

Abstract
The present study explored changes of the SIRT6/NF-κB pathway in myocardial hypoxia/reoxygenation induced injury and the effects on mitochondrial damage and myocardial damage by regulating SIRT6. SIRT6 expression decreased and NF-κB expression increased in H9c2 cells during hypoxic injury. Cell death and mitochondrial defects paralleled mPTP opening, and a decrease in ΔΨm occurred in hypoxic myocytes compared with normoxic control cells in annexin V and propidium iodide staining and TUNEL results. These effects were suppressed in cells overexpressing SIRT6, but reemerged in cells expressing the SIRT6 mutant. We also found that NF-κB p65 increased in both the cytoplasm and nuclei, which could be repressed by SIRT6 overexpression. The expression level of NF-κB was significantly and negatively correlated with the SIRT6 mRNA level. Our data demonstrated that SIRT6/NF-κB changed during hypoxic injury and SIRT6 overexpression averted mitochondrial defects through inhibition of NF-κB in hypoxic H9c2 cells. Activation of SIRT6 may be a potential method for hypoxia/reoxygenation injury therapy.
Keywords: SIRT6, hypoxia/reoxygenation, NF-κB, mitochondrial
Introduction
Myocardial hypoxia/reoxygenation (H/R) induced injury is significantly important in mediating the pathogenesis of acute myocardial infarction (AMI), and interest has therefore arisen concerning the mechanisms capable of limiting myocardial damage [1]. Methods to attenuate myocardial damage after H/R are being thoroughly investigated. The energy metabolism of cardiomyocytes is disrupted in AMI and is increasingly recognized as an effective therapeutic strategy.
Sirtuins are a family of NAD+-dependent protein deacetylases that mediate cellular functions. Among the sirtuins, Sirtuin6 (SIRT6) is involved in transcriptional proteins, stress tolerance, DNA repair, inflammation and life span [2-5]. Over expression of SIRT6 plays a protective role in hypertrophic cardiomyocytes and smooth muscle cells [6], as well as SIRT1 [7]. Functional studies showed that SIRT6 protects the heart from oxidative damage and metabolic imbalance through inhibiting NF-κB-dependent transcriptional activation [8-11].
In the pathological process of AMI, modulation of the mitochondrial proteins is vitally important. The heart is an oxygen-sensitive site of metabolic regulation in coronary heart disease. Mitochondria are the power houses of cells that function in cardiac energy metabolism. The occurrence of oxidative damage after AMI can mediate mPTP opening, ΔΨm loss [12-17], and a decrease of ATP synthesis [18,19], triggering cell apoptosis [20].
Studies suggest that mitochondrial enzymes are regulated through deacetylation by sirtuins and energy metabolism [3,21]. However, controversy surrounds the mechanism of sirtuins on mitochondria. Using purified hepatic mitochondria from mice, Hirschey et al. demonstrated that SIRT3 may modulate mitochondrial intermediary metabolism via long-chain acyl coenzyme A dehydrogenase (LCAD) during fasting [22]. Meanwhile, SIRT1 deacetylates PGC1α and is involved in mitochondrial biogenesis and activity [23-25]. To the best of our knowledge, it remains unclear whether mitochondria may mediate the interaction between SIRT6 and NF-κB during hypoxic stress. We presently intend to study the involvement of SIRT6 in regulating cardiomyocyte apoptosis, mitochondrial defects and further explore the potential mechanisms.
Materials and methods
Chemicals and reagents
SiRNA-SIRT6 and its reagents were purchased from Invitrogen (Carlsbad, CA, USA). The cDNA fragment encoding SIRT6 was isolated with the Takara RNA PCR kit (Takara, Japan). Rabbit anti-NF-κB p65 antibody was used (Bioworld Technology, USA). Other reagents were obtained (St. Louis, Missouri).
Cell culture and hypoxia-reoxygenation (H/R) model
An H9c2 cardiac myoblast cell line was obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in high glucose Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 25 mM Hepes in 0.5% CO2 at 37°C. Then, the cells were placed in an incubator at 37°C. N2 (95%) and CO2 (5%) were used to produce a hypoxic environment (less than 1% O2). After 2 hours of culture, H9c2 cells were reoxygenated under normoxia (21% O2) for 2 h.
Adenovirus-mediated overexpression of SIRT6 and siRNA treatment of H9c2 cells
H9C2 cardiac myoblast cells were infected with adenoviral vectors containing cDNAs for SIRT6 using a multiplicity of infection of 1000 viral particles per cell (20 infectious units per well). The infection efficiency (95-100%) was monitored 48 hours later by Western blot. Cells infected with adenovirus containing empty vectors served as controls.
Specific siRNA and negative control GFP siRNA targeting SIRT6 from Invitrogen were used. All of the treatments of H9c2 cells with SIRT6 siRNA or GFP siRNA oligonucleotides were at 20 μM for 72 h before the cells were harvested and the hypoxic assay and Western blot analyses were performed.
PCR
RNA was isolated from H9c2 cells using a Total RNA Kit, and then converted into cDNA. One-fourth of the cDNAs were subjected to qRT-PCR amplification. The relative mRNA levels of SIRT6 and NF-κB were measured with β-actin as the internal control.
Western blotting
Cell extraction was performed on ice, and protein concentration was determined by BCA assays. Samples (60 μg) were loaded onto 10% SDS-PAGE, transferred to membranes, and blocked with 5% nonfat milk. The blots were incubated with the primary antibody (anti-β-actin, anti-SIRT6 antibodies) for 2 h at 37°C, and then incubated with the secondary antibody. A chemiluminescence system was used to detect the immunoreactive bands.
Apoptosis assays
To investigate the mechanism of apoptosis, we performed an annexin V-FITC/PI double staining assay. Cells were washed twice with PBS and then re-suspended in a binding buffer, followed by staining with annexin V-FITC and propidium iodide for 15 min (dark, 37°C). FACS flow cytometry was utilized to analyze the stained cells. The apoptosis index (positive cells/total cells ×100%) was used as the apoptosis correlation indicator.
Cardiac myocyte apoptosis was also monitored by the TUNEL method. After fixing and permeabilizing the cells, they were incubated in a TUNEL reaction mixture for 1 h at 37°C. The fluorescence density was analyzed using immunofluorescent confocal microscopy (Leica Tcs sp2) (Ex = 485 nm; Em = 530 nm) as previously described. The cells were also stained by annexin V-FITC/PI. Annexin V positive cells were excited and detected at 495/519 nm by Leica confocal microscopy (Ex = 495 nm; Em = 519 nm), and the fluorescence was quantitated using Image J software.
Mitochondrial PTP and ΔΨm
To determine whether SIRT6 could damage the mitochondria during H/R, we assessed the mitochondrial PTP opening using the calcein-AM/cobalt method as reported previously [1,25]. H9c2 cells were loaded with 1 μmol/L calcein-acetoxymethyl ester (calcein-AM, Biotium) as molecular probes in the presence of 1 mmol/L CoCl2. After washing twice with warm DMEM without serum, the cells were observed by confocal microscopy (Ex = 490 nm; Em = 515 nm).
Cellular ΔΨm was monitored using tetramethylrhodamine ethyl ester (TMRE), which accumulates in the mitochondria in a potential-dependent manner. Cells were loaded with 50 nmol/L and visualized with confocal microscopy (Ex = 540 nm; Em = 600 nm). Sequential digital images were acquired and integrated the average fluorescence density.
NF-κB translocation assays
Immunocytochemistry was utilized to identify whether NF-κB translocates from the cytoplasm to the nucleus. After being washed and fixed, the cells were immersed in 3% H2O2 solution to inactivate the endogenous peroxidase. After being saturated on ice by 10% BSA for 30 min, the samples were thereafter incubated overnight with 1 μg/mL of anti-NF-κB p65, then incubated for one hour with an anti-rabbit IgG secondary antibody at room temperature. 3,3’-Diaminobenzidine (DAB) stained slices were acquired and observed by the Olympus-Tokyo inverted microscope. Cytoplasmic and nuclear cells with golden-brown staining were denoted as positive cells and counted separately. The images were processed with Image J software.
Statistical analysis
The data are presented as the mean ± SEM. The Tukey test in ANOVA and repeated measures ANOVA were utilized to detect statistical differences. P<0.05 is considered significantly different. Linear regression was used to analyze the correlation between SIRT6 and NF-κB expression levels.
Results
SIRT6 decreased in H9c2 cells after hypoxia damage
After H9c2 cells were under hypoxic conditions for 2 h, they were re-oxygenated for up to 2 h (0, 30, 60 and 120 min). Our data revealed that the SIRT6 protein and mRNA expression decreased after H/R, peaking at 2/1 h (P<0.05). Based on this result, all of the experiments were performed at the time of 2 h/1 h of hypoxia/reoxygenation in the following study (Figure 1A, 1B).
Figure 1.
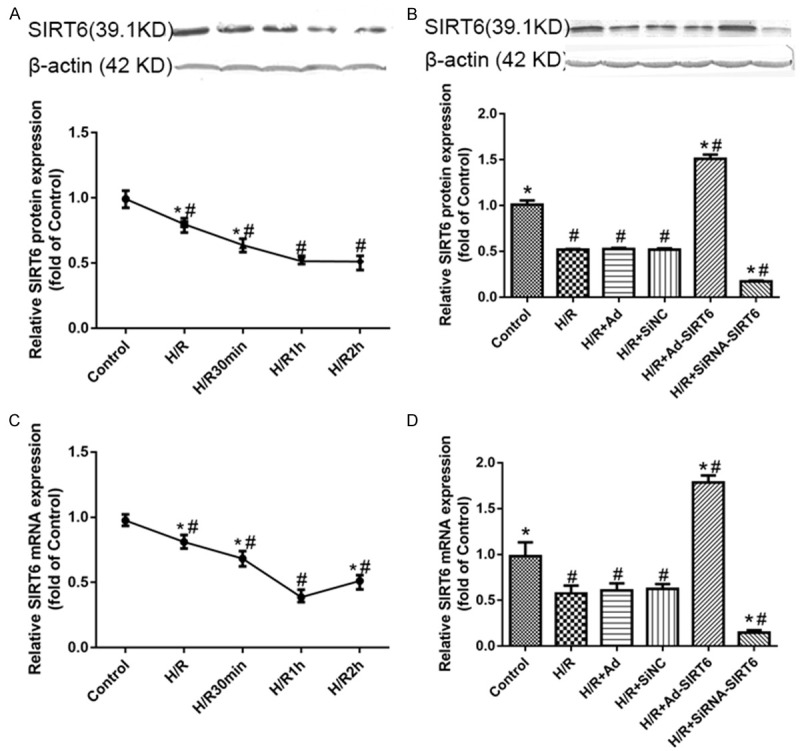
SIRT6 protein and mRNA levels decrease in H9c2 cell lines, especially at 1 h of reoxygenation (A, B). SIRT6 protein and mRNA expression for the (1) control, (2) H/R-treated, (3) H/R+Ad-GFP-treated, (4) H/R+SiNC-treated, (5) H/R+Ad-SIRT6-treated and (6) H/R+SiRNA-SIRT6-treated groups (C, D). Data are presented as the means ± S.E.M. #P<0.05 vs. control group. *P<0.05 vs. HR group.
The cells were divided into 6 groups: the control group (Control), H/R group (HR), H/R+ adenovirus blank vector group (H/R+Ad), H/R+siRNA negative control GFP group (H/R+SiNC), HR+adenovirus-mediated overexpression of SIRT6 group (H/R+Ad-SIRT6), and H/R+siRNA treated SIRT6 group (H/R+SiRNA-SIRT6).
The mRNA and protein levels of SIRT6 among the H/R, H/R+Ad and H/R+SiNC treated groups was not different (P>0.05). Overexpression of SIRT6 showed significantly increased rates of its protein and mRNA levels, especially during conditions of hypoxic stress (P<0.05). In contrast, the expression of SIRT6 was down-regulated in SIRT6-slienced cells (P<0.05).
SIRT6 inhibits H9C2 cell apoptosis
To observe whether SIRT6 influenced cell apoptosis, we performed FACS to detect apoptotic cells. We observed a significant increase in apoptosis in H9c2 cells, and this apoptosis was further exacerbated in cells lacking SIRT6 after H/R (P<0.05). Compared with the control group, apoptosis increased in H/R cells infected with siRNA-SIRT6 (P<0.05). In contrast, SIRT6 overexpression treatment resulted in down-regulation of TUNEL positive cells (P<0.05) (Figure 2A). Additionally, no differences were measured in H/R-treated, H/R+Ad-GFP-treated and H/R+SiNC-treated groups (P>0.05).
Figure 2.

SIRT6 inhibits apoptosis in H9c2 cells. A. Representative FACS analysis using the annexin V and FITC/PI staining for the (1) control, (2) H/R-treated, (3) H/R+Ad-GFP-treated, (4) H/R+SiNC-treated, (5) H/R+Ad-SIRT6-treated and (6) H/R+SiRNA-SIRT6-treated groups where the Q4 quadrant indicates early apoptotic cells and the percentage is shown inset. B. The representative scatterplot of fragmented DNA was detected by the TUNEL method using a confocal microscope. Immunofluorescence staining was performed by annexin V labeling on phosphatidylserine externalization. C. Mean fluorescence intensity was quantified using ImageJ 1.48 software (NIH, Bethesda, MD, USA). Values are given as the mean ± SEM (n = 5). #P<0.05 with respect to control; *P<0.05 with respect to the H/R-treated group.
To further observe apoptosis under the above conditions, we assessed annexin-V binding to externalized phosphatidylserine. Hypoxia led to a significant up-regulation of cell apoptosis after exposure to H/R treatment (P<0.05). Additionally, after pre-treatment of Ad-SIRT6, H/R induced cell apoptosis was significantly antagonized (P<0.05) (Figure 2B and 2C). These results indicate that SIRT6 negatively contributes to apoptosis in H9c2 cells.
Enhanced mitochondrial defects in SIRT6-deficient cells and attenuated mitochondrial defects in SIRT6-overexpression cells
Because perturbations to mitochondria intermediary metabolism were observed as a characteristic of hypoxia-induced mitochondrial death, we further observed whether SIRT6 influenced mitochondrial PTP and ΔΨm. As showed in Figure 3, the control group displayed dotted green fluorescence distribution, suggesting that PTP was in a closed state. In contrast, we observed a significant decrease (P<0.01) of the green fluorescence in the H/R group as an index of PTP opening. More importantly, hypoxia-induced PTP opening could be inhibited in H9c2 cells using adenovirus-mediated SIRT6 overexpression but not in the siRNA-SIRT6 group, consistent with the loss of mitochondrial ΔΨm (P<0.01).
Figure 3.

The confocal images of mPTP opening and ΔΨm in cultured H9c2 cells (A, B). Variation in relative fluorescence intensity of mitochondrial inner membrane potential (%, mean ± SEM) (C, D). Bar = 100 µm.
SIRT6 inhibits NF-κB signaling
NF-κB plays important roles in many human diseases. Normal H9c2 cells express NF-κB in the cytoplasm but not in the nuclei. To explore whether NF-κB is activated during hypoxia, an immunocytochemical analysis of NF-κB translocation was performed. After treatment with H/R, the numbers of cytoplasmic- and nuclear-positive cells were counted separately. As showed in Figure 4A-C, DAB staining increased in both the cytoplasm and nuclei of H/R groups compared with the control groups (P<0.05). SiRNA-SIRT6 groups displayed increased DAB staining in both the cytoplasm and nuclei compared to the H/R groups (P<0.05).
Figure 4.

Effects of SIRT6 on H/R-induced NF-κB p65 activity in H9C2 cells. DAB staining of NF-κB p65 translocation assay (A). The cytoplasm and nucleus mIOD of the H/R group both increased significantly compared with the control group. DAB staining increased in the SiRNA-SIRT6 groups while SIRT6 overexpression led to an opposite effect (B and C). Effects of SIRT6 on mRNA of NF-κB p65 by RT-PCR (D). Linear regression indicates the correlation between SIRT6 and NF-κB levels (E). #P<0.05 vs. control group. *P<0.05 vs. HR group.
We then detected NF-κB expression after H/R compared to normal H9c2 cells. As illustrated in Figure 4D, NF-κB mRNA expression remained increased after H/R and significantly increased in the siRNA-mediated SIRT6 knockout group. We explored whether NF-κB levels were correlated with SIRT6 mRNA levels in H9c2 cells, and the linear regression analysis indicated that the NF-κB level was significantly and negatively correlated with the SIRT6 mRNA level (Figure 4E).
Discussion
Despite improvements focusing on reperfusion in AMI treatment, the disease prognosis remains poor. Apoptosis is a key pathologic feature in acute myocardial infarction and heart failure. Mitochondrial impairment is a critical event leading to hypoxia-induced cell death. It is widely assumed that the mitochondrial respiratory chain is the predominant intracellular site of superoxide production, which is generated from electron leaks in the mitochondrial electron transport system [26].
Mitochondria play vital roles in apoptosis by releasing mitochondrial proteins into the cytoplasm. We presently confirmed that hypoxia may lead to cell apoptosis and mitochondrial defects (dysfunctions of mPTP and ΔΨm). The results strongly suggested that the opening of mPTP causes loss of ΔΨm and ultimately, adenosine triphosphate depletion and apoptosis.
SIRT6 has various physiological activities, including glucose and free fatty acid utilization, mitochondrial biogenesis, and inflammatory responses [27]. PCR and western blots at different time points indicate that SIRT6 decreased significantly at 2 h/1 h of H/R. Through overexpression and knockout in H9c2 cells, we further showed that SIRT6 overexpression inhibited apoptosis and the mitochondrial defects of H9c2 cells induced by H/R. The results indicate the subtle possibilities of therapies targeting SIRT6.
Possible explanations concerning SIRT6 protecting against mitochondrial perturbations during hypoxia are not well understood. Previous studies showed that SIRT6 protects the heart from oxidative damage and metabolic imbalance by inhibiting NF-κB mediated transcription activation [8-11]. NF-κB is widely accepted as an important transcription factor and its role has been deeply investigated in hypertrophic cardiomyopathy and heart failure. We also demonstrated that the up-regulation of NF-κB was observed after H/R, and NF-κB levels significantly and negatively correlate with SIRT6 mRNA levels. Based on the previous studies, we provide 3 possibilities that may explain the relationship between SIRT6 and NF-κB:
(1) After histone H3 lysine 9 and K56 of NF-κB target gene promoters were deacetylated by SIRT6, the NF-κB RELA promoter occupancy decreased accordingly, which subsequently promotes the translation and expression of p50/p65 and activation of NF-κB signaling [11].
Moreover, other sirtuin family members also regulate NF-κB signaling, which subsequently induce changes of stress- or age-associated genes [28,29]. We presently demonstrated that SIRT6 correlated with NF-κB expression after cardiac hypoxia/reoxygenation. We therefore speculate that the prevention of hypoxia-induced NF-κB activation is an important mechanism by which SIRT6 protects against apoptosis.
(2) NF-κB is an important anti-apoptotic factor and protects cardiomyocytes from hypertrophy. Previous studies demonstrated that NF-κB was associated with myocytes death. ROS and oxidative stress may lead to the activation of AP-1 and NF-κB, which subsequently increases downstream factor expression (cytokines, adhesion molecules and proinflammatory enzymes) [30]. Furthermore, mitochondrial-derived ROS may activate the NF-κB pathway, which then releases proinflammatory factors in aging [31]. Regula et al. presented that NF-κB activation may suppress apoptosis in ventricular myocytes during hypoxia by preventing mitochondrial defects that underlie PTP opening [32]. The results above indicated that NF-κB might affect metabolic disorders in different diseases. The different roles of NF-κB in different diseases are worth further research.
(3) The sirtuins play a key role in cardioprotection by inducing mitochondrial expansion and oxidative metabolism [33-35], mitigating the effects of ROS and inflammation [20,36,37]. Recently, several sirtuins have been shown to possess more efficient activity for the removal other acyl lysine modifications. The crystal structure of SIRT6 reveals a large hydrophobic pocket that can accommodate long chain fatty acyl groups. Long chain fatty acid, a previously ignored protein, can be hydrolyzed by SIRT6 and enhance SIRT6 acetylation activity by promoting the secretion of TNFα through lysine demyristoylation [21,38].
In this study, we showed that transgenic cells that overexpress exogenous mouse SIRT6 are protected against mitochondrial defects caused by hypoxia while mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation [33]. Further studies are required to evaluate the contribution of SIRT6 to long chain fatty acids, in addition to its effect on cardiac energy metabolism.
Conclusions
In conclusion, these findings confirmed a novel metabolic regulatory mechanism where SIRT6 attenuated hypoxia-induced apoptosis and mitochondrial defects via down-regulation and translocation of NF-κB p65. We therefore speculate that the prevention of hypoxia-induced NF-κB activation is an important mechanism by which SIRT6 protects against apoptosis. These findings will lead to further investigations of the SIRT6 pathways in preclinical and clinical settings in the future.
Acknowledgements
This work was supported by grants from the Natural Science Research Project of Jiangsu Provincial Department of Education (No. 15KJD320003), Xuzhou Science and Technology Innovation Project (No. KC15SH036) and National Natural Science Foundation of China (31100801 and 81200858).
Disclosure of conflict of interest
None.
References
- 1.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 2.Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 4.Guarente L. Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364:2235–2244. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- 5.Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443–461. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu SS, Cai Y, Ye JT, Pi RB, Chen SR, Liu PQ, Shen XY, Ji Y. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. Br J Pharmacol. 2013;168:117–128. doi: 10.1111/j.1476-5381.2012.01903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90:276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 8.Tian K, Liu Z, Wang J, Xu S, You T, Liu P. Sirtuin-6 inhibits cardiac fibroblasts differentiation into myofibroblasts via inactivation of nuclear factor kappaB signaling. Transl Res. 2015;165:374–386. doi: 10.1016/j.trsl.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Maksin-Matveev A, Kanfi Y, Hochhauser E, Isak A, Cohen HY, Shainberg A. Sirtuin 6 protects the heart from hypoxic damage. Exp Cell Res. 2015;330:81–90. doi: 10.1016/j.yexcr.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Sun T, Wu J, Kalionis B, Zhang C, Yuan D, Huang J, Cai W, Fang H, Xia S. Icariin intervenes in cardiac inflammaging through up-regulation of SIRT6 enzyme activity and inhibition of the NF-kappa B pathway. Biomed Res Int. 2015;2015:895976. doi: 10.1155/2015/895976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santos AC, Uyemura SA, Lopes JLC, Bazon JN, Mingatto FE, Curti C. Effect of Naturally Occurring Flavonoids on Lipid Peroxidation and Membrane Permeability Transition in Mitochondria. Free Radic Biol Med. 1998;24:1455–1461. doi: 10.1016/s0891-5849(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 13.van Dijk C, Driessen AJ, Recourt K. The uncoupling efficiency and affinity of flavonoids for vesicles. Biochem Pharmacol. 2000;60:1593–1600. doi: 10.1016/s0006-2952(00)00488-3. [DOI] [PubMed] [Google Scholar]
- 14.Dorta DJ, Pigoso AA, Mingatto FE, Rodrigues T, Prado IM, Helena AF, Uyemura SA, Santos AC, Curti C. The interaction of flavonoids with mitochondria: effects on energetic processes. Chem Biol Interact. 2005;152:67–78. doi: 10.1016/j.cbi.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Trumbeckaite S, Bernatoniene J, Majiene D, Jakstas V, Savickas A, Toleikis A. The effect of flavonoids on rat heart mitochondrial function. Biomed Pharmacother. 2006;60:245–248. doi: 10.1016/j.biopha.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Modriansky M, Gabrielova E. Uncouple my heart: the benefits of inefficiency. J Bioenerg Biomembr. 2009;41:133–136. doi: 10.1007/s10863-009-9212-z. [DOI] [PubMed] [Google Scholar]
- 17.Panickar KS, Anderson RA. Mechanisms underlying the protective effects of myricetin and quercetin following oxygen-glucose deprivation-induced cell swelling and the reduction in glutamate uptake in glial cells. Neuroscience. 2011;183:1–14. doi: 10.1016/j.neuroscience.2011.03.064. [DOI] [PubMed] [Google Scholar]
- 18.Carrasco-Pozo C, Gotteland M, Speisky H. Apple peel polyphenol extract protects against indomethacin-induced damage in Caco-2 cells by preventing mitochondrial complex I inhibition. J Agric Food Chem. 2011;59:11501–11508. doi: 10.1021/jf202621d. [DOI] [PubMed] [Google Scholar]
- 19.Sandoval-Acuna C, Lopez-Alarcon C, Aliaga ME, Speisky H. Inhibition of mitochondrial complex I by various non-steroidal anti-inflammatory drugs and its protection by quercetin via a coenzyme Q-like action. Chem Biol Interact. 2012;199:18–28. doi: 10.1016/j.cbi.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Sandoval-Acuna C, Ferreira J, Speisky H. Polyphenols and mitochondria: an update on their increasingly emerging ROS-scavenging independent actions. Arch Biochem Biophys. 2014;559:75–90. doi: 10.1016/j.abb.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 24.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M, Krawczyk M, Irusta PM, Martin-Montalvo A, Hubbard BP, Zhang Y, Lehrmann E, White AA, Price NL, Swindell WR, Pearson KJ, Becker KG, Bohr VA, Gorospe M, Egan JM, Talan MI, Auwerx J, Westphal CH, Ellis JL, Ungvari Z, Vlasuk GP, Elliott PJ, Sinclair DA, de Cabo R. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El Assar M, Angulo J, Rodriguez-Manas L. Oxidative stress and vascular inflammation in aging. Free Radic Biol Med. 2013;65:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Gao E, Lau WB, Wang Y, Liu G, Li JJ, Wang X, Yuan Y, Koch WJ, Ma XL. G-protein-coupled receptor kinase 2-mediated desensitization of adiponectin receptor 1 in failing heart. Circulation. 2015;131:1392–1404. doi: 10.1161/CIRCULATIONAHA.114.015248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang H, Zhang W, Pan H, Feldser HG, Lainez E, Miller C, Leung S, Zhong Z, Zhao H, Sweitzer S, Considine T, Riera T, Suri V, White B, Ellis JL, Vlasuk GP, Loh C. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS One. 2012;7:e46364. doi: 10.1371/journal.pone.0046364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu BP, Chung HY. Adaptive mechanisms to oxidative stress during aging. Mech Ageing Dev. 2006;127:436–443. doi: 10.1016/j.mad.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 31.Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 32.Regula KM, Baetz D, Kirshenbaum LA. Nuclear factor-kappaB represses hypoxia-induced mitochondrial defects and cell death of ventricular myocytes. Circulation. 2004;110:3795–3802. doi: 10.1161/01.CIR.0000150537.59754.55. [DOI] [PubMed] [Google Scholar]
- 33.Pougovkina O, te Brinke H, Ofman R, van Cruchten AG, Kulik W, Wanders RJ, Houten SM, de Boer VC. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum Mol Genet. 2014;23:3513–3522. doi: 10.1093/hmg/ddu059. [DOI] [PubMed] [Google Scholar]
- 34.Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, Uppala R, Fitch M, Riiff T, Zhu L, Zhou J, Mulhern D, Stevens RD, Ilkayeva OR, Newgard CB, Jacobson MP, Hellerstein M, Goetzman ES, Gibson BW, Verdin E. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Someya S, Yu W, Hallows WC, Xu JZ, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–428. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem. 2013;288:31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]