

Abstract
Objectives: Mandibular condylar chondrocyte apoptosis is mainly responsible for the development and progression of temporomandibular joint osteoarthritis (TMJ-OA). Interleukin-1β (IL-1β) generally serves an agent that induces chondrocyte apoptosis. Hyperbaric oxygen (HBO) treatment increases proteoglycan synthesis in vivo. We explore the protective effect of HBO on IL-1β-induced mandibular condylar chondrocyte apoptosis in rats and the potential molecular mechanisms. Methods: Chondrocytes were isolated from the TMJ of 3-4-week old Sprague-Dawley rats. The Cell Counting Kit-8 (CCK-8) assay was used to determine cell viability. The phosphorylated phosphoinositide-3 kinase (p-PI3K), phosphorylated AKT (p-Akt), type II collagen (COL2), and aggrecan (AGG) content was detected by immunofluorescence, immunocytochemistry and western blotting. The expression of Pi3k, Akt, Col2 and Agg mRNA was measured using real-time quantitative polymerase chain reaction (RT-qPCR). Results: HBO inhibited the cytotoxicity and apoptosis induced by IL-1β (10 ng/mL) in the mandibular condylar chondrocytes. HBO also decreased the IL-1β activity that decreased p-PI3K and p-AKT levels, and increased COL2 and AGG expression, with the net effect of suppressing extracellular matrix degradation. Conclusions: These data suggest that HBO may protect mandibular condylar chondrocytes against IL-1β-induced apoptosis via the PI3K/AKT signaling pathway, and that it may promote the expression of mandibular condylar chondrocyte extracellular matrix through the PI3K/AKT signaling pathway.
Keywords: Hyperbaric oxygen, IL-1β, PI3K/AKT signaling, extracellular matrix
Introduction
The temporomandibular joint (TMJ) is one of the most common sites affected by osteoarthritis (OA), a degenerative disease with age-related joint disorder characterized by the progressive loss of articular cartilage and degradation of the cartilage matrix. TMJ-OA is characterized by cartilage degradation, subchondral bone remodeling, chronic pain, and joint dysfunction [1,2].
Proinflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) play a critical role in the development of OA pathological changes by inducing the secretion of matrix metalloproteinases (MMPs) by chondrocytes, which degrade the extracellular matrix [3] and promote chondrocyte apoptosis [4]. For example, the expression of type I IL-1 receptor (IL-1R) is augmented in chondrocytes in OA, rendering these cells more sensitive to stimulation by IL-1β [5,6]. IL-1 in particular, has been proven to induce chondrocyte apoptosis [4]. Therefore, IL-1β is generally serves as an agent that induces chondrocyte apoptosis [7-9]. Moreover, previous reports have indicated that the major causes of OA are excessive chondrocyte apoptosis, inducing the loss of chondrocytes and the degeneration of cartilage tissue [10]. Mandibular condylar chondrocyte apoptosis plays an important role in the development of cartilage degeneration in TMJ-OA and it is increasingly considered one of the potential targets for TMJ-OA treatment [11-13]. Therefore, it is essential to elucidate the molecular mechanisms of chondrocyte apoptosis in OA.
As a potential signaling pathway for apoptosis inhibition, the phosphoinositide-3 kinase (PI3K)/AKT signaling pathway plays a critical role in inhibiting chondrocyte apoptosis [14]. 17β-Estradiol increases cell proliferation in rat OA model chondrocytes through the PI3K/AKT signaling pathway [13]. The activation of PI3K/AKT signaling regulates cell growth, proliferation, migration, and adhesion [15]. A recent study reported that in early chondrogenesis, the activation of PI3K/AKT signaling promoted chondrocyte proliferation and increased sulfated glycosaminoglycan (sGAG) deposition; inhibition of PI3K signaling resulted in decreased expression of the early chondrogenic marker genes for aggrecan (AGG), type II collagen α1 (COL2A1) and SRY-box 9 (SOX9) [16]. Several other studies have reported a variety of functions of PI3K/AKT signaling during chondrogenesis. The constitutively active form of AKT accelerated the chondrogenic differentiation of ATDC5 cells [17]. In contrast, the inhibition of PI3K signaling suppressed the expression of the early chondrocytic differentiation marker Col2A1 and the production of sulfated proteoglycans (PG) [18]. Likewise, in chondrocytic cell lines and primary articular chondrocytes, proliferation and the synthesis of sGAG is dependent on the PI3K/AKT signaling pathway [19].
Hyperbaric oxygen (HBO) therapy is a safe, noninvasive modality that increases the oxygen tension of tissues and the microvasculature [20]. Previous reports have suggested that HBO treatment increases PG synthesis in vivo [21,22]. In the present study, similar results suggested that HBO treatment upregulated Agg and Col2 mRNA expression in OA rat chondrocytes [23]. HBO treatment decreased the expression of IL-1β and increased the gene expression of Agg and Col2 [24,25]. However, the effect and potential molecular mechanism of HBO on IL-1β-induced chondrocytes in OA remain unclear.
In our research, we explored whether HBO could suppress apoptosis and induce the secretion of extracellular matrix in IL-1β-stimulated rat TMJ chondrocytes. We also studied the molecular mechanism of the protective effect of HBO on IL-1β-induced rat mandibular condylar chondrocytes by researching changes in the PI3K/AKT signaling pathway.
Materials and methods
Materials
Phosphate-buffered saline (PBS), 0.25% trypsin, 0.2% type II collagenase, 100 mg/mL penicillin, and 100 mg/mL streptomycin were purchased from Invitrogen (Grand Island, NY, USA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from HyClone (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Sciencell (Carlsbad, CA, USA). Dimethylsulfoxide (DMSO) was obtained from Sigma Chemical (St. Louis, MO, USA). The bicinchoninic acid (BCA) protein assay kit and Cell Counting Kit-8 (CCK-8) assay kit were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). The PI3K-specific inhibitor LY294002 was purchased from Cell Signaling Technology (Beverly, MA, USA). Recombinant rat IL-1β was purchased from PeproTech (Rocky Hill, NJ, USA).
Isolation and culture of rat mandibular condylar chondrocytes
Condylar TMJ cartilage tissues were harvested from 10 4-week-old Wistar rats. The tissues were washed thrice with PBS, minced finely, digested with 0.25% trypsin for 10 min, and subsequently digested with 0.1% collagenase II in DMEM growth medium, supplemented with 20% FBS, 100 mg/mL penicillin, and 100 mg/mL streptomycin. Following incubation at 37°C in a humidified atmosphere of 5% CO2, the chondrocytes were collected at 2-h intervals by centrifugation. Then, the cells were resuspended with the medium in 6-cm culture dishes. For the duration of the culture, the medium was changed every 3 days, and in the subsequent experiments, second passage (P2) cells were used. The morphology of the chondrocytes was observed under a microscope. To investigate the effect of HBO on chondrocytes under inflammatory conditions, we analyzed the effect of HBO on chondrocytes treated with recombinant rat IL-1β, a proinflammatory cytokine. To eliminate the influence of IL-1β from the serum in the culture medium, the medium was replaced with serum-free culture medium 20 h before HBO treatment. Finally, the culture medium was replaced with fresh serum-free medium or serum-free medium containing IL-1β (10 ng/mL) just before HBO treatment.
Immunohistochemistry
Chondrocytes were fixed in 4% paraformaldehyde for 30 min and washed with PBS three times. Two drops of 3% H2O2-methanol solution were added to the slide, which was incubated at room temperature for 10 min and then washed with PBS three times. Goat serum (50-100 μL) was added to the slide, which was then incubated at room temperature for 20 min. Collagen type II antibody (50-100 μL of a 1:200 dilution) was added to the slide, which was then incubated at 37°C for 2 h and washed with PBS three times before the addition of 50 μL intensifier. The slides were then incubated at room temperature for 30 min and washed with PBS three times. A universal immunoglobin G (IgG) antibody-Fab segment-horseradish peroxidase polymer (50 μL) was added to the slide, which was then incubated at 37°C for 30 min and washed with PBS three times. Fresh 3,3’-diaminobenzidine solution was used for color development. The slides were washed for 15 min with tap water and then once with distilled water. Hematoxylin staining was performed according to a standard protocol.
Immunofluorescence
Chondrocytes were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, and then, permeabilized and blocked in PBS containing 0.1% Triton X-100 and 5% FBS for 30 min. The fixed cells were washed with PBS and incubated overnight at 4°C with anti-collagen II antibody (1:200; lot no. ab34712; Abcam). The cells were washed and incubated with rhodamine- or fluorescein-conjugated secondary antibodies, washed again, and observed under a standard fluorescence microscope. Nuclei were identified with 4,6-diamidino-2-phenylindole staining.
Cell treatments
The cells were divided into four groups: control (no treatment); IL-1β treatment (cells were stimulated with 10 ng/mL IL-1β for 24 h); HBO treatment (cells were stimulated with 10 ng/mL IL-1β for 24 h and treated with HBO); HBO+ inhibitior (cells were pretreated with 25 μM LY294002 for 1 h and 10 ng/mL IL-1β for 24 h followed by HBO treatment).
Exposure to intermittent HBO
Cells in the control group and IL-1β group were maintained in 5% CO2/95% air (non-HBO) through the experimental protocol. All hyperoxic cells were exposed to 100% O2 for 25 min and then to air for 5 min at 1.5 atmospheres absolute (ATA) in a hyperbaric chamber (Billups-Rothenberg, Del Mar, USA) with a total treatment time of 90 min per 48 h.
Cell viability assay
The cells were plated in 96-well culture plates. After incubation with test medium for 24 h, the number of viable cells was determined using CCK-8 reagent according to the manufacturer’s instructions. Briefly, cells were seeded in 96-well plates at a concentration of 1×105/mL. After 48 h, 10 μL CCK-8 reagents was added to each well and incubated for 2 h. The absorbance at 450 nm was measured using a microplate reader. Each treatment was replicated in three wells. The data in each treatment group are expressed as a percentage of the control.
Western blotting
Cells were lysed using protein lysis buffer and protease inhibitor cocktail. The protein concentration of the cell lysates was quantified by the BCA assay kit, and equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The PVDF sheet was blocked with 5% non-fat dried milk in Tris-buffered saline containing 0.1% Tween 20 at room temperature for 1 h, and incubated with primary rabbit polyclonal antibodies against rat antigens. The following antibodies were used to detect the proteins: rabbit anti-COL2 polyclonal antibody (1:400; lot no. SAB4500366, Sigma), anti-AGG (1:1000; lot no. SAB4500662, Sigma), anti-total PI3K monoclonal antibody (1:1000; lot no. #4257, Cell Signaling Technology), and anti-phosphorylated (p-) PI3K polyclonal antibody (1:1000; lot no. SAB4504314, Sigma), anti-total AKT (1:1,000; lot no. #4691, Cell Signaling Technology), and anti-p-AKT (1:1,000; lot no. #4060, Cell Signaling Technology). Incubation with monoclonal mouse β-actin antibody (1:1000; Beyotime) was used as the loading sample control. The blots were developed using a horseradish peroxidase-conjugated secondary antibody (Beyotime) and enhanced chemiluminescence (ECL) using an ECL chemiluminescence kit (Beyotime). The blots were exposed to autoradiographic film for 1-2 min for detection.
Reverse transcription and real-time quantitative polymerase chain reaction (RT-qPCR) analysis
The total RNA from each group was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Reverse transcription and RT-qPCR were carried out using an Ultra SYBR Two Step RT-qPCR Kit (with ROX; CW Biotech, Beijing, China) according to the manufacturer’s instructions. RT-qPCR was carried out in an Eppendorf RealPlex 4 (Eppendorf AG, Hamburg, Germany) with the following settings: 10-min of pre-incubation at 95°C followed by 40 cycles of 20 s at 95°C and 60 s at 55°C. The 25-μL reaction volume contained 2×Ultra SYBR mixture (with ROX), forward and reverse primers (10 mM), and template complementary DNA (cDNA). Melting curve analysis was carried out using the default program. After each reaction, the cycle threshold (Ct) was recorded when the amplification curve reflected the exponential kinetic measurements. The 2-ΔΔCt method was adopted using glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as the reference gene.
The primers for rat Col2 (forward: 5’-AAGAGCAAGGAGAAGAAG-3’, reverse: 5’-TTACAGTGGTAGGTGATG-3’), Agg (forward: 5’-GCAGCACAGACACTTCAGGA-3’, reverse: 5’-CCCACTTTCTACAGGCAAGC-3’) and Gapdh (forward: 5’-ATGATTCTACCCACGGCAAG-3’, reverse: 5’-CTGGAAGATGGTGATGGGTT-3’) were designed with Primer Premier Version 5.0 software and their efficiency was confirmed by sequencing their conventional PCR products.
Immunofluorescence and immunohistochemistry
Immunofluorescence was used to determine the expression patterns of type II collagen in each group. Briefly, the cultured chondrocytes were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, and permeabilized and blocked in PBS containing 0.1% Triton X-100 and 5% FBS for 30 min. The fixed cells were washed with PBS and incubated overnight at 4°C with anti-collagen II antibody (1:200; lot no. ab34712; Abcam). The cells were washed and incubated with rhodamine- or fluorescein-conjugated secondary antibodies, washed again, and then observed under a standard fluorescence microscope. Nuclei were identified with 4,6-diamidino-2-phenylindole staining.
Immunohistochemistry was used to determine the expression patterns of AGG, p-PI3K, and p-AKT in each group. Immunohistochemical staining was carried out using the streptavidin-peroxidase (S-P) method. Endogenous peroxidase activity was inhibited by 3% hydrogen peroxide. The cells were reacted overnight at 4°C with the following rabbit polyclonal antibodies: anti-AGG (1:50), anti-p-PI3K (1:50) and anti-p-AKT (1:50). The secondary antibody, biotinylated anti-rabbit IgG, was applied for 30 min at room temperature. The cells were visualized using 3,3’-diaminobenzidine tetrahydrochloride (DAB). Digital images were analysed via Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
All data are expressed as the means ± standard error. Experimental data were analyzed by one-way analysis of variance (ANOVA). Relative indices were analyzed using SPSS version 13.0 software (SPSS, Chicago, IL, USA). The Student-Newman-Keuls q test was used to calculate differences between the groups. Data were graphically presented using GraphPad Prism 6 (San Diego, CA, USA). A P-value of less than 0.05 was considered statistically significant.
Results
Identification of normal mandibular condylar chondrocytes
Chondrocytes with abundant cytoplasm and round nuclei grew to complete confluence within 4-7 days. As shown in Figure 1, immunohistochemical and immunofluorescence staining for type II collagen was positive in the chondrocytes.
Figure 1.
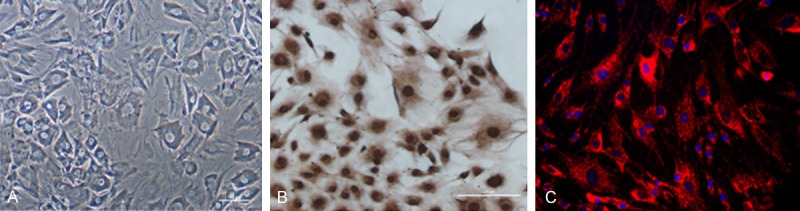
Identification of normal mandibular condylar chondrocytes. A: The morphology of the P2 chondrocytes was observed under a microscope. B: The immunohistochemical staining for type II collagen was positive in the chondrocytes. C: The immunofluorescence staining for type II collagen was positive in the chondrocytes.
HBO increased the proliferation and viability of IL-1β-induced chondrocytes through the PI3K/AKT signaling pathway
To investigate whether HBO has a positive effect on IL-1β-induced chondrocytes, the proliferation and viability of HBO-treatment chondrocytes was examined by CCK-8 assay. As shown in Figure 2, IL-1β significantly reduced cell proliferation and viability. However, the addition of HBO visibly increased the cell proliferation and viability decreased by IL-1β. To further observe the effect of PI3K/AKT signaling in HBO-mediated promotion of chondrocyte proliferation and viability induced by IL-1β, 25 μM LY294002 was added together with HBO, where LY294002 decreased the prior effect of HBO on IL-1β-decreased chondrocyte proliferation and viability.
Figure 2.
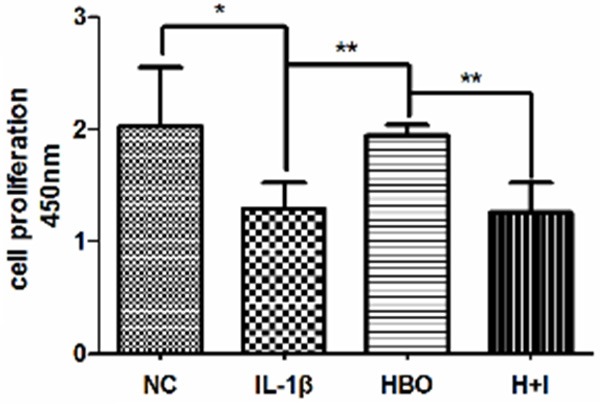
Effects of HBO on IL-1β-induced chondrocyte proliferation. CCK-8 assay was used to examine the cell proliferation of each group, the absorbance was measured at 450 nm (n=5 per group). Bars represent the mean and SEM of each group. NC, normal control; IL-1β, cells with 10 ng/mL interleukin-1β; HBO, hyperbaric oxygen treated cells with 10 ng/mL interleukin-1β; H+I, hyperbaric oxygen treated cells with 10 ng/mL interleukin-1β and 25 μM LY294002 (the PI3K inhibitor). ** < 0.01, * < 0.05.
Protein expression of COL2, AGG, PI3K, p-PI3K, AKT, and p-AKT
The immunofluorescence and immunohistochemistry results are shown in Figure 3 and Table 1. Compared with the control group, the integrated optical density (IOD) and the positive expression areas of COL2, AGG, p-PI3K, and p-AKT were significantly lower in the IL-1β group (P < 0.05). Compared with the IL-1β group, the IOD and positive expression areas of COL2, AGG, p-PI3K, and p-AKT were significantly higher in the HBO group (P < 0.05). Compared with the HBO group, the IOD and positive expression areas of COL2, AGG, p-PI3K, and p-AKT were significantly lower in the HBO+ inhibitor group (P < 0.05). To confirm the expression quantity in the chondrocytes, the above proteins were quantified by western blot analysis. As shown in Figure 4, compared with the control group, levels of COL2, AGG, p-PI3K, and p-AKT expression were significantly lower in the IL-1β group (P < 0.01). There were significantly increased levels of COL2, AGG, p-PI3K, and p-AKT expression in the HBO group compared with the IL-1β group (P < 0.05), and compared with the HBO group, levels of COL2, AGG, p-PI3K, and p-AKT expression were significantly lower in the HBO+ inhibitor group (P < 0.05).
Figure 3.
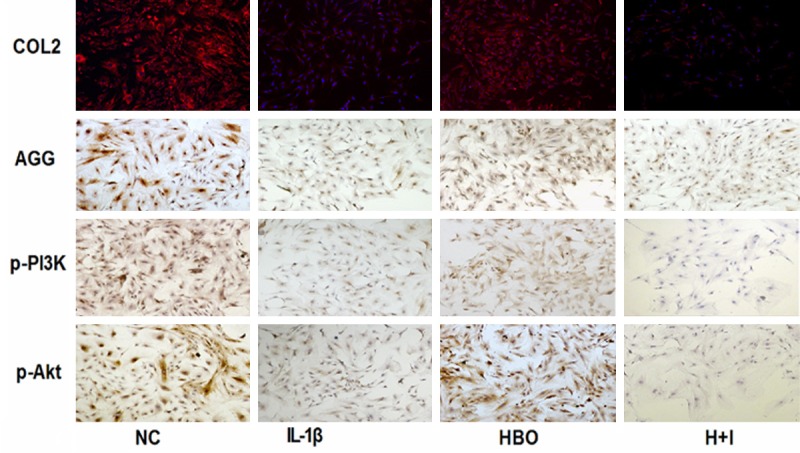
COL2, AGG, p-PI3K and p-AKT protein levels measured by immunohistochemistry and immunofluorescence. Comparison of the AGG, p-PI3K, p-AKT and COL2 protein levels in the different groups as determined by immunohistochemistry and immunofluorescence (n=6 per group).
Table 1.
The integrated optical density (IOD) and the positive expression areas of COL2, AGG, p-PI3K and p-AKT measured by immunohistochemistry and immunofluorescence
| NC | IL-1β | HBO | H+I | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| IOD | Positive areas (μm2) | IOD | Positive areas (μm2) | IOD | Positive areas (μm2) | IOD | Positive areas (μm2) | |
| COL2 | 97.8±7.6 | 70.6±10.2 | 42.1±6.1* | 23.5±4.2* | 78.5±16.7# | 83.7±12.2# | 50.5±8.4& | 30.1±7.3& |
| AGG | 104.5±9.8 | 68.2±9.4 | 50.2±7.7* | 34.2±5.9* | 80.9±11.7# | 71.2±10.8# | 66.1±12.2& | 39.7±8.7& |
| p-PI3K | 80.1±11.2 | 59.1±13.2 | 30.8±4.3* | 20.7±3.7* | 63.3±10.4# | 48.8±10.4# | 38.8±6.2& | 22.1±6.3& |
| p-AKT | 89.3±10.9 | 67.7±12.9 | 37.6±4.4* | 21.3±6.1* | 59.8±12.3# | 51.1±11.7# | 43.3±7.7& | 29.4±5.5& |
NC, normal control; IL-1β, cells with 10 ng/mL interleukin-1β; HBO, hyperbaric oxygen treated cells with 10 ng/mL interleukin-1β; H+I, hyperbaric oxygen treated cells with 10 ng/mL interleukin-1β and 25 μM LY294002 (the PI3K inhibitor).
P < 0.05, significantly different from the normal control group;
P < 0.05, significantly different from the IL-1β group.
P < 0.05, significantly different from the HBO group.
Data are represented as the M ± SEM of n=6. M, mean; SEM, Standard Error; n, sample size.
Figure 4.
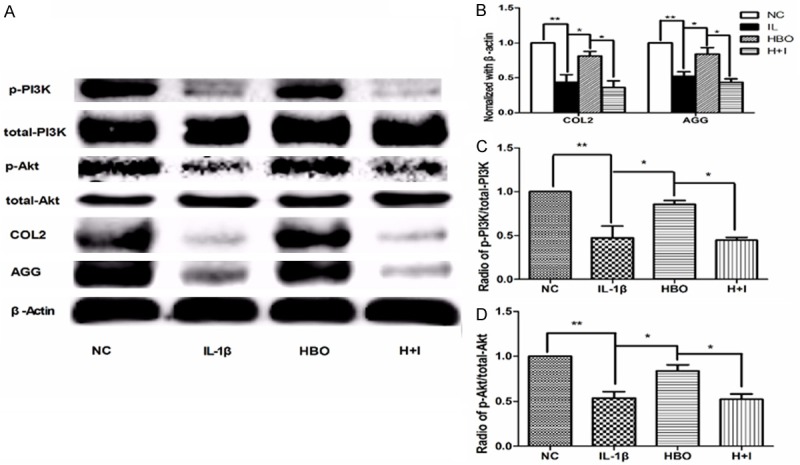
Protein expression of COL2, AGG, PI3K, p-PI3K, AKT, and p-AKT. Western blot technique was used to examine the possible mechanism by which HBO protect chondrocytes. A: Comparison of the COL2, AGG, PI3K, p-PI3K, AKT and p-AKT protein levels in the different groups as determined by Western blot. B: Mean relative protein levels of COL2 and AGG in different groups (n=6 per group). C: p-PI3K levels were normalized to t-PI3K levels in different groups (n=6 per group). D: p-AKT levels were normalized to t-AKT levels in different groups (n=6 per group). Bars represent the mean and SEM of each group. ** < 0.01, * < 0.05.
Col2 and Agg mRNA expression
As shown in Figure 5, there was significantly decreased mRNA expression of Col2 and Agg in the IL-1β group compared with the control group (P < 0.05). In contrast, Col2 and Agg mRNA expression was increased significantly in the HBO group compared with the IL-1β group (P < 0.05). In the HBO+ inhibitor group, Col2 and Agg mRNA expression was significantly decreased in contrast to the HBO group (P < 0.05).
Figure 5.
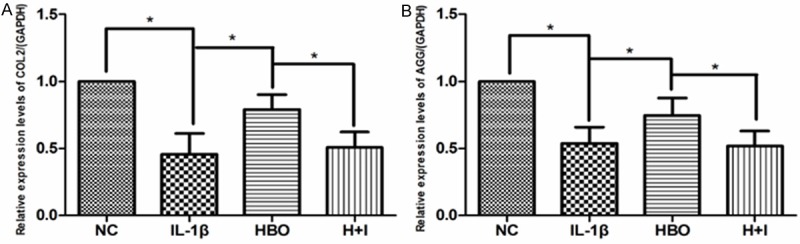
The 2-ΔΔCt method was adopted with GAPDH as the reference gene. Reverse transcription and real-time quantitative polymerase chain reaction technique was used to examine the mRNA expression COL2 and AGG in each group. A: The COL2 mRNA levels were normalized to GAPDH levels in different groups (n=6 per group). B: The AGG mRNA levels were normalized to GAPDH levels in different groups (n=6 per group). Bars represent the mean and SEM of each group. * < 0.05.
Discussion
Mandibular condylar chondrocyte apoptosis and extracellular matrix degradation play an important role in the development of cartilage degeneration in TMJ-OA. HBO is currently established for many clinical applications and has potential for use in regenerative therapy, yet its mechanism of action in most applications remains unknown. In addition, HBO treatment suppresses the apoptosis in degenerated disc cells [26] and osteoarthritic chondrocytes [22], suggesting a beneficial effect of HBO. However, the effects of HBO in OA have not been reported. Our results show that treatment with HBO increased cell proliferation and viability and inhibited apoptosis in IL-1β-stimulated rat chondrocytes, a model of OA chondrocytes, and increased COL2, AGG, p-PI3K, and p-AKT expression. Our results indicate that HBO treatment has the net effect of inhibiting the extracellular matrix degradation mediated by the promotion of PI3K/AKT signaling.
IL-1β, a proinflammatory cytokine that can induce chondrocyte apoptosis in rats and humans, is associated with the incidence and development of OA [27]. The present report provides the first evidence that HBO inhibits chondrocyte apoptosis induced by IL-1β and therefore may play a protective role against IL-1β-relevant incidence and development of OA. Apoptosis plays an important role in TMJ disc degeneration, cartilage degradation, and bone resorption. It is well documented that apoptosis is a potential contributor in the pathogenesis of TMJ disorder (TMD) [28,29]. The PI3K/AKT pathway has been identified as a major regulator of cellular proliferation, differentiation, and death in numerous cell types [30]. Specifically, normal expression of this pathway has an irreplaceable role in regulating the proliferation and differentiation of chondrocytes [31-33].
Previous studies have shown that the PI3K/AKT signaling pathway is responsible for sustaining chondrocyte survival and promoting extracellular matrix synthesis [19,30,34-37]. Conversely, the inhibition of PI3K/AKT signaling decreases PG synthesis in chondrocytes and increases chondrocyte survival [35-37]. In the present study, HBO treatment reversed the effects of IL-1β on chondrocyte apoptosis and promoted cell proliferation and viability. Moreover, the PI3K-specific inhibitor LY294002 significantly inhibited the anti-apoptotic effect of HBO. Consequently, the above results suggest that PI3K/AKT signaling is a critical molecular target of OA and that HBO may have a potential therapeutic action by activating this signaling pathway.
Furthermore, HBO treatment of OA not only inhibited chondrocyte apoptosis, but also promoted chondrocyte extracellular matrix synthesis. The present study showed that HBO treatment increased COL2 and AGG expression in extracellular matrix synthesis affected by IL-1β. Furthermore, the addition of LY294002 blocked the effects of HBO on the regulation of COL2 and AGG expression induced by IL-1β, indicating that PI3K/AKT signaling plays a critical role during this process. Therefore, we deduced that PI3K/AKT signaling, a target of HBO both inhibits chondrocyte apoptosis and decreases extracellular matrix degradation.
In summary, our results confirm that HBO inhibits chondrocyte apoptosis induced by IL-1β and promotes extracellular matrix synthesis in rat OA mandibular condylar chondrocytes via the PI3K/AKT signaling pathway. These findings suggest that HBO has a potential therapeutic function for treating TMJ-OA.
Acknowledgements
This study was supported by the Natural Science Foundation of Shandong Province of China (ZR2014HM041), Natural Science Foundation of China (61471384, 81400573), Science and Technology Development Plans of Shandong province (2014GSF118101), Army Health Breeding Programme (15QNP019) and Youth Science and Technology Star Plant (2013032). The authors are deeply thankful to Dr. Fabin Han of Liaocheng People’s Hospital of China (Liaocheng, China) for his earnest guidance.
Disclosure of conflict of interest
None.
References
- 1.Kapila S, Wang W, Uston K. Matrix metalloproteinase induction by relaxin causes cartilage matrix degradation in target synovial joints. Ann N Y Acad Sci. 2009;1160:322–328. doi: 10.1111/j.1749-6632.2009.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scrivani SJ, Keith DA, Kaban LB. Temporomandibular disorders. N Engl J Med. 2008;359:2693–2705. doi: 10.1056/NEJMra0802472. [DOI] [PubMed] [Google Scholar]
- 3.Aida Y, Maeno M, Suzuki N, Shiratsuchi H, Motohashi M, Matsumura H. The effect of IL-1beta on the expression of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in human chondrocytes. Life Sci. 2005;77:3210–3221. doi: 10.1016/j.lfs.2005.05.052. [DOI] [PubMed] [Google Scholar]
- 4.Zhou PH, Liu SQ, Peng H. The effect of hyaluronic acid on IL-1beta-induced chondrocyte apoptosis in a rat model of osteoarthritis. J Orthop Res. 2008;26:1643–1648. doi: 10.1002/jor.20683. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Armada MJ, Carames B, Lires-Dean M, Cillero-Pastor B, Ruiz-Romero C, Galdo F, Blanco FJ. Cytokines, tumor necrosis factor-alpha and interleukin-1beta, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthritis Cartilage. 2006;14:660–669. doi: 10.1016/j.joca.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Goldring MB. Osteoarthritis and cartilage: the role of cytokines. Curr Rheumatol Rep. 2000;2:459–465. doi: 10.1007/s11926-000-0021-y. [DOI] [PubMed] [Google Scholar]
- 7.Yasuhara R, Miyamoto Y, Akaike T, Akuta T, Nakamura M, Takami M, Morimura N, Yasu K, Kamijo R. Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem J. 2005;389:315–323. doi: 10.1042/BJ20041996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terkeltaub R, Johnson K, Murphy A, Ghosh S. Invited review: the mitochondrion in osteoarthritis. Mitochondrion. 2002;1:301–319. doi: 10.1016/s1567-7249(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 9.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 2007;39:443–455. doi: 10.1080/03602530701468516. [DOI] [PubMed] [Google Scholar]
- 10.Kim HA, Blanco FJ. Cell death and apoptosis in osteoarthritic cartilage. Curr Drug Targets. 2007;8:333–345. doi: 10.2174/138945007779940025. [DOI] [PubMed] [Google Scholar]
- 11.Huang JG, Xia C, Zheng XP, Yi TT, Wang XY, Song G, Zhang B. 17beta-Estradiol promotes cell proliferation in rat osteoarthritis model chondrocytes via PI3K/Akt pathway. Cell Mol Biol Lett. 2011;16:564–575. doi: 10.2478/s11658-011-0023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn K, D’Lima DD, Hashimoto S, Lotz M. Cell death in cartilage. Osteoarthritis Cartilage. 2004;12:1–16. doi: 10.1016/j.joca.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 13.Thomas CM, Fuller CJ, Whittles CE, Sharif M. Chondrocyte death by apoptosis is associated with the initiation and severity of articular cartilage degradation. Int J Rheum Dis. 2011;14:191–198. doi: 10.1111/j.1756-185X.2010.01578.x. [DOI] [PubMed] [Google Scholar]
- 14.Montaseri A, Busch F, Mobasheri A, Buhrmann C, Aldinger C, Rad JS, Shakibaei M. IGF-1 and PDGF-bb suppress IL-1beta-induced cartilage degradation through down-regulation of NF-kappaB signaling: involvement of Src/PI-3K/AKT pathway. PLoS One. 2011;6:e28663. doi: 10.1371/journal.pone.0028663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 16.Kita K, Kimura T, Nakamura N, Yoshikawa H, Nakano T. PI3K/Akt signaling as a key regulatory pathway for chondrocyte terminal differentiation. Genes Cells. 2008;13:839–850. doi: 10.1111/j.1365-2443.2008.01209.x. [DOI] [PubMed] [Google Scholar]
- 17.Hidaka K, Kanematsu T, Takeuchi H, Nakata M, Kikkawa U, Hirata M. Involvement of the phosphoinositide 3-kinase/protein kinase B signaling pathway in insulin/IGF-I-induced chondrogenesis of the mouse embryonal carcinoma-derived cell line ATDC5. Int J Biochem Cell Biol. 2001;33:1094–1103. doi: 10.1016/s1357-2725(01)00067-x. [DOI] [PubMed] [Google Scholar]
- 18.Fujita T, Fukuyama R, Enomoto H, Komori T. Dexamethasone inhibits insulin-induced chondrogenesis of ATDC5 cells by preventing PI3K-Akt signaling and DNA binding of Runx2. J Cell Biochem. 2004;93:374–383. doi: 10.1002/jcb.20192. [DOI] [PubMed] [Google Scholar]
- 19.Oh CD, Chun JS. Signaling mechanisms leading to the regulation of differentiation and apoptosis of articular chondrocytes by insulin-like growth factor-1. J Biol Chem. 2003;278:36563–36571. doi: 10.1074/jbc.M304857200. [DOI] [PubMed] [Google Scholar]
- 20.Korhonen K. Hyperbaric oxygen therapy in acute necrotizing infections. With a special reference to the effects on tissue gas tensions. Ann Chir Gynaecol. 2000;89(Suppl 214):7–36. [PubMed] [Google Scholar]
- 21.Yuan LJ, Ueng SW, Lin SS, Yeh WL, Yang CY, Lin PY. Attenuation of apoptosis and enhancement of proteoglycan synthesis in rabbit cartilage defects by hyperbaric oxygen treatment are related to the suppression of nitric oxide production. J Orthop Res. 2004;22:1126–1134. doi: 10.1016/j.orthres.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Ueng SW, Yuan LJ, Lin SS, Niu CC, Chan YS, Wang IC, Yang CY, Chen WJ. Hyperbaric oxygen treatment prevents nitric oxide-induced apoptosis in articular cartilage injury via enhancement of the expression of heat shock protein 70. J Orthop Res. 2013;31:376–384. doi: 10.1002/jor.22235. [DOI] [PubMed] [Google Scholar]
- 23.Yuan LJ, Niu CC, Lin SS, Yang CY, Chan YS, Chen WJ, Ueng SW. Effects of low-intensity pulsed ultrasound and hyperbaric oxygen on human osteoarthritic chondrocytes. J Orthop Surg Res. 2014;9:5. doi: 10.1186/1749-799X-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inamoto Y, Okuno F, Saito K, Tanaka Y, Watanabe K, Morimoto I, Yamashita U, Eto S. Effect of hyperbaric oxygenation on macrophage function in mice. Biochem Biophys Res Commun. 1991;179:886–891. doi: 10.1016/0006-291x(91)91901-n. [DOI] [PubMed] [Google Scholar]
- 25.Roberts GP, Harding KG. Stimulation of glycosaminoglycan synthesis in cultured fibroblasts by hyperbaric oxygen. Br J Dermatol. 1994;131:630–633. doi: 10.1111/j.1365-2133.1994.tb04973.x. [DOI] [PubMed] [Google Scholar]
- 26.Niu CC, Lin SS, Yuan LJ, Chen LH, Wang IC, Tsai TT, Lai PL, Chen WJ. Hyperbaric oxygen treatment suppresses MAPK signaling and mitochondrial apoptotic pathway in degenerated human intervertebral disc cells. J Orthop Res. 2013;31:204–209. doi: 10.1002/jor.22209. [DOI] [PubMed] [Google Scholar]
- 27.Csaki C, Mobasheri A, Shakibaei M. Synergistic chondroprotective effects of curcumin and resveratrol in human articular chondrocytes: inhibition of IL-1beta-induced NF-kappaB-mediated inflammation and apoptosis. Arthritis Res Ther. 2009;11:R165. doi: 10.1186/ar2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loreto C, Almeida LE, Trevilatto P, Leonardi R. Apoptosis in displaced temporomandibular joint disc with and without reduction: an immunohistochemical study. J Oral Pathol Med. 2011;40:103–110. doi: 10.1111/j.1600-0714.2010.00920.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang XD, Kou XX, He DQ, Zeng MM, Meng Z, Bi RY, Liu Y, Zhang JN, Gan YH, Zhou YH. Progression of cartilage degradation, bone resorption and pain in rat temporomandibular joint osteoarthritis induced by injection of iodoacetate. PLoS One. 2012;7:e45036. doi: 10.1371/journal.pone.0045036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beier F, Loeser RF. Biology and pathology of Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in chondrocytes. J Cell Biochem. 2010;110:573–580. doi: 10.1002/jcb.22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malemud CJ. Protein kinases in chondrocyte signaling and osteoarthritis. Clin Orthop Relat Res. 2004:S145–151. doi: 10.1097/01.blo.0000143802.41885.50. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi R, Ryo A, Komitsu N, Mikuni-Takagaki Y, Fukui A, Takagi Y, Shiraishi T, Morishita S, Yamazaki Y, Kumagai K, Aoki I, Saito T. Low-intensity pulsed ultrasound activates the phosphatidylinositol 3 kinase/Akt pathway and stimulates the growth of chondrocytes in three-dimensional cultures: a basic science study. Arthritis Res Ther. 2008;10:R77. doi: 10.1186/ar2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulici V, Hoenselaar KD, Gillespie JR, Beier F. The PI3K pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev Biol. 2008;8:40. doi: 10.1186/1471-213X-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cravero JD, Carlson CS, Im HJ, Yammani RR, Long D, Loeser RF. Increased expression of the Akt/PKB inhibitor TRB3 in osteoarthritic chondrocytes inhibits insulin-like growth factor 1-mediated cell survival and proteoglycan synthesis. Arthritis Rheum. 2009;60:492–500. doi: 10.1002/art.24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Starkman BG, Cravero JD, Delcarlo M, Loeser RF. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem J. 2005;389:723–729. doi: 10.1042/BJ20041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin W, Park JI, Loeser RF. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J Biol Chem. 2009;284:31972–31981. doi: 10.1074/jbc.M109.056838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Wu G, Zhu G, Ma C, Zhao H. Chronic sleep restriction induces changes in the mandibular condylar cartilage of rats: roles of Akt, Bad and Caspase-3. Int J Clin Exp Med. 2014;7:2585–2592. [PMC free article] [PubMed] [Google Scholar]