

Abstract
The physiological level of nitric oxide (NO) released by brain microvascular endothelial cells (BMECs) at normoxia can block the degradation of hypoxia-inducible factor-1α (HIF-1α) in astrocytes and initiate the compensatory response to hypoxia. However, it is unclear whether this occurs at mild hypoxia. This study was to investigate the expression of HIF-1α, VEGF and LDHA and the lactic acid production in astrocytes with or without co-culture with BMECs after mild hypoxia exposure. During mild hypoxia (5% O2), exogenous NO blocked the degradation of HIF-1α in astrocytes but up-regulated the transcription of VEGF and LDHA, accompanied by elevated expression of VEGF protein and increased production of lactic acid. This was further confirmed by silencing of HIF-1α expression in astrocytes. In astrocytes co-cultured with primary rat BMEC under mild hypoxia, NO was released by the BMECs and prevented the degradation of HIF-1α in astrocytes, leading to the up-regulated mRNA expression of VEGF and LDHA, elevated VEGF protein expression and increased production of lactic acid. In BMECs, NO was derived from intracellular eNOS. Based on these findings, we hypothesize that, under mild hypoxia, even though astrocytes do not respond to hypoxia, NO produced by BMECs may transmit a hypoxia signal to astrocytes, triggering their adaptive response via HIF-1α.
Keywords: Brain microvascular endothelial cells, astrocytes, nitric oxide, hypoxia-inducible factor-1α, hypoxia
Introduction
Hypoxia is caused by a lack of oxygen in the blood while the supply of other nutrients remains normal, resulting in decreased arterial oxygen partial pressure and reduced oxygen in tissues [1]. Major causes of hypoxia include low atmospheric oxygen partial pressure, such as in high altitude environments [2], and external respiratory dysfunction, such as respiratory stenosis and obstructive lung diseases [3]. The brain is very sensitive to hypoxia, and the pathology of hypoxia induced injury to the central nervous system (CNS) is complex, including the brain edema and brain cell injury [4]. After varying degrees of hypoxia, different degrees of neurological symptoms will be present. In acute severe hypoxia, symptoms include headaches, irritability, convulsions, coma and even death, while chronic moderate hypoxia will cause fatigue, drowsiness, difficult concentration, and memory impairment [5-7]. Since the brain has a certain threshold to the hypoxia, mild hypoxia outside this range may cause only compensatory adaptive responses without causing any neurological symptoms.
The neurovascular unit (NVU) is the basic organizational structure of the blood-brain barrier (BBB). In the NVU, astrocytes contact with more than 99% of the total brain microvasculature, and may directly interact with brain microvascular endothelial cells (BMEC) [8,9]. On the other side of the vasculature, the astrocytes may bridge blood vessels and neurons for substance transportation and signal transmission [10,11]. Oxygen diffuses from the peripheral circulation into tissues based on the oxygen partial pressure gradient, and the farther the distance of the tissue to the blood vessels, the lower the oxygen partial pressure of the cells is [4,12]. The physiological oxygen level of the cerebral capillaries in the NVU ranges 30 mmHg to 100 mmHg. However, the oxygen level around glial cells and neurons is 23.8-33.3 mmHg [13]. Therefore, we hypothesize that there is a difference in the hypoxia threshold among BMEC, astrocytes, and neurons.
The brain compensatory adaptive response to mild hypoxia can provide adequate oxygen to neurons as much as possible in order to preserve the normal neuronal function, which primarily depends on the compensatory response of the brain vasculature and glial cells. The most important compensatory responses of vascular system include vasodilation and angiogenesis, leading to increased local oxygen supply [14,15]. The most important compensatory responses of the astrocyte system include the reduced aerobic oxidation, increased glycolysis, and reduced oxygen consumption, thus providing more oxygen to neurons [16-18]. Nitric oxide (NO) is a potent vasodilator in the brain [19]. It is involved in the regulation of the stability of hypoxia inducible factor-1α (HIF-1α), a core molecule initiating the compensatory response to hypoxia [20]. Studies have shown that NO can block degradation of HIF-1α under normoxic and hypoxic conditions [21-24], and coculture models of the BBB have found that physiological levels of NO released by BMEC can also block HIF-1α degradation in astrocytes, but have no effect on neurons [25,26]. However, it is unclear whether the physiological levels of NO released by BMEC under mild hypoxia can still block HIF-1α degradation in astrocytes. In the present study, we investigated whether primary cultured rat BMECs were more likely to recognize hypoxia than astrocytes, and whether, under mild hypoxia (5% O2), the physiological levels of NO released by BMECs could initiate the compensatory response to hypoxia in astrocytes.
Methods
Materials
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT), dimethyl sulfoxide (DMSO), 2,2’-(Hydroxynitrosohydrazino) bis-ethanamine (DETA), NG-Nitro-L-arginine Methyl Ester (L-NAME), 2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO), L-arginine (LArg), fibronectin, collagenase II, deoxyribonuclease I (DNase I), and puromycin were purchased from Sigma Chemicals (St. Louis, MO, USA). Collagenase/Dispase was from Roche (Basel, Switzerland). Percoll was from Amersham Biosciences (Piscataway, NJ, USA). Antibodies against HIF-1α (ab2185) and β-actin (ab3280) were from Abcam (Cambridge, MA, USA), and anti-VEGF antibody (sc-7269) was from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Horseradish peroxide (HRP)-conjugated goat anti-rabbit IgG antibody (ab6721) and HRP-conjugated rabbit anti-mouse IgG antibody (ab6728) were from Abcam. The Lactate Colorimetric Assay Kit (K627-100) and the Nitric Oxide Fluorometric Assay Kit (K252-200) were from Biovision (Milpitas, CA, USA). Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) was from Dingguo Biotech (Beijing, China) and fetal bovine serum (FBS) from Sijiqing Biotech Co., Ltd (Hangzhou, China). Endothelial cell medium (ECM, Catalog #1001) containing FBS, endothelial cell growth supplement (ECGS) and penicillin/streptomycin solution (P/S) was from ScienCell (Carlsbad, CA, USA). HIF-1α small interfering RNA (siRNA, sc-45919), control siRNA-A (sc-37007), siRNA Transfection Reagent (sc-29528), and siRNA transfection medium (sc-36868) were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Pierce enhanced chemiluminescence (ECL) detection reagent (32016) was from Thermo Fisher Scientific (Waltham, MA, USA). All the other chemicals were from Sigma.
Primary culture of rat cortical astrocytes
The care and use of animals complied with the Provisions and General Recommendation of the Chinese Experimental Animals Administration Legislation and this study was approved by the Animal Ethics Committee of Southwest Hospital, the Third Military Medical University, Chongqing. Astrocytes were prepared from the cortex of 1 to 2-day-old neonatal Wistar rats as previously described [27]. The rats were sacrificed by cervical decapitation and the forebrain was rapidly collected, the cerebellum and olfactory bulbs were removed, and the meninges and blood vessels were carefully removed by using a stereomicroscope. The remaining tissues were cut into 1-3 mm3 blocks in pre-cooled PBS (1 ml), and then incubated with 4 ml of 0.25% trypsin at 37°C for 15 min. Then 5 ml of complete DMEM/F12 medium (containing 10% FBS, 50 units/ml penicillin and 50 µg/ml streptomycin) was added to terminate the digestion. Cells after digestion were gently pipetted, and then filtered through a 70-µm mesh. The filtrate was centrifuged at 1000 × g for 5 min at 4°C. The supernatant was removed, and an appropriate amount of complete DMEM/F12 medium was added to prepare single cell suspension at 4 × 104 cells/cm2. Cells were grown at 37°C in a humidified environment with 5% CO2. The culture medium was refreshed every three days. After culture for 7-9 days, cells were incubated at 37°C under constant shaking at 200 rpm/min for 18 h to purify the astrocytes by removing microglial cells. Then, the medium was removed. The remaining cells were washed with DPBS, harvested by trypsinization and suspended at a density of 4 × 104 cells/cm2.
Culture of primary rat BMECs
Rat BMECs were prepared from 3-week-old Wistar rats as previously described [28]. Briefly, rats were sacrificed by cervical decapitation and the forebrain was rapidly collected and stored in DPBS on ice. The forebrain was cut into 1-3 mm3 blocks, and then digested in DMEM/F12 containing 1 g/L type II collagenase and 15 mg/L DNAse I at 37°C under constant shaking at 200 rpm/min for 1.5 h. After centrifugation at 1000 × g at 4°C for 8 min, the supernatant was removed, and the pellet were re-suspended in DMEM/F12 containing 20% BSA. The suspension was centrifuged at 1000 × g for 20 min at 4°C, and the pellets containing microvessels were preserved. Microvessel pellets were digested in DMEM/F12 containing 1 g/L collagenase/dispase and 15 mg/L DNAse I at 37°C under constant shaking at 200 rpm/min for 1 h, followed by centrifugation at 1000 × g at 4°C for 8 min. Then, the supernatant was removed, and the resulting pellets were re-suspended in DMEM/F12 and cell suspension was added on the 33% Percoll gradient, followed by centrifugation at 1000 × g for 10 min at 4°C. The resulting microvascular fragments were re-suspended with ECM medium containing 4 µg/ml puromycin, seeded in flasks pre-coated with fibronectin, and maintained at 37°C in a humidified environment with 5% CO2. After culture for 48 h, the medium was refreshed with ECM medium without puromycin. Cells were grown until 80% cell confluence was observed, trypsinized and then seeded at a density of 4 × 104 cells/cm2. This was the first passage of cells. Cells of the third passage were used in the following experiment, in which L-Arg in ECM medium was supplemented at a final concentration of 500 µM.
siRNA transfection
Astrocytes were seeded into 6-well plates at a density of 6 × 104 cells/cm2 in 2 ml antibiotic-free DMEM/F12 medium supplemented with FBS and then maintained at 37°C for 24 h until the cell confluence reached 80%. Transfection of siRNA against HIF-1α was performed in the presence of siRNA Transfection Reagent according to the manufacturer’s instructions. Transfection mixtures (800 µl/well) included 160 pmol siRNA and 24 µl of siRNA Transfection Reagent. Cells were incubated for 5 h at 37°C in an environment with 5% CO2. Subsequently, the siRNA transfection medium was refreshed with antibiotic-free DMEM/F12 medium and cells were incubated for another 20 h at 37°C in an environment with 5% CO2. In a negative control group, a control siRNA-A (Santa Cruz) was used.
Hypoxia exposure
Hypoxia exposure was performed in a commercial Heal Force HF100 tri-gas incubator (Shanghai, China). In the normoxic condition, the incubator was flushed with 21% O2 (21% O2, 5% CO2, 74% N2), and the hypoxia conditions were mimicked with 9% O2 (9% O2, 5% CO2, 86% N2), 7% O2 (7% O2, 5% CO2, 88% N2), 5% O2 (5% O2, 5% CO2, 90% N2), 3% O2 (3% O2, 5% CO2, 92% N2), or 1% O2 (1% O2, 5% CO2, 94% N2). Cells were grown in the condition with predetermined oxygen level. When the cell confluence reached 80%, the medium was refreshed with the hypoxic medium (the medium was exposed to hypoxia for equilibration), and incubation continued.
Co-culture of BMECs and astrocytes
BMECs were cocultured were with astrocytes as previously described [25]. In brief, BMEC were seeded into the lower chamber of 6-well transwell chamber at a density of 6 × 104 cells/cm2, and then maintained for approximately 3 days until 80% confluence was achieved. Astrocytes were seeded into the upper chamber of another 6-well transwell chamber (BD, 0.4-µm pore size) at a density of 6 × 104 cells/cm2, and then maintained for 2 days until 80% confluence was achieved. Then, the upper chamber with astrocytes was placed into the transwell chamber containing BMECs and then the supernatant was collected for the detection of lactic acid content at specific time point. In addition, astrocytes in the upper chamber were lysed for the extraction of total RNA, followed by detection mRNA expression of VEGF and LDHA. In addition, total protein was extracted from cells after lysis, and the protein expression of HIF-1α and VEGF was detected.
Treatments
In cell viability assay, primary astrocytes were treated with 0.4, 0.6, 0.8, 1.0, 1.2 and 1.4 mM DETA. In the following experiments, primary astrocytes were treated with 1.0 mM DETA. In addition, primary astrocytes were pretreated for 30 min with 100 µM eNOS inhibitor L-NAME, 100 or 200 µM NO scavenger (CPTIO) before the treatment with 1.0 mM DETA for 12 h.
Cell viability assay
Cell viability was evaluated by MTT assay. Astrocytes were seeded into 96-well plates at a density of 3 × 104 cells/cm2 and maintained at 37°C for 24 h. Cells were exposed to various concentrations of DETA (0.4, 0.6, 0.8, 1.0, 1.2 and 1.4 mM). After 24-h incubation, MTT solution (0.5 mg/ml in DPBS) was added to each well, followed by incubation for another 4 h. Then, 150 µl of DMSO was added to each well to dissolve the formazan crystals, and the absorbance was measured at 490 nm using the Thermo Scientific Varioskan Flash microplate reader. The cell viability was determined according the absorbance and then compared with that of untreated cells. All experiments were performed in triplicate.
NO assay
Astrocytes or BMEC were seeded into 96-well plates at a density of 6 × 104 cells/cm2 and maintained at 37°C for 24 h. In order to detect the NO, the medium was refreshed and then collected 12 h later. The nitrate/nitrite concentration of the medium was used as an indicator of NO production and measured as previously described [29] using a commercially available Nitric Oxide Fluorometric Assay Kit according to the manufacturer’s instructions. The medium was added to a 96-well plate (75 µl/well) followed by addition of 15 µl of reaction mix (5 µl of enzyme cofactor working solution, 46 µl of lactate assay buffer, and 5 µl of nitrate reductase) and subsequent incubation for 4 h at room temperature. Then, 5 µl of Enhancer solution was added to each well, followed by incubation for 30 min at room temperature. After the addition of DAN Reagent (5 µl/well), incubation was done for 10 min at room temperature. Finally, 5 µl of NaOH was added to each well, and the mixture was incubated for 10 minutes at room temperature. The fluorescence was measured at excitation wavelength of 360 nm/emission wavelength of 450 nm using the Thermo Scientific Varioskan Flash fluorescence reader. The fluorescence reflects the concentration of sodium nitrite. The standard curve of sodium nitrite concentration was delineated, from which the concentration of nitrite was calculated. All experiments were performed in triplicate.
Lactate release assay
Astrocytes were seeded into 96-well plates or 6-well transwell plates at a density of 6 × 104 cells/cm2 and maintained at 37°C for 24 h. In order to detect the lactic acid in the supernatant, the medium was refreshed before drug treatment, hypoxia exposure or coculture. The supernatant was collected at 12 and 24 h for the detection of lactate content. In control group, the supernatant was collected from cells exposed to mild hypoxia (5% O2) alone. Lactate level of the supernatant was assessed as previously described [30] using a commercially available Lactate Colorimetric Assay Kit according to the manufacturer’s instructions. The supernatant was added to a 96-well plate (50 µl/well), followed by addition of 50 µl of reaction mix (46 µl lactate assay buffer, 2 µl lactate substrate mix, and 2 µl lactate enzyme mix) and subsequent incubation for 30 min at room temperature. The absorbance was measured at 450 nm using the Thermo Scientific Varioskan Flash microplate reader. The lactate level was determined according to the absorbance value and then compared with that of control group. All experiments were performed in triplicate.
Quantitative real-time reverse transcriptase PCR
Astrocytes were seeded into 12-well plates at a density of 6 × 104 cells/cm2 and maintained at 37°C for 24 h before drug treatment, hypoxia exposure or coculture. Cells were collected at 3, 6, and 12 h, and total RNA was extracted. For siRNA transfection, the medium was refreshed, and cells were collected at 24 h followed by extraction of total RNA with a Takara MiniBEST Universal RNA Extraction Kit (Dalian, China). Both the amount and purity of total RNA were determined by measuring the absorbance at 260 nm and 280 nm. Total RNA (1 μg) was reversely transcribed into cDNA using the Takara PrimeScript™ RT reagent Kit (Dalian, China). Quantitative real-time reverse transcriptase PCR (RT-PCR) was performed to detect the mRNA expression of HIF-1α, VEGF, LDHA, and β-actin using a Roche LightCycler® 480 System. Quantitative PCR was conducted in 0.2-ml PCR tubes with the corresponding primers and Takara SYBR® Premix Ex Taq™ II working solution (Dalian, China), using a custom PCR master mix under the following conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. The primers used were as follows: HIF-1α, 5’-GGGGAGGACGATGAACATCAA-3’ (F) and 5’-GGGTGGTTTCTTGTACCCACA-3’ (R); VEGF, 5’-CTGCCGTCCGATTGAGACC-3’ (F) and 5’-CCCCTCCTTGTACCACTGTC-3’ (R); LDHA, 5’-TGTCTCCAGCAAAGACTACTGT-3’ (F) and 5’-GACTGTACTTGACAATGTTGGGA-3’ (R); and β-actin, 5’-GGCTGTATTCCCCTCCATCG-3’ (F) and 5’-CCAGTTGGTAACAATGCCATGT-3’ (R). β-actin was used as the internal reference. The mRNA expression of each target gene was normalized to that of β-actin by using the 2-ΔΔCT method.
Western blotting
Astrocytes were seeded into 6-well plates at a density of 6 × 104 cells/cm2 and maintained at 37°C for 24 h, before drug treatment, hypoxia exposure or co-culture. Cells were collected at 6, 12, and 24 h, washed with ice-cold DPBS and re-suspended in 200 μl of lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS supplemented with protease inhibitor cocktail [Sigma, USA]) followed by incubation at 4°C for 5 min. Then, cells were sonicated for 15 s on ice and centrifuged at 13,000 × g for 10 min at 4°C. The supernatant was collected, and the protein content was determined using an enhanced BCA protein assay kit from Beyotime (Haimen, China). The total protein from each sample was mixed with loading buffer and then heated at 100°C for 5 min. Total protein (20 μg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene fluoride (PVDF) membranes using a Bio-Rad Trans-Blot® transblot module. Non-specific protein binding of PVDF membranes was blocked by incubation with 5% bovine serum albumin (BSA) at room temperature for 1 h. Then, these membranes were treated with primary antibodies against HIF-1α (1:500), VEGF (1:200), or β-actin (1:2000) overnight at 4°C. After three washes with Tris-buffered saline solution containing 0.1% Tween 20 (TBST), the membranes were incubated at room temperature for 1 h with HRP-conjugated goat anti-rabbit IgG antibody (1:5,000) or HRP-conjugated rabbit anti-mouse IgG antibody (1:5,000). Visualization was done using ECL detection reagent according to the manufacturer’s instructions. The optical density of each band was measured using the Bio-Rad ChemiDocTM XRS luminescent image analyzer and Gel-Pro analyzer 4.0 software.
Statistical analysis
All the data are presented as mean ± standard error (S.E.M). One-way ANOVA followed by the least-significant difference (LSD) was used for the comparisons among groups. A value of P<0.05 was considered statistically significant.
Results
Hypoxia threshold difference between in vitro primary cultured astrocytes and BMEC
Cells in different tissues are exposed to oxygen at different partial pressure, and the farther the distance between the tissue and blood vessels, the lower the oxygen partial pressure of the tissue is. Thus, the threshold of different types of cells recognizing hypoxia is also different [12,31,32]. In the in vitro culture, cells are exposed to 21% oxygen (oxygen partial pressure of the medium is about 90 mmHg), which ensures sufficient oxygen supply to different types of cells, but this manipulation ignores the different thresholds of different cell types recognizing hypoxia. In order to investigate the threshold of primary astrocytes and primary BMEC recognizing hypoxia in vitro, cells were exposed to oxygen at different concentrations (21%, 9%, 7%, 5%, 3%, and 1%), and the corresponding oxygen partial pressure of the medium is 45, 35, 25, 15, and 5 mmHg, respectively. Then, the protein expression of HIF-1α was detected in whole cell lysate after 12-h culture. After culture in 3% or 1% oxygen, the HIF-1α protein expression increased significantly compared with cells exposed to 21% oxygen (control group) (P<0.001), while exposure to 9%, 7%, or 5% oxygen failed to significantly alter the HIF-1α protein expression compared with control group (Figure 1A). When BMECs were exposed to 5%, 3%, or 1% oxygen, the HIF-1α protein expression increased significantly compared with control group (P<0.001), while exposure to 9% or 7% oxygen failed to significantly alter the HIF-1α protein expression compared with control group (Figure 1B). These suggest that, when compared with BMECs, astrocytes are more potent to tolerate hypoxia, or BMEC are more sensitive to hypoxia than astrocytes, as 5% oxygen (oxygen partial pressure of 25 mmHg) is probably the threshold for BMECs to recognize hypoxia, while astrocytes has the threshold of 3% oxygen. Therefore, 5% oxygen concentration was used as the mild hypoxia, and then applied in following experiments.
Figure 1.
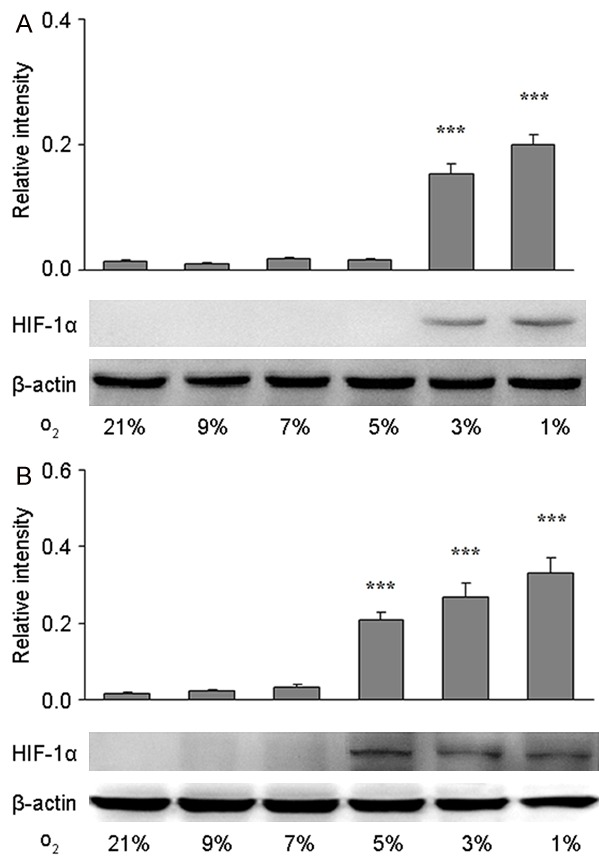
Threshold of hypoxia recognition of primary astrocytes and brain microvascular endothelial cells (BMEC) in vitro. HIF-1α protein expression was detected by Western blotting in primary astrocytes (A) and primary BMECs (B) treated with 21%, 9%, 7%, 5%, 3%, or 1% O2 for 12 h. β-actin served as a loading control. The optical density of HIF-1α band was normalized to that of β-actin, and quantification was performed from two to three independent experiments. Data are shown as mean ± S.E.M. *P<0.05, **P<0.01, ***P<0.001 vs. control group (21% O2).
Exogenous NO blocked HIF-1α degradation in astrocytes under mild hypoxia
In order to determine if exogenous NO was able to block HIF-1α degradation in astrocytes under mild hypoxia, DETA was used as an exogenous NO donor. First, MTT assay was performed to assess the toxicity of varying concentrations of DETA (0.4, 0.6, 0.8, 1.0, 1.2, and 1.4 mM) on astrocytes under mild hypoxia (5% oxygen). Low concentrations (0.4-1.2 mM) of DETA had no significant impact on the viability of astrocytes while DETA at 1.4 mM significantly inhibited the viability of astrocytes (Figure 2A, P<0.05). In order to investigate the degradation of HIF-1α in astrocytes under mild hypoxia (5% oxygen), astrocytes were treated with different concentrations of DETA (0.4, 0.6, 0.8, and 1.0 mM) for 6, 12, or 24 h. With 5% oxygen, there was no significant difference in HIF-1α protein expression in astrocytes between the 0.4 or 0.6 mM DETA group at 12 h and the control group (no DETA), while treatment with 0.8 or 1.0 mM DETA significantly increased the HIF-1α protein expression when compared with control group, and the HIF-1α protein expression was comparable to that seen in astrocytes exposed to 1% oxygen (Figure 2B, P<0.001). After treatment with 1.0 mM DETA for 6, 12, or 24 h, HIF-1α protein expression significantly increased when compared with control group (P<0.001), but was comparable to that in cells exposed to 1% oxygen (Figure 2C). In order to clarify whether NO was able to block HIF-1α degradation in astrocytes under mild hypoxia, an NO scavenger was used. First, astrocytes were pretreated with the NO scavenger CPTIO (100 or 200 µm), and then exposed to 1.0 mM DETA for 12 h under mild hypoxia (5% oxygen). HIF-1α protein expression of astrocytes in CPTIO treated groups decreased significantly when compared with control group (CPTIO-, Figure 2D, P<0.05 and P<0.001, respectively), but there was still a significant difference in HIF-1α protein expression between the 100 and 200 µm CPTIO groups and the control group (DETA-, P<0.001 and P<0.01, respectively).
Figure 2.

DETA blocked HIF-1α degradation in astrocytes under mild hypoxia. (A) Cell viability of primary astrocytes in the presence or absence of DETA (0.4, 0.6, 0.8, 1.0, 1.2 or 1.4 mM) after exposure to 5% O2 for 24 h (MTT assay). DETA had no cytotoxicity on astrocytes at the concentration of up to 1.2 mM. (B-D) HIF-1α protein expression was detected by Western blotting in primary astrocytes treated with 0.4, 0.6, 0.8, or 1.0 mM DETA for 12 h (B), with 1.0 mM DETA for 6, 12, or 24 h (C), or pretreated with 100 or 200 µM of the NO scavenger CPTIO for 30 min before the 1.0 mM DETA treatment for 12 h (D). β-actin served as a loading control. The optical density of HIF-1α band was normalized to that of β-actin, and quantification was performed in two to three independent experiments. Data are expressed as mean ± S.E.M. *P<0.05, **P<0.01, ***P<0.001 vs. control group (5% O2 exposure alone); #P<0.05, ##P<0.01, and ###P<0.001 vs. control group (1.0 mM DETA treatment and exposure to 5% O2).
Effects of exogenous NO on the angiogenesis and expression of glycolysis-related genes in astrocytes under mild hypoxia
In order to investigate the effect of exogenous NO on the angiogenesis and transcription and expression of glycolysis-related genes in astrocytes under mild hypoxia, DETA was used to stimulate primary astrocytes under 5% oxygen, and total RNA and protein were independently extracted for the detection of mRNA and protein expression of VEGF as well as the mRNA expression of LDHA. In addition, the production of lactate under 5% oxygen was also measured in astrocytes after treatment with 1.0 mM DETA for 6 or 12 h. Results showed mRNA expression of VEGF was significantly higher than in control group (DETA-, Figure 3A, P<0.01, P<0.001); the increased mRNA expression of VEGF was comparable to that in cells under the extreme hypoxia (1% oxygen). After treatment with DETA for 12 or 24 h, VEGF protein expression was significantly higher than in control group (DETA-, Figure 3C, P<0.01), but was similar to that under extreme hypoxia (1% oxygen). Under 5% oxygen, after treatment with 1.0 mM DETA for 6 or 12 h, the mRNA expression of LDHA was significantly higher than in control group (DETA-, Figure 3B, P<0.05 and P<0.001, respectively), and the increased mRNA expression of LDHA after DETA treatment for 12 h was similar to that after exposure to extreme hypoxia (1% oxygen). After DETA treatment for 24 h, the lactate in the supernatant significantly increased when compared with control group (DETA-, Figure 3D, P<0.001), but was comparable to that after exposure to extreme hypoxia (1% oxygen).
Figure 3.
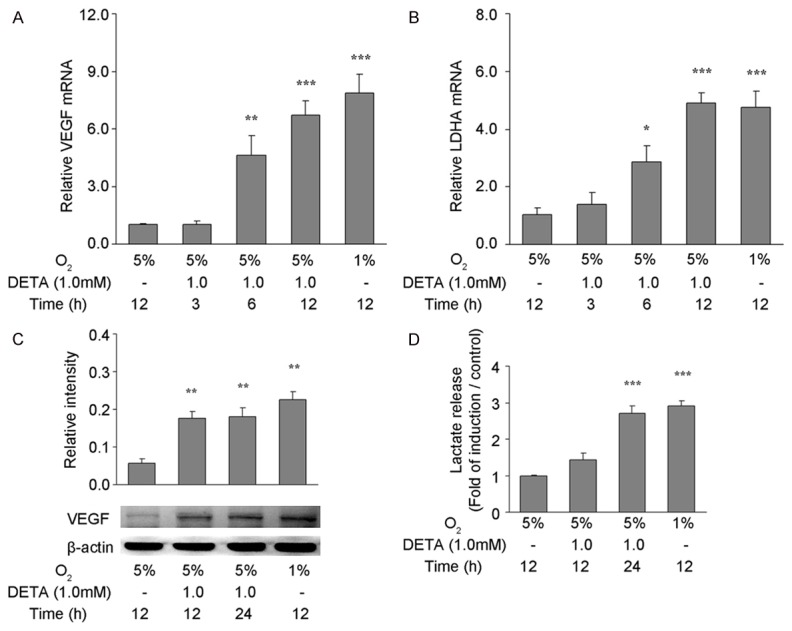
Influence of DETA on the angiogenesis and expression of glycolysis related genes in astrocytes under mild hypoxia. Primary astrocytes with or without 1.0 mM DETA treatment were exposed to 5% or 1% O2 for 3, 6, or 12 h. qRT-PCR was performed to detect the mRNA expression of VEGF (A) and LDHA (B). The mRNA expression of target genes was normalized to that of β-actin by using the 2-ΔΔCT method. Primary astrocytes with or without 1.0 mM DETA treatment were exposed to 5% or 1% O2 for 12 or 24 h. Western blotting was performed to detect VEGF protein expression in astrocytes (C) and colorimetric measurement was done for the detection of lactate in the medium (D). Data are expressed as mean ± S.E.M. of triplicate samples from one experiment. The experiment was repeated three times on different cell preparations. *P<0.05, **P<0.01, ***P<0.001 vs. control group treated (5% O2 exposure alone).
In order to determine whether exogenous NO could regulate angiogenesis and expression of glycolysis-related genes in astrocytes by blocking HIF-1α degradation under mild hypoxia, HIF-1α specific siRNA was used to silence HIF-1α expression in astrocytes. After silencing, HIF-1α mRNA expression decreased by 85% as compared to negative control group (Figure 4A). After treatment with 5% oxygen and 1.0 mM DETA for 12 h, HIF-1α protein expression (Figure 4B) as well as VEGF and LDHA mRNA expression (Figure 4C, 4D) decreased significantly in cells treated with HIF-1α siRNA as compared to negative control group (P<0.01 or P<0.001). When HIF-1α siRNA-transfected astrocytes were treated with 1.0 mM DETA for 12 h under hypoxia, VEGF protein expression (Figure 4E) and lactate release (Figure 4F) decreased significantly when compared with negative control group (P<0.001). Interestingly, after silencing HIF-1α expression, VEGF protein expression still increased although there was DETA treatment in astrocytes (Figure 4E, P<0.05).
Figure 4.

HIF-1α siRNA silencing prevents NO-induced angiogenesis and expression of glycolysis-related genes in astrocytes under mild hypoxia. Primary astrocytes were transfected with HIF-1α siRNA or negative control siRNA (NC) 24 h before DETA treatment, and the transfection efficiency was confirmed by qRT-PCR (A) and Western blotting (B). mRNA expression of VEGF (C) and LDHA (D) was performed in astrocytes by qRT-PCR after 12-h exposure to 5% O2 in the presence or absence of 1.0 mM DETA treatment. The mRNA expression of target genes was normalized to that of β-actin by using the 2-ΔΔCT method. Western blotting was used to detect VEGF protein expression (E), and colorimetric measurement was done to for the detection of lactate (F) in the medium after 12 or 24-h exposure to 5% O2 in the presence or absence of 1.0 mM DETA treatment. Data are expressed as mean ± S.E.M. of triplicate samples from one experiment. The experiment was repeated three times on different cell preparations. *P<0.05, **P<0.01, ***P<0.001 vs. the indicated group.
Effects of BMEC-derived NO on the angiogenesis and expression of glycolysis-related genes in astrocytes under mild hypoxia
In order to determine the effects of endogenous BMEC-derived NO on the degradation of HIF-1α protein in astrocytes under mild hypoxia, BMECs were co-cultured with astrocytes. Under mild hypoxia (5% oxygen), BMECs could release NO [33], thereby blocking the degradation of HIF-1α protein. In order to confirm the source of NO, the NO content was measured in the supernatant of BMECs and astrocytes exposed to 5% oxygen. After exposure to 5% oxygen, the content of NO released by BMECs was significantly higher than that released by astrocytes (Figure 5A, P<0.001). When treated with an eNOS inhibitor L-NAME, the content of NO released by BMECs was significantly reduced (P<0.001). Then, Transwell co-culture system was used for the coculture of BMEC and astrocytes, and the HIF-1α protein expression was detected in astrocytes. Under 5% oxygen, HIF-1α protein expression in astrocytes cocultured with BMECs was significantly higher than in the control group (BMECs-, Figure 5B, P<0.01). However, after pretreatment with eNOS inhibitor L-NAME, HIF-1α protein expression in astrocytes cocultured with BMECs and exposed to 5% oxygen was significantly reduced (P<0.01), and was similar to that in control group. After coculture of BMEC and astrocytes transfected with negative control siRNA, the HIF-1α protein expression in astrocytes increased significantly (Figure 5C, P<0.001). In contrast, after silencing HIF-1α expression in astrocytes by HIF-1α siRNA, the HIF-1α protein expression in astrocytes was decreased significantly (P<0.001) following co-culture with BMECs and exposure to 5% oxygen, but was still higher than in control group (BMEC-, P<0.01).
Figure 5.

Effects of BMEC-derived NO on the angiogenesis and expression of glycolysis-related genes in astrocytes under mild hypoxia. (A) NO content in primary BMEC and primary astrocytes was assessed after 12-h exposure to 5% O2 with or without pretreatment with 100 µM L-NAME. (B) HIF-1α protein expression was assessed by Western blotting in astrocytes alone or cocultured with BMECs after 12-h exposure to 5% O2 with or without pretreatment with 100 µM L-NAME. (C) Primary astrocytes with or without transfection with HIF-1α siRNA or negative control siRNA (NC) 24 h before coculture with BMECs were exposed to 5% O2. After 12-h exposure, HIF-1α protein expression was detected by Western blotting. In addition, mRNA expression of VEGF (D) and LDHA (E) was detected by qRT-PCR from astrocytes with or without co-culture with BMECs after exposure to 5% O2 for 12 h in the presence or absence of pretreatment with 100 µM L-NAME. The mRNA expression of target genes was normalized to that of β-actin by using the 2-ΔΔCT method. VEGF expression was detected by Western blotting (F). Colorimetric measurement was done for the detection of lactate in the medium (G) of astrocytes with or without coculture with BMECs after exposure to 5% O2 in the presence of absence of pretreatment with 100 µM L-NAME. Data are expressed as mean ± S.E.M. of triplicate samples from one experiment. The experiment was repeated three times on different cell preparations. *P<0.05, **P<0.01, ***P<0.001 vs. indicated group.
Then, the effects of endothelial-derived NO on the angiogenesis and expression of glycolysis-related genes were further investigated in astrocytes under mild hypoxia. Under 5% oxygen, the VEGF and LDHA mRNA expression in astrocytes co-cultured with BMECs was significantly increased when compared with control group (BMEC-, Figure 5D and 5E, P<0.001). This effect was blocked by the pretreatment with eNOS inhibitor L-NAME; the VEGF and LDHA mRNA expression in astrocytes was significantly decreased (P<0.001). There was no significant difference in the VEGF mRNA expression between astrocytes co-cultured with BMECs and astrocytes without co-culture (BMEC-), while the LDHA mRNA expression in astrocytes co-cultured with BMECs was higher than in control group (P<0.05). VEGF protein expression in astrocytes and lactate in the supernatant were significantly higher in astrocytes co-cultured with BMECs after exposure to 5% oxygen than in control group (Figure 5F and 5G, P<0.001 and P<0.01, respectively). However, after pretreatment with eNOS inhibitor L-NAME, VEGF protein expression in astrocytes and lactate in the supernatant were significantly decreased (P<0.001, P<0.01); but the lactate in the supernatant was similar to that in control group, while VEGF protein expression in astrocytes co-cultured with BMECs was markedly higher than in control group (P<0.01).
Discussion
Hypoxia triggers a compensatory adaptive response that is protective and may avoid the damage of local or systemic hypoxia on cells, organs or organisms [34]. Following hypoxia exposure, the body should recognize the reduced oxygen partial pressure, and one of the most important mechanisms has involvement of HIF-1 [20]. HIF-1 is a heterodimer composed of an α subunit and a β subunit: HIF-1α is the major regulatory and active subunit, and its activity is strictly regulated by oxygen [35]. HIF-1α is regulated by oxygen via prolyl hydroxylase (PHD) and the whole process needs the involvement of oxygen, Fe2+, oxoglutarate (2-OG), and ascorbate as cofactors [36,37]. PHD is activated in normoxia to hydroxylate the proline residue of HIF-1α, which may promote the HIF-1α degradation through the ubiquitin-proteasome pathway [38]. In hypoxia, PHD is inactivated, and thus the HIF-1α degradation is blocked. Then, HIF-1α translocates into the nucleus to form dimers with HIF-1β. After biding to hypoxic response elements (HRE) of specific genes, it may regulate their transcription, causing a series of compensatory adaptive response in cells to hypoxia [34]. Approximately one hundred of the genes can be regulated directly by HIF-1, and the changes in the transcriptome and proteome secondary to the activation of these genes are closely related to the angiogenesis, glycolysis and cell survival [14,16-18].
Generally, oxygen is transported by circulation and may diffuse into tissues and cells according to the oxygen partial pressure gradient. In different tissues, due to the difference in the distribution of blood vessels, the cells are exposed to oxygen at different partial pressures in their local environment [4,12]. The farther the distance between the tissue and the blood vessels, the lower the oxygen partial pressure of the tissue is; the physiological oxygen partial pressure of vascular endothelial cells is 30-100 mmHg, while that of organs or tissues is 8-72 mmHg [31]. The range of physiological oxygen partial pressure in the brain is 23.8-33.3 mmHg [13]. It can be speculated that when the oxygen partial pressure is less than 30 mmHg, BMECs will experience hypoxia. However, when the oxygen partial pressure is less than 23.8 mmHg, glial cells and neurons in the brain will experience hypoxia. Thus, it is hypothesized that there is a difference in the hypoxia recognition threshold between BMECs and other brain cells (such as glial and neurons).
In in vitro culture, cells are exposed to 21% oxygen (the oxygen partial pressure of the medium is approximately 90 mmHg). This oxygen partial pressure ensures sufficient oxygen supply, but this manipulation ignores the variation in hypoxia recognition threshold among different cell types. In the present study, primary rat astrocytes and primary BMEC were exposed to oxygen at different concentration, the response to hypoxia was evaluated by detecting HIF-1α expression, and the hypoxia recognition threshold was assessed in these two cell types. Results showed that, when compared with BMECs, astrocytes were more tolerant to hypoxia, or BMECs were more sensitive to hypoxia than astrocytes; 5% oxygen (oxygen partial pressure of the medium was approximately 25 mmHg) was probably the recognition threshold of these two cell types for hypoxia, which was consistent with previous findings that BMECs would recognize hypoxia when the oxygen partial pressure was less than 30 mmHg, while glial cells and neurons in brain tissues could recognize hypoxia when the oxygen partial pressure was less than 23.8 mmHg [13]. Under different hypoxic conditions, the different responses of different cell types in hypoxia recognition may be related to the cofactors (Fe2+, 2-OG, ascorbate) essential for PHD enzymatic catalysis, or to the newly produced NO or superoxide in cells. These can regulate the activity of PHD [21,37], thereby regulating the stability of HIF-1α [36-38], but the specific mechanism should be explored in future studies.
Hypoxia in BMECs and astrocytes may activate HIF to promotes the transcription and expression of VEGF, which leads to the HIF dependent post-hypoxic angiogenesis. This increases the vasculature density and thereby increases the oxygen supply [39]. In astrocytes, HIF may also regulate the transcription and expression of key enzymes in the anaerobic glycolysis, such as pyruvate dehydrogenase kinase 1 (PDK1) and lactate dehydrogenase (LDH) [25,40]. Mitochondrial pyruvate dehydrogenase complex (PDC) can catalyze the oxidative decarboxylation of pyruvate into NADH and acetyl coenzyme A that can produce large amounts of ATP [41]. In hypoxia, activated HIF can induce the over-expression of PDK1, which inhibits the PDC activity and reduces oxygen consumption of the citric acid cycle, leading to the entry of pyruvate into the anaerobic glycolysis pathway [42]. LDH is an enzyme that catalyzes pyruvic acid into lactic acid, and is composed of four subunits [43]. The two most common subunits are LDH-M and LDH-H, which are encoded by the LDHA and LDHB genes, respectively. In hypoxia, activated HIF can induce the over-expression of LDHA to further catalyze pyruvic acid that can’t enter the citric acid cycle into lactic acid, which is also known as the anaerobic glycolytic pathway of glucose metabolism [44,45]. It is essential to protect the brain against hypoxia through initiating the compensatory adaptive response in the vascular system and glial cells during hypoxia [46-48].
During hypoxia, the compensatory responses of the vascular system include the vasodilation of small arteries, which is directly regulated by NO [19] and also has involvement of HIF-1α stability [49-51]. In addition to oxygen, the activation of PHD needs Fe2+ as a cofactor to catalyze HIF-1α hydroxylation, entering the ubiquitin degradation pathway [36-38]. Studies have shown that NO can inhibit the activity of PHD by interfering with PHD-bound iron, thereby blocking HIF-1α degradation [52]. This was also confirmed in cells under normoxic and hypoxic conditions [21-24]. However, NO released by NO donor is capable of blocking HIF-1α degradation in astrocytes under normoxic conditions but has no effect on neurons [25,26]. Meanwhile, in BBB models, the physiological level of NO released by BMECs also can block HIF-1α degradation in astrocytes [25]. Similar findings were also found in our study. Under normoxia (21% O2) and hypoxia (9%, 7%, and 5% O2), NO released by an exogenous NO donor blocked HIF-1α degradation in primary astrocytes. Under mild hypoxia (5% O2), NO up-regulated the mRNA expression of VEGF and LDHA the protein expression of VEGF and the production of lactic acid. Through silencing HIF-1α expression in astrocytes, we demonstrated that NO achieved this effect by regulating the HIF-1α stability. Under mild hypoxia (5% O2), in coculture of primary rat BMEC and astrocytes, the physiological level of NO released by BMECs also blocked HIF-1α degradation in astrocytes, increased the mRNA expression of VEGF and LDHA, up-regulated protein expression of VEGF and increased production of lactic acid.
Our studies proved that NO that can initiate HIF degradation in astrocytes is from BMECs rather than astrocytes, and NO produced by BMECs is produced by the catalysis of intracellular eNOS. However, when the eNOS was inhibited, the HIF-1α protein expression compared was comparable to that in control group while LDHA mRNA expression and VEGF protein expression still increased, which suggests there may be other mechanisms. We speculate that HIF-2α may play a role in the physiological response to hypoxia. HIF has two subtypes in mammalian cells, HIF-1α and HIF-2α, which are stable under hypoxic conditions and have very similar regulatory effects on downstream target genes. Compared with extreme hypoxia, mild hypoxia is more likely to affect HIF-2α and HIF-2α has been considered as an initiatior of physiological response to hypoxia [31,53,54]. Therefore, under mild hypoxia, HIF-2α and HIF-1α may regulate a variety of downstream target genes cooperatively, or by feedback, but the specific mechanism is still unclear.
In conclusion, during mild hypoxia (5% O2), exogenous NO and NO released from BMEC blocked the degradation of HIF-1α in astrocytes but up-regulated the transcription of VEGF and LDHA, accompanied by elevated expression of VEGF protein and increased production of lactic acid. In BMECs, NO was derived from intracellular eNOS. Based on these findings, we hypothesized that under mild hypoxia (5% O2), even though astrocytes do not recognize the decline of oxygen partial pressure in the environment, NO produced by BMECs may enter the astrocytes by diffusion, transmitting the hypoxia signal to initiate the compensatory adaptive response to hypoxia in a HIF-1α dependent manner (Figure 6). The key physiological significance of our studies is that NO may be one of factors initiating the hypoxia signal transmission between cells in the neurovascular unit without recognizing low oxygen partial pressure. Despite the different thresholds of hypoxia recognition among different cell types, this response allows the glial cells in the NVU to adapt to mild hypoxia independent of the local oxygen partial pressure, thereby providing adequate oxygen supply to neurons as much as possible so as to maintain normal physiological function.
Figure 6.
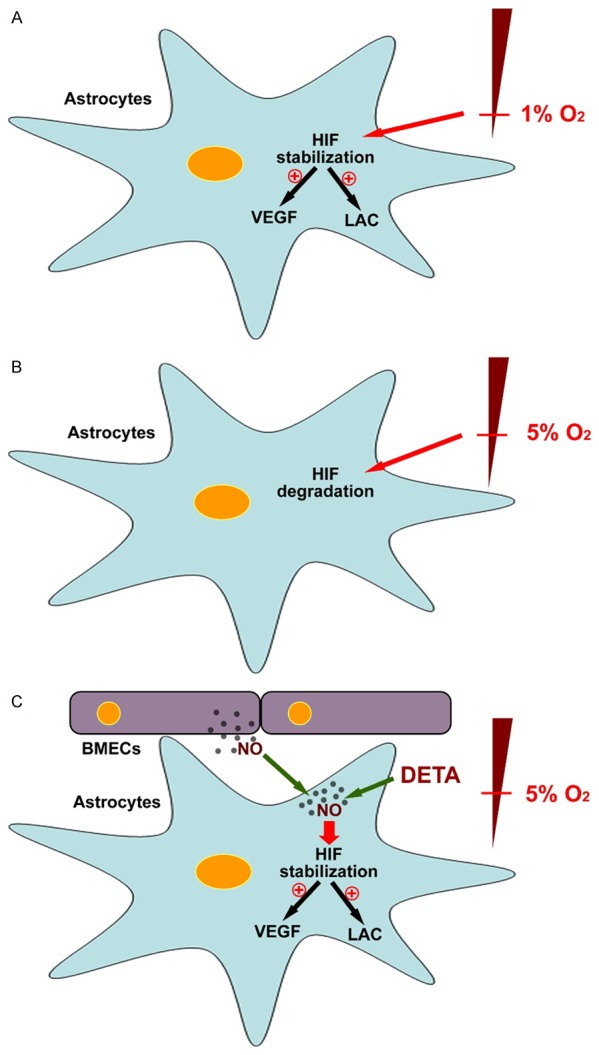
NO acts as a hypoxia signal to initiate a compensatory response in astrocytes under mild hypoxia in a HIF-1α dependent manner. A. Under extreme hypoxia (1% O2), HIF-1α degradation in rat primary astrocytes was blocked, and the mRNA expression of VEGF and LDHA was up-regulated, thereby increasing the protein expression of VEGF and the production of lactate. B. Under mild hypoxia (5% O2), HIF-1α in astrocytes was degraded. C. However, under mild hypoxia, increased NO (either from an NO donor or from BMEC) was capable of blocking HIF-1α degradation in primary astrocytes, increasing the mRNA expression of VEGF and LDHA, thereby increasing the protein expression of VEGF and the production of lactic acid.
Acknowledgements
This work was supported by the National Science Foundation of China (No. 81301134).
Disclosure of conflict of interest
None.
References
- 1.Korner P. Circulatory adaptations in hypoxia. Physiol Rev. 1959;39:687–730. doi: 10.1152/physrev.1959.39.4.687. [DOI] [PubMed] [Google Scholar]
- 2.Petrassi FA, Hodkinson PD, Walters PL, Gaydos SJ. Hypoxic hypoxia at moderate altitudes: review of the state of the science. Aviat Space Environ Med. 2012;83:975–984. doi: 10.3357/asem.3315.2012. [DOI] [PubMed] [Google Scholar]
- 3.May AM, Mehra R. Obstructive sleep apnea: role of intermittent hypoxia and inflammation. Semin Respir Crit Care Med. 2014;35:531–544. doi: 10.1055/s-0034-1390023. [DOI] [PubMed] [Google Scholar]
- 4.Erecinska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128:263–276. doi: 10.1016/s0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- 5.Amann M, Kayser B. Nervous system function during exercise in hypoxia. High Alt Med Biol. 2009;10:149–164. doi: 10.1089/ham.2008.1105. [DOI] [PubMed] [Google Scholar]
- 6.Neubauer JA, Melton JE, Edelman NH. Modulation of respiration during brain hypoxia. J Appl Physiol (1985) 1990;68:441–451. doi: 10.1152/jappl.1990.68.2.441. [DOI] [PubMed] [Google Scholar]
- 7.Schurr A, Rigor BM. The mechanism of neuronal resistance and adaptation to hypoxia. FEBS Lett. 1987;224:4–8. doi: 10.1016/0014-5793(87)80411-8. [DOI] [PubMed] [Google Scholar]
- 8.Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K, Lo EH. Cell-cell signaling in the neurovascular unit. Neurochem Res. 2007;32:2032–2045. doi: 10.1007/s11064-007-9342-9. [DOI] [PubMed] [Google Scholar]
- 10.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 11.Kim JH, Park JA, Lee SW, Kim WJ, Yu YS, Kim KW. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol. 2006;39:339–345. doi: 10.5483/bmbrep.2006.39.4.339. [DOI] [PubMed] [Google Scholar]
- 12.Pittman RN. Oxygen gradients in the microcirculation. Acta Physiol (Oxf) 2011;202:311–322. doi: 10.1111/j.1748-1716.2010.02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dings J, Meixensberger J, Jager A, Roosen K. Clinical experience with 118 brain tissue oxygen partial pressure catheter probes. Neurosurgery. 1998;43:1082–1095. doi: 10.1097/00006123-199811000-00045. [DOI] [PubMed] [Google Scholar]
- 14.Bolanos JP, Almeida A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim Biophys Acta. 1999;1411:415–436. doi: 10.1016/s0005-2728(99)00030-4. [DOI] [PubMed] [Google Scholar]
- 15.Plate KH. Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol. 1999;58:313–320. doi: 10.1097/00005072-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci. 2015;9:112. doi: 10.3389/fnins.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci. 2008;28:1988–1993. doi: 10.1523/JNEUROSCI.5323-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marrif H, Juurlink BH. Astrocytes respond to hypoxia by increasing glycolytic capacity. J Neurosci Res. 1999;57:255–260. doi: 10.1002/(SICI)1097-4547(19990715)57:2<255::AID-JNR11>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 19.Akgoren N, Dalgaard P, Lauritzen M. Cerebral blood flow increases evoked by electrical stimulation of rat cerebellar cortex: relation to excitatory synaptic activity and nitric oxide synthesis. Brain Res. 1996;710:204–214. doi: 10.1016/0006-8993(95)01354-7. [DOI] [PubMed] [Google Scholar]
- 20.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 21.Brune B, Zhou J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1alpha (HIF-1alpha) Curr Med Chem. 2003;10:845–855. doi: 10.2174/0929867033457746. [DOI] [PubMed] [Google Scholar]
- 22.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 23.Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D’Acquisto F, Addeo R, Makuuchi M, Esumi H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood. 2000;95:189–197. [PubMed] [Google Scholar]
- 24.Sogawa K, Numayama-Tsuruta K, Ema M, Abe M, Abe H, Fujii-Kuriyama Y. Inhibition of hypoxia-inducible factor 1 activity by nitric oxide donors in hypoxia. Proc Natl Acad Sci U S A. 1998;95:7368–7373. doi: 10.1073/pnas.95.13.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brix B, Mesters JR, Pellerin L, Johren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J Neurosci. 2012;32:9727–9735. doi: 10.1523/JNEUROSCI.0879-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosafio K, Pellerin L. Oxygen tension controls the expression of the monocarboxylate transporter MCT4 in cultured mouse cortical astrocytes via a hypoxia-inducible factor-1alpha-mediated transcriptional regulation. Glia. 2014;62:477–490. doi: 10.1002/glia.22618. [DOI] [PubMed] [Google Scholar]
- 28.Calabria AR, Weidenfeller C, Jones AR, de Vries HE, Shusta EV. Puromycin-purified rat brain microvascular endothelial cell cultures exhibit improved barrier properties in response to glucocorticoid induction. J Neurochem. 2006;97:922–933. doi: 10.1111/j.1471-4159.2006.03793.x. [DOI] [PubMed] [Google Scholar]
- 29.Nussler AK, Glanemann M, Schirmeier A, Liu L, Nussler NC. Fluorometric measurement of nitrite/nitrate by 2,3-diaminonaphthalene. Nat Protoc. 2006;1:2223–2226. doi: 10.1038/nprot.2006.341. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg JC, Rush BF. An enzymatic-spectrophotometric determination of pyruvic and lactic acid in blood. Methodologic aspects. Clin Chem. 1966;12:299–307. [PubMed] [Google Scholar]
- 31.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15:1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carreau A, Kieda C, Grillon C. Nitric oxide modulates the expression of endothelial cell adhesion molecules involved in angiogenesis and leukocyte recruitment. Exp Cell Res. 2011;317:29–41. doi: 10.1016/j.yexcr.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood. 2004;104:2073–2080. doi: 10.1182/blood-2004-02-0744. [DOI] [PubMed] [Google Scholar]
- 34.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 35.Huang LE, Bunn HF. Hypoxia-inducible factor and its biomedical relevance. J Biol Chem. 2003;278:19575–19578. doi: 10.1074/jbc.R200030200. [DOI] [PubMed] [Google Scholar]
- 36.Fandrey J, Gorr TA, Gassmann M. Regulating cellular oxygen sensing by hydroxylation. Cardiovasc Res. 2006;71:642–651. doi: 10.1016/j.cardiores.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 37.Brune B, Zhou J. Nitric oxide and superoxide: interference with hypoxic signaling. Cardiovasc Res. 2007;75:275–282. doi: 10.1016/j.cardiores.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 40.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 41.Rich PR. The molecular machinery of Keilin’s respiratory chain. Biochem Soc Trans. 2003;31:1095–1105. doi: 10.1042/bst0311095. [DOI] [PubMed] [Google Scholar]
- 42.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Holmes RS, Goldberg E. Computational analyses of mammalian lactate dehydrogenases: human, mouse, opossum and platypus LDHs. Comput Biol Chem. 2009;33:379–385. doi: 10.1016/j.compbiolchem.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warrell DA, White NJ, Veall N, Looareesuwan S, Chanthavanich P, Phillips RE, Karbwang J, Pongpaew P, Krishna S. Cerebral anaerobic glycolysis and reduced cerebral oxygen transport in human cerebral malaria. Lancet. 1988;2:534–538. doi: 10.1016/s0140-6736(88)92658-x. [DOI] [PubMed] [Google Scholar]
- 45.Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 47.Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci. 2011;31:7477–7485. doi: 10.1523/JNEUROSCI.0415-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zilberter Y, Zilberter T, Bregestovski P. Neuronal activity in vitro and the in vivo reality: the role of energy homeostasis. Trends Pharmacol Sci. 2010;31:394–401. doi: 10.1016/j.tips.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Sandau KB, Fandrey J, Brune B. Accumulation of HIF-1alpha under the influence of nitric oxide. Blood. 2001;97:1009–1015. doi: 10.1182/blood.v97.4.1009. [DOI] [PubMed] [Google Scholar]
- 50.Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL, Hirota K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem. 2004;279:2550–2558. doi: 10.1074/jbc.M308197200. [DOI] [PubMed] [Google Scholar]
- 51.Park YK, Ahn DR, Oh M, Lee T, Yang EG, Son M, Park H. Nitric oxide donor, (+/-)-S-nitroso-N-acetylpenicillamine, stabilizes transactive hypoxia-inducible factor-1alpha by inhibiting von Hippel-Lindau recruitment and asparagine hydroxylation. Mol Pharmacol. 2008;74:236–245. doi: 10.1124/mol.108.045278. [DOI] [PubMed] [Google Scholar]
- 52.Berchner-Pfannschmidt U, Tug S, Kirsch M, Fandrey J. Oxygen-sensing under the influence of nitric oxide. Cell Signal. 2010;22:349–356. doi: 10.1016/j.cellsig.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Ben-Shoshan J, Maysel-Auslender S, Luboshits G, Barshack I, Polak-Charcon S, Tzahor E, Keren G, George J. Hypoxia-inducible factor-1alpha and -2alpha additively promote endothelial vasculogenic properties. J Vasc Res. 2009;46:299–310. doi: 10.1159/000181546. [DOI] [PubMed] [Google Scholar]
- 54.Lofstedt T, Fredlund E, Holmquist-Mengelbier L, Pietras A, Ovenberger M, Poellinger L, Pahlman S. Hypoxia inducible factor-2alpha in cancer. Cell Cycle. 2007;6:919–926. doi: 10.4161/cc.6.8.4133. [DOI] [PubMed] [Google Scholar]