, Superti-Furga A, Tenni R, Forlino A, Rossi A. N-acetylcysteine treatment ameliorates the skeletal phenotype of a mouse model of diastrophic dysplasia. Hum Mol Genet. 2015;24:5570–80. doi: 10.1093/hmg/ddv289. [DOI] [PubMed] [Google Scholar]
- 20.Bulacio RP, Anzai N, Ouchi M, Torres AM. Organic Anion Transporter 5 (Oat5) Urinary Excretion Is a Specific Biomarker of Kidney Injury: Evaluation of Urinary Excretion of Exosomal Oat5 after N-Acetylcysteine Prevention of Cisplatin Induced Nephrotoxicity. Chem Res Toxicol. 2015;28:1595–1602. doi: 10.1021/acs.chemrestox.5b00176. [DOI] [PubMed] [Google Scholar]
- 21.Cazzola M, Calzetta L, Page C, Jardim J, Chuchalin AG, Rogliani P, Gabriella Matera M. Influence of N-acetylcysteine on chronic bronchitis or COPD exacerbations: a meta-analysis. Eur Respir Rev. 2015;24:451–461. doi: 10.1183/16000617.00002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]