

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a prevalent obesity-related disease that affects large populations throughout the world due to excessive calorie intake and an increasingly sedentary lifestyle. Fibroblast growth factor 21 (FGF21) has recently emerged as a promising therapeutic candidate for the treatment of obesity and diabetes. FGF21 is a starvation-induced pleiotropic hormone with various beneficial metabolic effects, and pharmacological treatment in rodents has been shown to improve insulin sensitivity and decrease simple fatty liver disease. However, its effects on reversing the symptoms of advanced liver disease have yet to be validated. Here, we investigated the protective effects of the LY2405319 compound, an engineered FGF21 variant, in a non-alcoholic steatohepatitis (NASH) model using leptin-deficient ob/ob mice and a methionine- and choline-deficient (MCD) diet to induce steatohepatitis. LY2405319 treatment in ob/ob mice corroborated previous results showing that improvements in the metabolic parameters were due to increased mitochondrial oxygen consumption rate and fatty acid oxidation. LY2405319 treatment in ob/ob mice on an MCD diet significantly reduced the symptoms of steatohepatitis, as confirmed by Masson’s trichrome staining intensity. Serum levels of AST and ALT were also reduced, suggesting an attenuation of liver injury, while detection of inflammatory markers showed decreased mRNA expression of TGF-β1 and type-I collagen, and decreased phosphorylation of NF-kB p65, JNK1/2, and p38. Collectively, these data show that LY2405319 treatment attenuated MCD diet-induced NASH progression. We propose that the LY2405319 compound is a potential therapeutic candidate for the treatment of advanced liver disease.
Keywords: LY2405319, FGF21, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, mitochondria, inflammation
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease in many parts of the world [1,2]. NAFLD encompasses a spectrum of liver pathologies with different clinical manifestations, from the simple accumulation of triglycerides within hepatocytes to steatohepatitis, cirrhosis, and primary liver cancer [3-5]. Several hypotheses have been proposed regarding the pathogenesis of NAFLD, from a “double hit” model to a “multi-hit” model, which involves lipotoxicity, oxidative stress, endoplasmic reticulum stress, chronic inflammatory stress and mitochondrial dysfunction [6,7]. In both hypotheses, insulin resistance is regarded as the key pathogenic factor of NAFLD, suggesting that NAFLD is a manifestation of metabolic syndrome [8,9]. Consequently, metformin and thiazolidinedione have been introduced as treatments for NAFLD, with modest benefits. Nevertheless, effective therapeutic drugs focusing on the liver pathophysiology have not been developed.
Fibroblast growth factor 21 (FGF21) is a circulating protein with pleiotropic metabolic actions [10]. Physiologically, FGF21 expression is induced in the liver by prolonged fasting and in white adipose tissue by feeding [11,12]. In mouse models of diabetes and obesity, pharmacological administration of FGF21 normalized plasma glucose levels, improved insulin sensitivity, reduced plasma triglyceride and cholesterol levels, reversed hepatic steatosis, and increased energy expenditure [13-15]. Furthermore, similar beneficial effects were observed in non-human primates and obese humans with type 2 diabetes [16,17]. These findings support its development as a novel therapy for the treatment of NAFLD and other metabolic disorders [18]. However, the mechanism of FGF21 action in NAFLD has not been fully addressed. White adipose tissue expresses high levels of fibroblast growth factor receptor 1 and β-klotho and is considered the predominant site of FGF21 action, suggesting that the beneficial effects of FGF21 in vivo may result, in part, from its direct effects on adipose tissue [19]. However, Fisher et al. showed that FGF21 activated signaling and transcriptional events within the liver and peripheral tissues [20]. Therefore, a direct effect of FGF21 on the liver cannot be ruled out [21].
LY2405319 is an engineered FGF21 protein with a disulfide bridge (L118C-A134C) in the C-terminus and a S167A substitution to improve the conformational stability and productivity of the recombinant protein, respectively [22]. LY2405319 is indistinguishable from native human FGF21 in cell-based, rodent, and non-human primate studies with respect to potency, selectivity, biological activity and efficacy. In a clinical trial, LY2405319 significantly reduced body weights, fasting insulin levels, fasting triglycerides, and total and low density lipoprotein cholesterol levels and robustly increased serum adiponectin and high density lipoprotein cholesterol levels [17]. However, the therapeutic effects of either native FGF21 or LY2405319 on NAFLD have only been reported as reducing hepatic steatosis. The reversal of steatohepatitis, fibrosis, and hepatic steatosis needs to be confirmed for LY2495319 to be an attractive therapeutic modality for NAFLD.
In this study, we reported that the pharmacological effects of LY2405319 on non-alcoholic steatohepatitis (NASH) were to improve hepatic steatosis, reduce inflammation, and reverse fibrosis in the liver. Furthermore, we found that LY2405319 directly restored mitochondrial function in primary hepatocytes cultured from the experimental model.
Materials and methods
Animals and treatments
All experimental procedures were completed in accordance with the Institutional Animal Care and Use Committee of the Chungnam National University School of Medicine (Daejeon, South Korea). Six-week-old male ob/+control mice and ob/ob C57BL/6J Lep(-/-) mice were purchased from Harlan (Indianapolis, IN, USA). These mice were maintained under standard conditions (22°C and 12-h light/dark cycle) and fed a normal chow diet. The mice were divided into three groups (n = 10 each group), including the ob/+control, vehicle-treated (ob/ob-Vehicle) and LY2405319-treated groups (ob/ob-LY), to test the therapeutic effects of LY2405319 (Eli Lilly Indianapolis, IN, USA) on the livers of ob/ob mice. LY2405319 was provided by Eli Lilly and Co. and generated as previously described [22]. ob/ob mice were intraperitoneally administered vehicle or LY2405319 (5 mg/kg) for 3 weeks. Body weights were recorded every 5 days during the experimental period. For the fibrosis studies, 6-week-old male ob/ob mice were fed either a methionine- and choline-deficient (MCD) diet (#A02082002B; Research Diets, Inc., New Brunswick, NJ, USA) or a control diet for 4 weeks. ob/ob mice were divided into a control group that was fed a normal chow diet (ob/ob-vehicle) and an MCD group that was fed the MCD diet plus vehicle (ob/ob-MCD-vehicle) or LY2405319 (ob/ob-MCD-LY; 5 mg/kg) for 4 weeks. Body weights were recorded at the start and end of the experimental period. Liver aliquots were snap frozen in liquid nitrogen and stored at -80°C until analysis. A portion of each liver was fixed in 10% neutral formalin for histology.
Intraperitoneal glucose tolerance and insulin tolerance tests
For the intraperitoneal glucose tolerance test (IPGTT), mice were fasted for 16 h and then 2 g/kg or 1 g/kg glucose was injected into the intraperitoneal cavity of each mouse. Blood glucose levels were measured at 0, 15, 30, 60, 90 and 120 min using a glucometer (ACCU-CHEK, Roche Diagnostics Corporation, IN). The intraperitoneal insulin tolerance test (IPITT) was performed by measuring the blood glucose levels after a 6 h fast, followed by an intraperitoneal insulin injection (0.75 U/kg; Humalog).
Indirect calorimetry
Mice were fed standard chow and placed into individual metabolic chambers, with free access to food and water. Oxygen consumption (VO2), carbon dioxide production (VCO2) and energy expenditure (EE) were measured using a Physical cage system (Oxylet, Panlab, Cornella, Spain). The data were analyzed using METABOLISM software version 2.2. The average values obtained during the light and dark periods were calculated. Student’s t-test was used to calculate the p values.
Serum analysis
Blood was collected from the heart under general anesthesia. The blood was centrifuged at 10,000 rpm for 5 min, and the supernatant was collected. Serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), triglyceride (TG) and total cholesterol were measured using a Hitachi 7180 autoanalyzer and Wako reagents (Wako Pure Chemical Industries, Osaka, Japan). Serum and hepatic tumor necrosis factor-α (TNF-α) levels were measured using ELISA kits (R&D Systems, Minneapolis, MN).
Assessment of liver histology and immunohistochemistry
The livers were fixed in 10% (v/v) neutral formalin and embedded in paraffin to evaluate steatosis and inflammation. Four micrometer-thick sections were cut and stained with hematoxylin-eosin (H&E) and Masson’s trichrome. For the detection of neutral lipids, 10 um-thick cryosections were stained with 0.5% (v/v) Oil Red O for 60 min at room temperature and then counterstained with hematoxylin. Immunohistochemistry of paraffin-embedded liver sections was performed using an anti-8-OHdG antibody (#ab62623, Abcam) to characterize oxidative damage. For the immunohistochemical analysis, the liver sections were incubated with a primary anti-F4/80 antibody (#ab6640, Abcam), followed by incubation with the corresponding secondary antibody. Positive staining was detected using a 3,3’-Diamin-obenzidine (DAB) Kit (Dako, Ely, Cambridgeshire, UK). Images were acquired at 200× or 400× magnification, and the percentage of the positively stained area was analyzed.
Analysis of thiobarbituric acid-reactive substances (TBARS)
Lipid peroxidation in liver tissues was analyzed by measuring the levels of thiobarbituric acid-reactive substances (TBARS) using a commercial TBARS assay kit (Cayman Chemical, Ann Arbor, MI, USA), according to the manufacturer’s specifications. Briefly, after treatment, the liver tissue was suspended in RIPA buffer, sonicated and then centrifuged at 2,000×g for 10 min at 4°C. Supernatants were collected, added to sodium dodecyl sulfate, mixed with the color reagent, and boiled for 30 min. The TBARS levels were quantified spectrophotometrically using a plate reader, and the results were normalized to the protein concentrations.
Measurement of the oxygen consumption rate (OCR)
The oxygen consumption rate was measured in 24-well plates using a Seahorse XF24 analyzer (Seahorse Bioscience Inc., North Billerica, MA, USA) as previously described [23]. The cells were seeded at a density of 20,000-50,000 cells per well 18 h prior to the analysis. The day before the OCR measurement, the sensor cartridge was calibrated with calibration buffer at 37°C in a non-CO2 incubator. Immediately before the measurement, the cells were washed and incubated with media lacking sodium bicarbonate at 37°C. Three readings were taken after the addition of each mitochondrial inhibitor, but before the injection of the subsequent inhibitor. Oligomycin (2 μg/ml), carbonylcyanide m-chlorophenyl hydrazine (CCCP, 5 μM) and rotenone (2 μM) were used as the mitochondrial inhibitors. The OCR was automatically calculated and recorded by the sensor cartridge and Seahorse XF-24 software.
Isolation of primary hepatocytes
Mouse primary hepatocytes were prepared from 8-week-old male C57BL/6J mice using the collagenase (0.8 mg/ml) perfusion method [24]. Isolated cells were further purified by a Percoll density gradient and plated at an appropriate density in Medium 199 (Sigma-Aldrich, St. Louis, MO, USA) in culture plates. After the cells were allowed for attach for 3-6 h, the medium was changed to remove unattached cells. All cells were tested for mycoplasma contamination using the MycoAlert PLUS mycoplasma detection kit (Lonza, Basel, Switzerland).
Western blot analysis
Western blot analysis was performed using standard methods with commercially available antibodies. Anti-phospho-NF-κB p65 (#3039), anti-phospho-JNK1/2 (#9255), anti-phospho-p38 MAPK T180/Y182 (#9211), anti-p38 MAPK (#9212) and anti-GAPDH (#2118) antibodies were obtained from Cell Signaling Technology. Secondary antibodies (goat anti-mouse and goat anti-rabbit) were obtained from Cell Signaling Technology (Beverley, MA, USA). The images were scanned using ODYSSEY and quantified using the Image Studio Digits software (LI-COR Biosciences, NE).
RNA isolation and analysis by quantitative Real-Time PCR (qRT-PCR)
RNA isolation and qRT-PCR were performed as previously described [24]. Briefly, total RNA was prepared using TRIzol reagent, and cDNAs were synthesized from the total RNA using M-MLV reverse transcriptase (invitrogen). The resulting cDNAs were amplified using the 7500 Fast Real-Time PCR System and software (Applied Biosystems, Foster City, CA). Real-time PCR was performed in triplicate with individual time-matched vehicle-treated or control mice using 2× SYBR Green Mix (Applied Biosystems). All quantitative calculations were performed using the dCT method and normalized to GAPDH. The primer sequences were: Tnf-α: 5’-AGCACAGAAAGCATGATCCG-3’ and 5’-GCCACAAGCAGGAATGAGAA-3’; Collagen 1a: 5’-ATGTTCAGCTTTGTGGACCTC-3’ and 5’-TCCCTCGACTCCTACATCTTC-3’; TGF-β1: 5’-CAACATGTGGAACTCTACCA-3’ and 5’- GTATTCCGTCTC-CTTGGTTC-3’; and Gapdh: 5’-GACATGCCGCCTGGAGAAAC-3’ and 5’-AGCCCAGGATGCCCTTTAGT-3’.
Statistical analysis
Statistical analyses were performed using Stat Graph Prism (5.0) (GraphPad; San Diego, CA, USA). The data are reported as the mean ± standard error of the mean (SEM). A two-tailed Student’s t-test was used to determine differences between the groups. One-way analysis of variance was used to evaluate differences between groups with appropriate post hoc tests. Statistical significance was set at a P value of less than 0.05.
Results
An engineered FGF21 variant, LY2405319, reversed hepatic steatosis in ob/ob mice
We intraperitoneally administered LY2405319 (5 mg/kg) or vehicle to 6-week-old male ob/ob mice for 3 weeks to verify the improvement in NAFLD by LY2405319. Compared with the livers of ob/+control mice, the livers of ob/ob mice showed an accumulation of micro- and macrovesicular intracytoplasmic fat droplets in liver parenchymal cells. H&E staining of the liver sections showed that hepatic steatosis was substantially attenuated in ob/ob mice following the systemic administration of LY2405319 (Figure 1A). In addition, lipid droplets confirmed by Oil Red O staining were reduced in the livers of LY2405319-treated ob/ob mice compared with vehicle-treated ob/ob mice. This change was accompanied by a marked reduction in the serum levels of the liver injury markers AST and ALT (Figure 1B). The hepatic triglyceride and cholesterol levels were also significantly decreased in the LY2405139-treated group (Figure 1C). These data show that the administration of LY2405139 alleviated hepatic steatosis in ob/ob mice.
Figure 1.
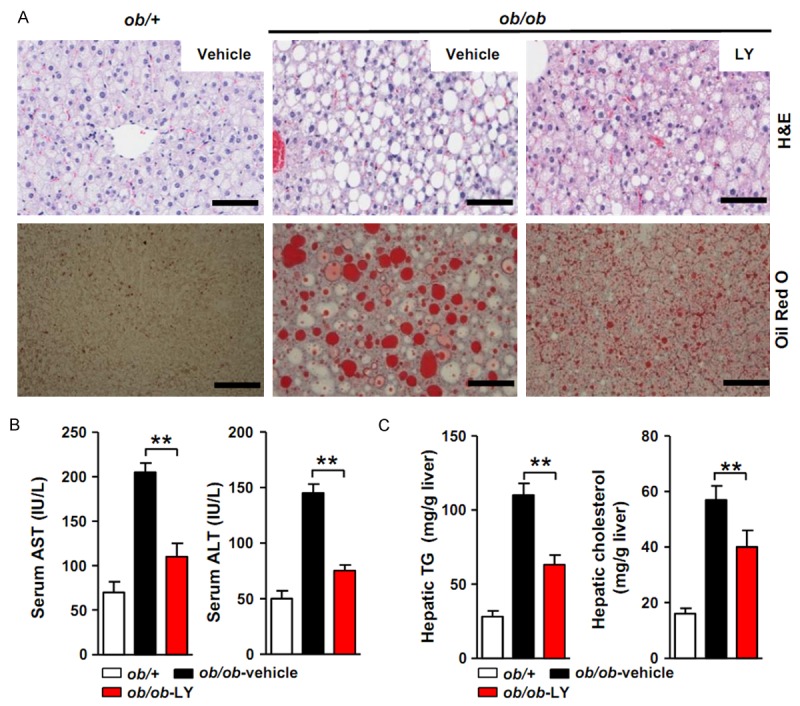
Effect of the LY2405319 treatment on hepatic steatosis in ob/ob mice. (A) Hepatic steatosis was evaluated with H&E and Oil Red O staining. Vehicle or LY2405319 (5 mg/kg) was administered to ob/ob mice by IP injection for 3 weeks. H&E and Oil Red O staining of the livers showed a marked improvement of steatosis. Vehicle vs. LY2405319. Magnification 400×. (B) The serum AST and ALT levels were reduced in the LY2405319 group compared with the vehicle group. **P < 0.01. Vehicle vs. LY2405319 (n = 10 per group). (C) Hepatic TG and cholesterol levels were significantly reduced in the LY2405319 group compared with the vehicle group (n = 10 per group). All data are presented as the mean ± sem, **P < 0.01.
LY2405319 prevented oxidative stress and lipid peroxidation in the livers of the ob/ob mice
Oxidative stress plays a central role in the pathophysiology of NAFLD as well as type 2 diabetes [25]. We performed immunohistochemical staining of 8-hydroxy-2’-deoxyguanosine (8-OHdG), a predominant form of free radical-induced oxidative lesion, to evaluate the effect of LY2405139 on oxidative stress in the livers of ob/ob mice [26]. Consistent with previous studies, the number of 8-OHdG-positive hepatocytes in vehicle-treated ob/ob mice was greater that of than ob/+mice. However, the LY2405319 treatment reduced the number of 8-OHdG-positive hepatocytes (ob/+control mice, 1.1 ± 0.9%; ob/ob-vehicle mice, 32 ± 8.3%; ob/ob-LY mice, 9.4 ± 4.3%) (Figure 2A and 2B).
Figure 2.
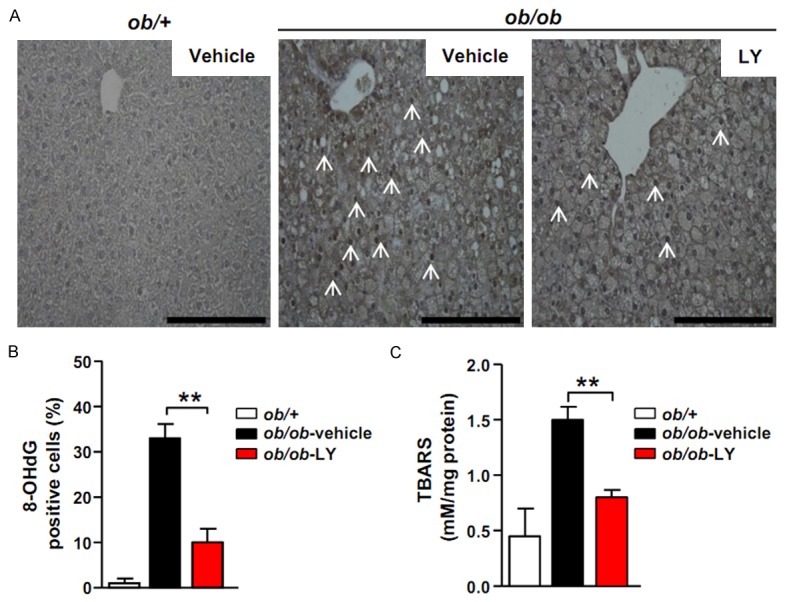
LY2405319 prevents oxidative stress and lipid peroxidation. (A) Immunohistochemical analysis of 8-OHdG in the livers of ob/+control, vehicle- and LY2405319-treated ob/ob mice. The arrows indicate 8-OHdG-positive cells. Magnification, 400×. (B) 8-OHdG expression in the livers of ob/+control, vehicle- and LY2405319-treated ob/ob mice (n = 10 per group). **P < 0.01. (C) Lipid peroxidation was determined by assaying the TBARS content in liver homogenates from ob/+control, vehicle- and LY2405319-treated ob/ob mice. All data are presented as the mean ± sem, **P < 0.01.
An increase in the reactive oxygen species levels in cells engorged with fat enhances lipid peroxidation. Therefore, we performed the TBARS assay to measure lipid peroxidation in the livers of these mice [27]. The TBARS levels were elevated in the livers of vehicle-treated ob/ob mice compared with ob/+mice. However, the livers of LY240319-treated ob/ob mice showed significantly reduced TBARS levels (ob/+control mice, 0.4 ± 0.3 nmol/mg; ob/ob-vehicle mice, 1.4 ± 0.8 nmol/mg; ob/ob-LY mice, 0.8 ± 0.2 nmol/mg) (Figure 2C). These data indicate that the LY240319 treatment prevented oxidative damage and hepatic lipid peroxidation.
LY2405319 reduced inflammatory signaling in the livers of ob/ob mice
Inflammation is also a critical factor in the progression from hepatic steatosis to steatohepatitis. The inflammatory process is provoked by the activation of various kinases, such as NF-kB, p38 mitogen-activated protein kinases (MAPK), and c-Jun N-terminal kinases (JNKs). NF-kB, p38 MAPK and JNK phosphorylation were observed by western blot analysis to determine the effect of LY2405319 on inflammatory signaling. The levels of phosphorylated NF-κB p65 were upregulated in the livers of vehicle-treated ob/ob mice compared with ob/+mice. In addition, the levels of phosphorylated p38 and JNK 1/2 were also elevated. However, the LY2405319 treatment resulted in a significant decrease in the phosphorylation of NF-κB p65, p38 and JNK 1/2 (Figure 3A). These data suggest that LY2405319 inhibited NAFLD-related inflammatory signaling.
Figure 3.
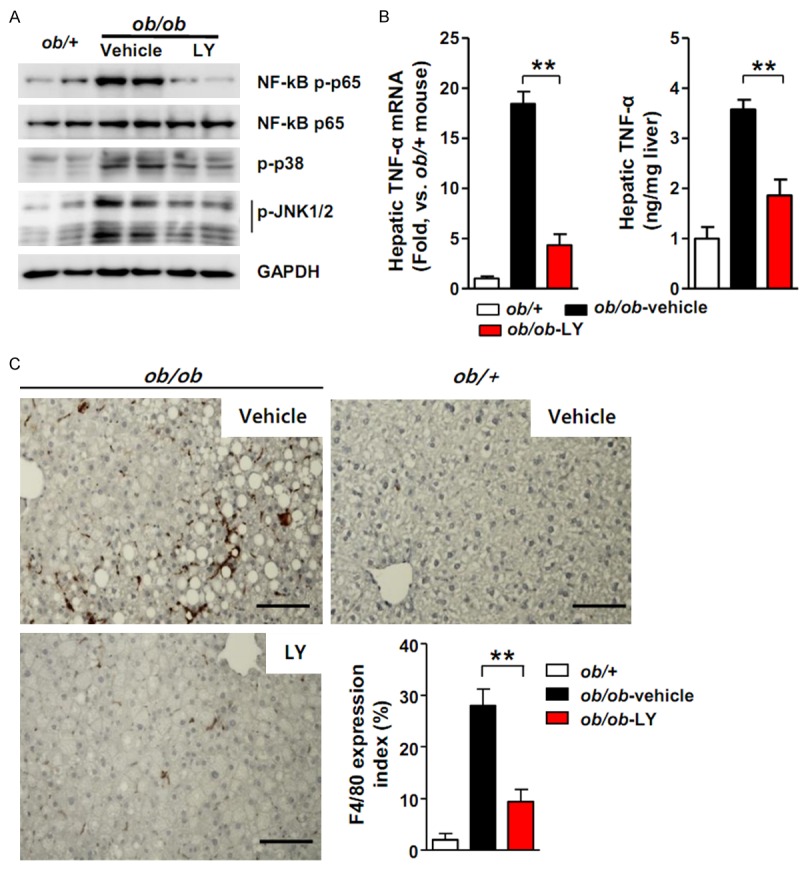
LY2405319 decreases inflammatory signaling and macrophage infiltration in ob/ob mouse livers. (A) Effect of LY2405319 on inflammatory signaling in the livers of ob/ob mice treated with or without LY2405319. Western blots were performed on liver extracts to detect NF-kB p65, p38 MAPK and JNK 1/2 phosphorylation. The blots are representative of three independent experiments. (B) Hepatic TNF-α mRNA and serum TNF-α levels in ob/+control and ob/ob mice treated with or without LY2405319. (n = 7-9 per group). **P < 0.01. Vehicle vs. LY2405319. (C) Immunohistochemistry with an anti-F4/80 macrophage-specific antibody in the livers of vehicle- or LY2405319-treated ob/ob mice. **P < 0.01. Vehicle vs. LY2405319, (n = 7-9 per group). Magnification, 400X. All data are presented as the mean ± sem, **P < 0.01.
TNF-α is a key inflammatory mediator of the pathogenesis of NAFLD [28]. The livers of vehicle-treated ob/ob mice exhibited a significant increase in TNF-α mRNA expression compared with ob/+mice; however, the LY2405319 treatment inhibited TNF-α mRNA expression in the liver. Hepatic TNF-α secretion was decreased in LY2405319-treated animals, as confirmed by ELISA (Figure 3B). The proinflammatory cytokine TNF-α amplifies local immune responses by recruiting macrophages. Consequently, increased F4/80 reactivity, an antibody that detects macrophages, was observed in the livers of vehicle-treated ob/ob mice; however, the LY2405319 treatment reduced F4/80 reactivity in the livers of ob/ob mice (Figure 3C). These results indicate that LY2405319 attenuated inflammatory signaling and macrophage infiltration in the livers of ob/ob mice.
LY2405319 prevented weight gain, improved insulin sensitivity and increased energy expenditure in ob/ob mice
Fibroblast growth factor 21 is known to affect systemic metabolism and NAFLD. Accordingly, we assessed the systemic effects of LY2405319 on body weight, glucose homeostasis and energy expenditure. Figure 4A shows the representative body weights of ob/ob mice that were treated with vehicle or LY2405319. Consistent with previous studies using recombinant FGF21 [13], the LY2405319 treatment reduced the body weights of ob/ob mice by 21 ± 3.7% after the 3-week treatment compared to the vehicle-treated controls (Figure 4A). An intraperitoneal glucose tolerance test and insulin tolerance test were performed after the 3-week LY2405319 or vehicle treatment. The fasting glucose concentration was not different between the vehicle and LY2405319 treatment groups. However, the glucose excursion after glucose challenge was improved by the LY2405319 treatment (Figure 4B). The insulin-mediated glucose disposal was also significantly increased by the LY2405319 treatment (Figure 4C).
Figure 4.
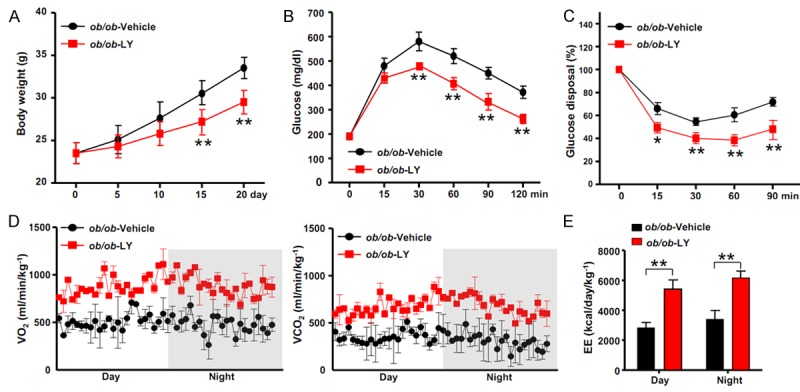
LY2405319 improves the metabolic parameters. (A) Body weights of ob/ob mice treated with or without LY2405319 (n = 10 per group). (B) IPGTT. At 9 weeks of age, mice were fasted overnight and glucose was injected intraperitoneally at a dose of 1 g/kg. (n = 6-8 per group). (C) The IPITT was performed on 9-week-old mice. Mice were fasted for 6 h and then the blood glucose levels were measured and glucose disposal was calculated at the indicated time points before and after intraperitoneal injections of insulin (0.75 U/kg) (n = 6-8 per group). All data are presented as the mean ± sem, *P < 0.05, **P < 0.01. (D, E) Oxygen consumption (VO2, left), carbon dioxide generation (VCO2, right) and energy expenditure (EE, E) of 9-week-old ob/ob mice treated with or without LY2405319. Mice were analyzed by indirect calorimetry over a period of one 12 h light/12 h dark cycle (n = 5 per group). The data are presented as the mean ± sem, **P < 0.01.
We used indirect calorimetry to calculate energy expenditure and substrate oxidation by measuring O2 consumption (VO2) and CO2 production (VOC2) after LY2405319 or vehicle treatment to clarify the effects of LY2405319 on energy metabolism. We detected a significant increase in O2 consumption and CO2 production in ob/ob mice that were treated with LY2405319 (Figure 4D). Energy expenditure was increased in LY2405319-treated ob/ob mice during both the day and night periods (ob/ob-vehicle mice during day, 3200 ± 180 kcal/day/kg-1; ob/ob-LY mice during day, 5400 ± 470 kcal/day/kg-1; ob/ob-vehicle mice during night, 3700 ± 260 kcal/day/kg-1; ob/ob-LY mice during night, 6100 ± 480 kcal/day/kg-1) (Figure 4E). Therefore, the LY2405319 treatment prevented weight gain, improved insulin sensitivity, and increased energy expenditure.
LY2405319 improved mitochondrial function by increasing the OCR and beta-oxidation
Decreased oxidative capacity and mitochondrial aberrations are known to be major contributors to the development of insulin resistance and accumulation of fatty acids in nonadipose tissues [29]. Accumulating and emerging evidence also indicates that hepatic mitochondria play a critical role in the development and pathogenesis of NAFLD [30]. Therefore, we analyzed the effects of LY2405319 on mitochondrial beta-oxidation. Primary hepatocytes isolated from the livers of WT mice were treated with LY2405319 or vehicle and the OCR was measured. The results indicate that LY2405319 increased the OCR in a dose-dependent manner (Figure 5A). We treated primary hepatocytes with palmitate-BSA and LY2405319 or vehicle to measure fatty acid oxidation. The addition of palmitate-BSA resulted in a dose-dependent increase in the OCR in the LY2405319-treated primary hepatocytes (Figure 5B). Therefore, LY2405319 increased the oxidative capacity and fatty acid oxidation, indicating that mitochondrial function was improved. Consistent with these in vitro studies, the expression of peroxisome proliferator-activated receptor-γ coactiveator-1 α (Pgc-1α), a transcriptional coactivator that is a central inducer of mitochondrial biogenesis, was upregulated in LY2403519-treated ob/ob mice. In addition, the expression of genes involved in electron transport chain complexes, such as Ndufa9, Cox1, and Atpase, was increased in the qRT-PCR analysis. The expression of mitochondrial ribosomal proteins related to mitochondrial translation (Mtg1, Mrpl3, Mrpl18, Mrpl47, Mrpl55, Mrpl35) was also upregulated. Furthermore, the expression of both carnitine palmitoyltransferase 1 (CPT1), the primary rate-controlling enzyme in fatty acid oxidation, and other beta-oxidation controlling genes, such as Pparα, Acadl, and Acadm, were increased by the LY2403519 treatment in vivo (Figure 5C-E). Taken together, the data indicate that LY2405319 increased both mitochondrial biogenesis and beta-oxidation, thus restoring mitochondrial function.
Figure 5.
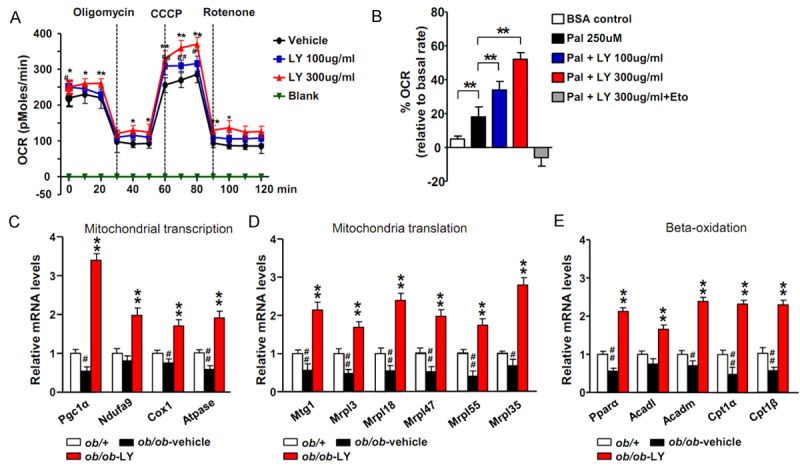
LY2405319 improves mitochondrial function by increasing the OCR and beta-oxidation. (A) OCR measured in primary cultured hepatocytes treated with the indicated concentrations of LY2405319. *P < 0.05, **P < 0.01. Vehicle vs. LY2405319 (B) Fatty acid oxidation in primary hepatocytes using palmitate-BSA as substrate after pretreatment with different concentrations of LY2405319 in the absence or presence of the CPT1 inhibitor, etomoxir (at 40 μM). (C-E) Expression of genes related to mitochondrial transcription (Pgc1α, Ndufa9, Cox1 and Atpase), mitochondrial translation (Mtg1, Mrpl3, Mrpl18, Mrpl47, Mrpl55 and Mrpl35) and beta-oxidation (Pparα, Acadl, Acadm, Cat1α and Cpt1β) in the livers of ob/ob-vehicle and ob/ob-LY2403519 mice (n = 6 per group). The data are presented as the mean ± sem. #P < 0.05, ##P < 0.01. ob/+vs. ob/ob-vehicle; *P < 0.05, **P < 0.01. ob/ob-vehicle vs. ob/ob-LY2405319.
LY2405319 inhibited liver fibrosis in MCD-fed ob/ob mice
Ob/ob mice possess a spontaneous mutation in the leptin gene and are protected against fibrosis and thus fail to progress from steatosis to the more severe steatohepatitis. Secondary insults, such as an MCD diet, are needed to trigger steatohepatitis in ob/ob mice and circumvent this limitation [31]. We fed 6-week-old ob/ob mice an MCD diet for 4 weeks to clarify whether LY2405319 reversed steatohepatitis. As shown in Figure 6A, Masson’s trichrome staining showed the development of pericellular fibrosis in MCD diet-fed mice. In contrast, when the fibrotic mice were treated with LY2405319, the intensity of Masson’s trichrome staining was significantly reduced compared with vehicle-treated mice. The expression of both the type-1a collagen and TGF-β mRNAs was also significantly decreased by the LY2405319 treatment (Figure 6B).
Figure 6.
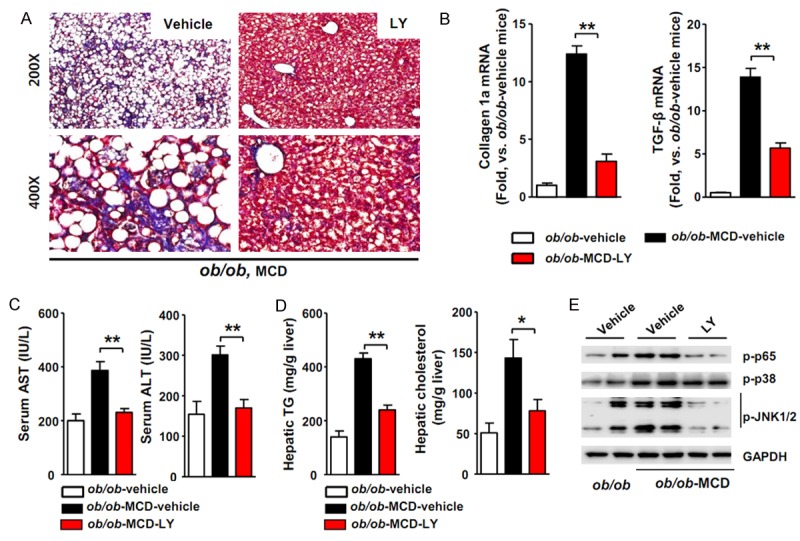
LY2405319 inhibits liver fibrosis in MCD diet-fed ob/ob mice. (A) Representative photomicrographs showing the effect of the MCD diet on collagen deposition in MCD-vehicle-fed and MCD-LY2405319-fed ob/ob mice. Paraffin-embedded sections from MCD-vehicle-fed ob/ob and MCD-LY2405319-fed ob/ob mice were stained with Masson’s trichrome stain. The arrows indicate pericellular fibrosis in MCD-vehicle-fed ob/ob mice (n = 10 per group). (B) Analysis of collagen type-1α and TGF-β1 mRNA expression by qRT-PCR. **P < 0.01 (C) Serum AST and ALT levels in MCD-vehicle-fed or MCD-LY2405319-fed ob/ob mice. **P < 0.01 Vehicle vs. LY2405319 (n = 6-8 per group). (D)Hepatic TG and cholesterol levels in MCD-vehicle-fed or MCD-LY2405319-fed ob/ob mice (n = 6-8 per group). *P < 0.05, **P < 0.01. (E) Effect of LY2405319 on inflammatory signaling in the livers of the MCD diet-fed ob/ob mice treated with or without LY2405319. Western blot analyses of liver extracts were performed to detect NF-kB p65, p38 and JNK 1/2 phosphorylation. GAPDH was used as a loading control. The blots are representative of three independent experiments.
The serum AST and ALT levels were significantly increased in MCD-fed ob/ob mice, but the LY2405319 treatment decreased the serum levels of these liver injury markers (Figure 6C). In addition, MCD-fed ob/ob mice that were treated with LY2405319 exhibited an approximately 50% decrease in the hepatic TG and cholesterol levels (Figure 6D). In the western blot assay, MCD-fed ob/ob mice showed a significant increase in NF-kB p65, JNK 1/2 and p38 phosphorylation compared with ob/ob mice that were fed a normal chow diet. However, the LY2405319 treatment inhibited the phosphorylation of these inflammatory markers in the livers of MCD-fed ob/ob mice (Figure 6E). Collectively, these data show that the MCD diet amplified inflammatory signaling and liver fibrosis in vehicle-treated ob/ob mice, whereas the LY2405319 treatment reversed the progression of NASH and its related pathogenesis. Hence, we propose that LY2405319 is a potentially effective therapeutic candidate for the treatment of NASH.
Discussion
According to the hypothesis for the development of NAFLD, impaired oxidative phosphorylation and increased ROS generation play a central role in the pathophysiology of NASH. Mitochondrial dysfunction contributes to the pathogenesis of NAFLD because it affects hepatic lipid homeostasis and promotes ROS production, lipid peroxidation, cytokine release, and cell death [32]. Indeed, mitochondrial respiratory chain activity is decreased in patients with NASH and in ob/ob mice with NAFLD [33,34]. Rector et al. also showed that mitochondrial dysfunction precedes both insulin resistance and hepatic steatosis and consequently contributes to the natural history of NAFLD in an obese rodent model [35]. Metformin and thiazolidinedione, available drugs with proven modest benefits for NAFLD, also increase mitochondrial fatty acid oxidation by activating 5’-adenosine monophosphate (AMP)-activated protein kinase [36-40]. In our study, LY2405319 upregulated the expression of mitochondrial transcription-, translation- and beta-oxidation related genes; increased OCR and fatty acid oxidation; and subsequently decreased oxidative stress and lipid peroxidation. Therefore, LY2405319 could be an effective therapeutic drug for NASH by modulating its diverse pathogenesis in the “multi-hit” model, namely, lipotoxicity, oxidative stress, chronic inflammatory stress and mitochondrial dysfunction.The metabolic effects of exogenously administered FGF21 have been thought to be mediated by the white adipose tissue through increased browning, energy expenditure and glucose disposal. In our study, chronic administration of an FGF21 variant also increased energy expenditure, prevented weight gain and improved insulin sensitivity, as expected (Figure 4). Therefore, we must also consider that the systemic effect of LY2405319, an FGF21 variant, resulted in the reversal of NAFLD. However, there are several lines of evidence that highlight the liver as a key site of action for the metabolic effects of FGF21. Zhang et al. showed that the overexpression of FGF21 repressed the transcription of sterol regulatory element binding protein 1c, the gene required for de novo lipogenesis in human liver-derived HepG2 cells [41]. In addition, decreased expression of Pparα was also reported in hepatocytes transfected with an FGF21 siRNA [42]. In addition, FGF21 activates AMP-activated protein kinase and sirtuin 1 and deacetylates its downstream target, Pgc-1α, in adipocytes [43]. In our study, the expression of both Pparα and Pgc-1α was upregulated in LY2405319-treated ob/ob mice. Moreover, we showed a dose-dependent increase in the OCR and fatty acid oxidation in LY2405319-treated primary hepatocytes obtained from ob/ob mice (Figure 5). Therefore, we suggest that LY2405319 suppress hepatic lipogenesis, increases lipolysis, and subsequently increases fatty acid metabolism in the mitochondria. In addition, LY2405319 also improved mitochondrial function; therefore, mitochondria could handle the increased fatty acids without producing more ROS in the liver. Consequently, we showed the direct effect of LY2405319 on the liver, in addition to its systemic effects, particularly on the adipose tissue. Furthermore, we should consider that the NAFLD-associated hepatic insulin resistance could be a cause or precursor of the metabolic syndrome [44]. FGF21 administration was shown to improve hepatic insulin resistance [45]. Inflammation is a required pathological feature that defines NASH. Compared to simple steatosis, NASH is a significant risk factor for hepatic cirrhosis and hepatocellular carcinoma. Therefore, the therapeutic drugs for NAFLD must also reverse the inflammation in the liver. As shown in Figure 3, LY2405319 decreased the hepatic levels of TNF-α, a proinflammatory cytokine secreted by injured hepatocytes. It also decreased the phosphorylation of NF-kB, p38 MAPK and JNK, the main inflammatory signaling pathways that are activated by proinflammatory cytokines, such as TNF-α. In addition, the histological manifestations of inflammation were also decreased in the livers of LY2405319-treated ob/ob mice. Moreover, LY2405319 reversed MCD diet-induced hepatic fibrosis in both histology with Masson’s trichrome staining and the expression of fibrosis marker genes (Figure 6). Collectively, LY2405319 reversed all of the main pathological consequences of NASH, namely, hepatic steatosis, inflammation, and fibrosis. Accordingly, LY2405319 showed good potential as an effective therapeutic drug for the treatment of NASH.
Several experimental studies of native FGF21 or FGF21 analogues have implicated FGF21 in the treatment of NAFLD. Intraperitoneal administration of 10 mg/kg recombinant murine FGF21 for 6 weeks reversed hepatic steatosis in diet-induced obese mice, as determined by histological examinations of liver sections and biochemical analyses of the hepatic TG and cholesterol content [13]. ob/ob mice that received 1 mg/kg recombinant human FGF21 for 2 weeks via continuous subcutaneous administration exhibited a decreased liver weight, without histological examination or biochemical analyses of the liver [14]. A polyethylene glycol-conjugated FGF21 developed with the aim of extending the half-life of FGF21 also reduced the hepatic TG levels in db/db mice that were subcutaneously injected for 12 days [46]. However, these studies did not show the effect of FGF21 on hepatic inflammation and fibrosis; therefore, they could not confirm the therapeutic effect on NASH. We showed that the FGF21 variant LY2405319 reversed hepatic steatosis, inflammation and fibrosis in ob/ob mice. Although ob/ob mice are not an ideal animal model in which to study inflammation and fibrosis in NASH because of their own characteristics, including leptin deficiency, they exhibited hepatic lipid accumulation, oxidative stress and mitochondrial dysfunction [24]. Furthermore, the MCD diet increased oxidative stress to progress them to a stage of the disease that resembles classical NASH features [31].
In conclusion, our study showed that the therapeutic effects of LY2405319 on NASH were to reduce inflammation and reverse fibrosis in the liver, as well as to improve hepatic steatosis. Additional studies may be required in other well-established liver models, such as diet-induced obese rodents, to solidify the beneficial effects of LY2405319 on NASH. Furthermore, we found that LY2405319 restored mitochondrial function, which could be a mechanism by which LY2405319 exerted its therapeutic effects on the liver, as well as its systemic effects.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2015R1A2A1A13000951, NRF-2014R1A1A1006176 and NRF-2014R1A1A3053786), Korea. H.K.C and K.S.K were supported by NRF grants from the Ministry of Education (NRF-2013R1A1A2058889 and NRF-2015R1D1-A1-A01060529), Korea.
Disclosure of conflict of interest
None.
References
- 1.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 2.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 3.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 4.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 6.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 7.Yao HR, Liu J, Plumeri D, Cao YB, He T, Lin L, Li Y, Jiang YY, Li J, Shang J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am J Transl Res. 2011;3:284–291. [PMC free article] [PubMed] [Google Scholar]
- 8.Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91:4753–4761. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 9.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59:713–723. doi: 10.1002/hep.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Zhang J, Jia W. Fibroblast growth factor 21: a novel metabolic regulator from pharmacology to physiology. Front Med. 2013;7:25–30. doi: 10.1007/s11684-013-0244-8. [DOI] [PubMed] [Google Scholar]
- 11.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. Endocrine Regulation of the Fasting Response by PPARα-Mediated Induction of Fibroblast Growth Factor 21. Cell Metabolism. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Dutchak Paul A, Katafuchi T, Bookout Angie L, Choi Jang H, Yu Ruth T, Mangelsdorf David J, Kliewer Steven A. Fibroblast Growth Factor-21 Regulates PPARγ Activity and the Antidiabetic Actions of Thiazolidinediones. Cell. 2012;148:556–567. doi: 10.1016/j.cell.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, Chen JL, Jung DY, Zhang Z, Ko HJ, Kim JK, Veniant MM. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, Moller DE, Kharitonenkov A. Fibroblast Growth Factor 21 Corrects Obesity in Mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 15.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kharitonenkov A, Wroblewski VJ, Koester A, Chen YF, Clutinger CK, Tigno XT, Hansen BC, Shanafelt AB, Etgen GJ. The Metabolic State of Diabetic Monkeys Is Regulated by Fibroblast Growth Factor-21. Endocrinology. 2007;148:774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 17.Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–340. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Xu Y, Hu Y, Wang G. The role of fibroblast growth factor 21 in the pathogenesis of non-alcoholic fatty liver disease and implications for therapy. Metabolism. 2015;64:380–390. doi: 10.1016/j.metabol.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Adams AC, Yang C, Coskun T, Cheng CC, Gimeno RE, Luo Y, Kharitonenkov A. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Mol Metab. 2012;2:31–37. doi: 10.1016/j.molmet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher FM, Estall JL, Adams AC, Antonellis PJ, Bina HA, Flier JS, Kharitonenkov A, Spiegelman BM, Maratos-Flier E. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology. 2011;152:2996–3004. doi: 10.1210/en.2011-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Li Y. Fibroblast growth factor 21, the endocrine FGF pathway and novel treatments for metabolic syndrome. Drug Discov Today. 2014;19:579–589. doi: 10.1016/j.drudis.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Kharitonenkov A, Beals JM, Micanovic R, Strifler BA, Rathnachalam R, Wroblewski VJ, Li S, Koester A, Ford AM, Coskun T, Dunbar JD, Cheng CC, Frye CC, Bumol TF, Moller DE. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS One. 2013;8:e58575. doi: 10.1371/journal.pone.0058575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryu MJ, Kim SJ, Kim YK, Choi MJ, Tadi S, Lee MH, Lee SE, Chung HK, Jung SB, Kim HJ, Jo YS, Kim KS, Lee SH, Kim JM, Kweon GR, Park KC, Lee JU, Kong YY, Lee CH, Chung J, Shong M. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet. 2013;9:e1003356. doi: 10.1371/journal.pgen.1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung HK, Kim YK, Park JH, Ryu MJ, Chang JY, Hwang JH, Lee CH, Kim SH, Kim HJ, Kweon GR, Kim KS, Shong M. The indole derivative NecroX-7 improves nonalcoholic steatohepatitis in ob/ob mice through suppression of mitochondrial ROS/RNS and inflammation. Liver Int. 2015;35:1341–1353. doi: 10.1111/liv.12741. [DOI] [PubMed] [Google Scholar]
- 25.Chang YC, Chuang LM. The role of oxidative stress in the pathogenesis of type 2 diabetes: from molecular mechanism to clinical implication. Am J Transl Res. 2010;2:316–331. [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 27.Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- 28.Seo YY, Cho YK, Bae JC, Seo MH, Park SE, Rhee EJ, Park CY, Oh KW, Park SW, Lee WY. Tumor Necrosis Factor-alpha as a Predictor for the Development of Nonalcoholic Fatty Liver Disease: A 4-Year Follow-Up Study. Endocrinol Metab (Seoul) 2013;28:41–45. doi: 10.3803/EnM.2013.28.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrauwen P, Hesselink MK. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 30.Nassir F, Ibdah JA. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int J Mol Sci. 2014;15:8713–8742. doi: 10.3390/ijms15058713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Ruiz I, Rodriguez-Juan C, Diaz-Sanjuan T, del Hoyo P, Colina F, Munoz-Yague T, Solis-Herruzo JA. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology. 2006;44:581–591. doi: 10.1002/hep.21313. [DOI] [PubMed] [Google Scholar]
- 35.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, Mikus CR, Laye MJ, Laughlin MH, Booth FW, Ibdah JA. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52:727–736. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998–1003. doi: 10.1038/79697. [DOI] [PubMed] [Google Scholar]
- 37.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet. 2001;358:893–894. doi: 10.1016/s0140-6736(01)06042-1. [DOI] [PubMed] [Google Scholar]
- 38.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38:1008–1017. doi: 10.1053/jhep.2003.50420. [DOI] [PubMed] [Google Scholar]
- 40.Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem Biophys Res Commun. 2004;314:580–585. doi: 10.1016/j.bbrc.2003.12.120. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Lei T, Huang JF, Wang SB, Zhou LL, Yang ZQ, Chen XD. The link between fibroblast growth factor 21 and sterol regulatory element binding protein 1c during lipogenesis in hepatocytes. Mol Cell Endocrinol. 2011;342:41–47. doi: 10.1016/j.mce.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Lee J, Hong SW, Park SE, Rhee EJ, Park CY, Oh KW, Park SW, Lee WY. Exendin-4 regulates lipid metabolism and fibroblast growth factor 21 in hepatic steatosis. Metabolism. 2014;63:1041–1048. doi: 10.1016/j.metabol.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci U S A. 2010;107:12553–12558. doi: 10.1073/pnas.1006962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lonardo A, Ballestri S, Marchesini G, Angulo P, Loria P. Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig Liver Dis. 2015;47:181–190. doi: 10.1016/j.dld.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Camporez JP, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, Zhang D, Kahn M, Samuel VT, Jurczak MJ, Shulman GI. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology. 2013;154:3099–3109. doi: 10.1210/en.2013-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mu J, Pinkstaff J, Li Z, Skidmore L, Li N, Myler H, Dallas-Yang Q, Putnam AM, Yao J, Bussell S, Wu M, Norman TC, Rodriguez CG, Kimmel B, Metzger JM, Manibusan A, Lee D, Zaller DM, Zhang BB, DiMarchi RD, Berger JP, Axelrod DW. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes. 2012;61:505–512. doi: 10.2337/db11-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]