

Abstract
Aim: A wealth of studies have demonstrated that abnormal cellular lipid metabolism plays an important role in prostate cancer (PCa) development. Therefore, manipulating lipid metabolism is a potential PCa therapy strategy. In this study, our goal is to investigate the role of farnesoid X receptor (FXR) in regulating the proliferation and lipid metabolism of human PCa cells following its ligand chenodexycholic acid (CDCA) treatment. Methods: Oil Red O was used to stain lipid contents in PCa cells, and siRNA knockdown was performed to deplete FXR expression. To study the cell proliferation when treated by CDCA or FXR knockdown, cell counting kit 8 (CCK8) was adopted to evaluate tumor cell growth. Western blot was used for protein analysis. Results: Our data suggest that activation of FXR by CDCA reduces lipid accumulation and significantly inhibits cells proliferation in prostate tumor cells. Instead, CDCA treatment doesn’t affect normal prostate epithelial RWPE-1 cells growth in vitro. FXR activation decreases mRNA and protein levels of sterol regulatory element binding protein 1 (SREBP1) and some other key regulators involved in lipid metabolism. Depletion of FXR by siRNA attenuates the inhibitory effects. Conclusion: Our study indicates that activation of FXR inhibits lipid metabolism via SREBP1 pathway and further suppresses prostate tumor growth in vitro.
Keywords: Prostate cancer, lipogenesis, FXR, SREBP1
Introduction
Prostate cancer (PCa) is the most common non-cutaneous type of cancer and the second leading cause of cancer deaths among men in the United States with nearly a million new cases diagnosed worldwide per year [1]. Currently, the standard systemic treatment for advanced PCa is based on androgen deprivation, but patients who initially responded to the treatment eventually became castrate resistant, and have a median survival of less than 2 years. New effective treatments and agents are urgently needed. Age, family history, genetic background, lifestyle, environmental influences, and diet are some of the most important risky factors associated with PCa, among which lipid metabolism affects several aspects of tumor biological processes, including cell proliferation and differentiation. It has become clear that both de novo and dietary lipids play an important role in the development and progression of PCa [2,3]. Epidemiologic evidence also supports a relationship between obesity and PCa progression, indicating that obesity is an adverse prognostic factor. Farnesoid X receptor (FXR), a chenodeoxycholic acid (CDCA) sensor, plays an essential role in maintaining lipid and glucose homeostasis [4]. Studies have shown that FXR inhibits fatty acid synthetase (FAS) expression and reduces fatty acid and triglyceride synthesis. The mechanism is the suppression of sterol regulatory element-binding protein-1c (SREBP-1c) by FXR via a SHP-mediated inhibition of co-activator recruitment to the SREBP1c promoters [5]. SREBP-1 is a major transcriptional regulator of the enzymes involved in lipid synthesis such as ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), fatty-acid synthase (FASN) [6]. It is a critical link between oncogenic signaling and tumor metabolism. Overexpression of SREBP1 is sufficient to increase tumorigenicity and invasiveness of PCa cells, while inhibition of SREBP1 can decrease de novo fatty acid synthesis and inhibit PCa cells proliferation [7]. Developing a SREBP1 inhibitor is a new strategy for PCa treatment.
So far, the function of FXR on the lipid regulation in PCa is still uncertain. Activation or overexpression of FXR has been shown to suppress PCa cell proliferation [8]. However, the mechanism of FXR in regulating PCa cell proliferation in prostate cancer cells remains unknown. We therefore hypothesize that activation of FXR inhibits PCa growth by modulating lipid metabolism. We screened FXR expression in prostate cancer tissues and compared them to normal prostate tissue. Our results indicate that FXR activation inhibits lipid accumulation and suppresses tumor cell proliferation in PCa cells by regulating SREBP1 and its down-stream factor expression.
Materials and methods
Cell lines and reagents
LNCaP and DU145 cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% CO2. RWPE-1 cell line was purchased from ATCC and maintained in keratinocyte growth medium with 5 ng/ml human recombinant epidermal growth factor and 0.05 mg/ml bovine pituitary extract. Chenodeoxycholic acid (CDCA) was purchased from Selleck Chemicals and dissolved in DMSO. SYBR Green PCR Master Mix kit was purchased from Applied Biosystems (Foster City, CA). Antibodies for FXR and SREBP1 were obtained from Abcam (Cambidge, MA). FXR, FASN, ACC, phosphor-ACC and actin antibody were purchased from Cell Signaling Technologies.
Knockdown of FXR by siRNA
For FXR knockdown, siRNA targeting to FXR was chemically synthesized (Gene Pharma, China). The siRNA sequence for human FXR depletion is 5’-GAGGAUGCCUCAGGAAAUA-3’. Scramble siRNA 5’-AAAGCGUCUGGAAAAGUCG-3’ was used as a control. LNCaP cells were transfected with siRNA using Lipofectamine2000 according to the manufacturer’s instructions (Invitrogen, USA). Efficiency of knockdown was performed through Western blot analysis.
Oil Red O (ORO) staining
ORO staining was performed to analyze lipid content such as neutral triglycerides and cellular cholesterol esters in tumor cells. RWPE-1, DU145 and LNCaP cells were seeded at 50,000 cells/well in a 6-well plate. After treatment, cells were fixed with 10% PBS buffered formalin for 15 minutes at room temperature, washed twice with distilled water and then with 60% isopropanol for 5 minutes. After the plate completely dried, cells were stained with ORO (0.3% ORO in 100% isopropanol, diluted with distilled water in the ratio of 3:2) for 30 minutes, and then washed with distilled water 5 times. Images were captured at 100 or 200 × magnification with a microscope. To quantify the lipid content, 500 μl of 100% isopropanol was added to each well and the optical density was measured by spectrophotometer at 520 nm.
Reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR was used to evaluate the expression of FXR, SREBP1, FAS and ACC. Total RNA was extracted from tumor cells using TRIzol® Reagent (Invitrogen). cDNA was prepared from 3 µg of total RNA by MMLV First-Strand Synthesis Kit ( Life technologies). qPCRs were performed in triplicate in a final volume of 20 µl with ABI 7500 real-time PCR thermal cycler (Applied Biosystems). Dissociation curves were run to detect nonspecific amplification and to confirm amplification of single products in each reaction. GAPDH was used as an internal control. The data were analyzed by 2-ΔΔCt method. The sequences of primers were used as the following: FXR (Forward primer: ATTCCTCATTCTGGGGCTTT; Reverse primer: TATGCTGGGTGTCATCTCCA), SREBP1 (Forward primer: ACAGCCATGAAGACAGACGG; Reverse primer: ATAGGCAGC TTCTCCGCATC). Fatty acid synthase (Forward primer: GAAACTGCAGGAGCTGTC; Reverse primer: CACGGAGTTGAGCCGCAT). ACC (Forward primer: CTGTAGAAACCCGGACAGTAGAAC; Reverse primer: GGTCAGCATACATCTCCATGTG). GAPDH (Forward primer: GACAGTCAGCCGCATCTTCT; Reverse primer: TTAAAAGCAGCCCTGGTGAC).
Cell proliferation assay
Cell Counting Kit 8 (CCK8) was used to evaluate cell proliferation according to the manufacturer’s instructions. Cells were seeded in 96-well plates at a density of 5 × 104/well and maintained at 37°C for 24 h. The cells were subsequently placed with fresh medium with or without CDCA (50 µM) and continued to be cultured for 24 h, 48 h and 72 h. 10 μL of CCK-8 reagent was added to each well, and then incubated at 37°C for another 3 hours. The absorbance was determined at 450 nm with microplate reader (Bio-TEK).
Western blot assay
Whole cell extracts were prepared and immunoblotting was performed following described previously [9]. 50 μg of total protein lysates were separated by 8-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and samples were transferred to nitrocellulose membranes. After blocking with 5% nonfat milk for 1 hour at room temperature, membranes were incubated with primary antibodies. Membranes were washed and exposed to peroxidase-conjugated secondary antibodies and visualized by ECL detection system according to manufacturer’s instructions.
Statistical analysis
All of the data were expressed as mean ± SEM. Comparisons between two groups were analyzed by Student’s t test for paired and unpaired analyses. P value less than 0.05 was considered statistically significant.
Results
Lipid accumulation in PCa cell lines
To evaluate the lipid accumulation in prostate cancer cells, we performed ORO staining and compared the endogenous lipid levels in androgen dependent LNCaP cells, androgen independent cells DU145, as well as normal prostate epithelial cell line RWPE-1. As shown in Figure 1A, prostate tumor cells exhibit much higher lipid content as compared with RWPE1 cells. The lipid content is androgen-dependent since LNCaP exhibited the highest lipid accumulation in all PCA cells (Figure 1A). These results suggest that PCa cells have abnormal lipids accumulation.
Figure 1.
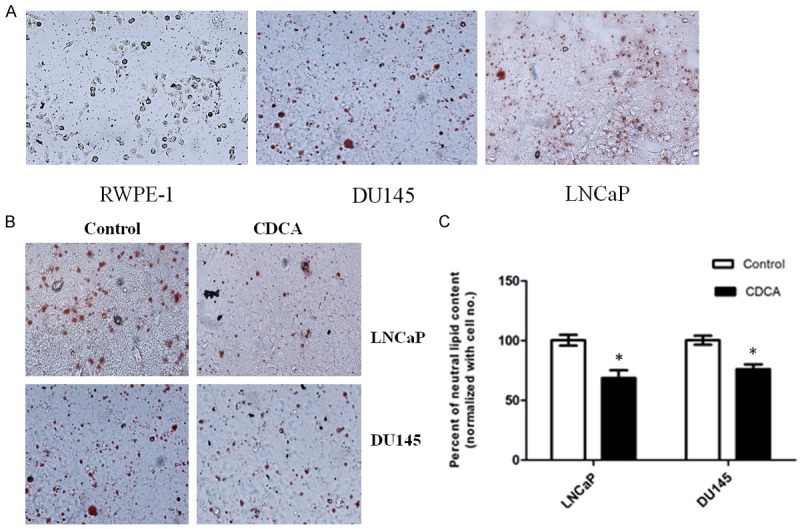
CDCA inhibits lipids synthesis in PCa cells. A. PCa cells exhibit abnormal neutral lipid accumulation as stained by ORO. B. CDCA (50 μM) treatment inhibits lipid droplet accumulation in LNCaP and DU145 cells. C. Intracellular lipid levels were measured by Oil red O staining and quantified by Spectrophotometric method and normalized with respective to cell number for each group. *P<0.05.
CDCA reduces cellular lipid content and inhibits PCa cell proliferation
Since PCa cells exhibit abnormal lipid accumulation, we then ask whether activation of FXR inhibits lipids metabolism in PCa cells. When cells were treated with FXR activator CDCA (50 µM) for 24 hours, the ORO staining indicated that FXR activation significantly reduces the neutral lipid level in LNCaP and DU145 cells. On the contrary, CDCA treatment has no effects on RWPE-1 cells (Figure 1B). Those data suggest that CDCA specifically affects tumor cells lipid accumulation.
Interfering with lipid metabolism of cancer cells has become a therapeutic approach and several small molecular inhibitors for lipid synthesis pathway have been identified with anti-tumor activity in PCa. Since activation of FXR has been shown to decrease lipid content in PCA cells, we then examined whether activation of FXR also inhibits PCa cells proliferation. LNCaP and RWPE-1 cells were treated with or without FXR agonist CDCA for 24 h, 48 h and 72 h. Cell proliferation was analyzed by CCK8 assay. As shown in Figure 2, CDCA treatment significantly inhibits LNCaP tumor growth (Figure 2A). However, CDCA treatment has no effect on non-tumorigenic RWPE-1 cells, which suggests that CDCA selectively suppresses PCa growth in vitro.
Figure 2.
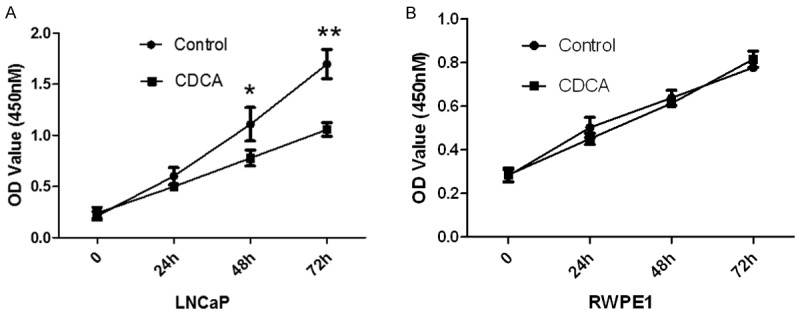
Activation of FXR inhibits PCa cells proliferation, but not normal prostate cells. The CCK8 assay used to evaluate the proliferation of PCa cells after CDCA (50 μM) treatment for 24 h, 48 h, 72 h. A. LNCaP cells. B. RWPE-1 cells. *P<0.05; **P<0.01.
CDCA decreases neutral lipid via down-regulating SREBP1 expression and its target genes in PCa cells
SREBP1 has been reported to decrease fatty acid synthesis and inhibit PCa cells proliferation [10]. To investigate whether or not the effect of FXR is associated with SREBP1, we then analyzed the role of CDCA on SREBP1 expression levels. Quantitative PCR analysis indicates that SREBP1 mRNA expression in LNCaP cells decreased following CDCA treatment (Figure 3A). Similarly, SREBP1 protein level was inhibited by CDCA treatment (Figure 3B), which suggests that FXR activation reduces the mRNA and protein levels of SREBP1.
Figure 3.
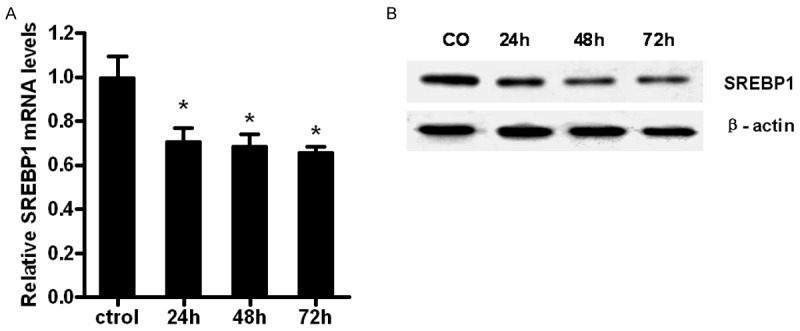
CDCA treatment inhibits SREBP1 mRNA and protein levels in LNCaP cells. A. LNCaP cells were treated with CDCA (50 μM) for 24 h, 48 h or 72 h., and mRNA of SREBP1 was analyzed by RT-PCR. *P<0.05; B. Western blot assay for SREBP1 protein levels after treated with CDCA (50 μM) for 24 h, 48 h or 72 h. Actin was used as control.
SREBP1 is the major transcriptional factor of the lipogenic enzymes FASN, ACLY, ACC. Therefore, we examined whether FXR activation affects those SREBP1-controlled lipogenic enzymes. As shown in Figure 4A, CDCA treatment dramatically inhibits FASN, ACC and ACLy mRNA levels in LNCaP cells. Similarly, decreased FASN and phosphorylated ACC protein levels were also observed after CDCA treatment. In conclusion, these results indicate that FXR modulates PCa lipid metabolism by regulating SREBP1 and its downstream genes expression.
Figure 4.
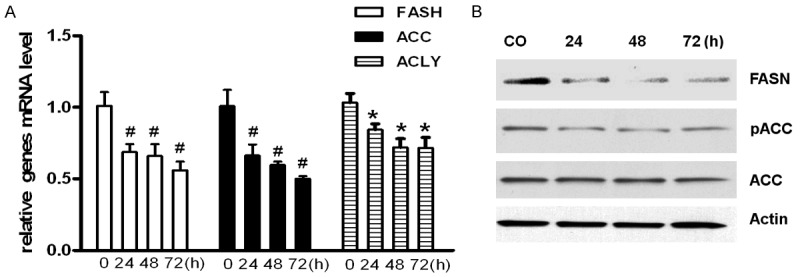
CDCA treatment regulates the expression or phosphorylation of key molecules involved in lipogenesis. A. LNCaP cells were treated with CDCA (50 μM) for 24 h, 48 h, or 72 h, and mRNA levels were analyzed by RT-PCR. *P<0.05, #P<0.01. B. Total cell lysates were prepared after treated with CDCA (50 μM) for 24 h, 48 h, or 72 h, and FASN and ACC phosphorylation were examined by Western blot. Actin was used as control.
Depletion of FXR attenuates the inhibition of CDCA on SREBP1
To directly test whether or not FXR plays an essential role in SREBP1 mediated down-regulation of lipid accumulation, we first transfected LNCaP cells with FXR specific siRNA or scramble siRNA. Then we treated the cells with or without CDCA and analyzed SREBP1 levels and LNCaP tumor growth. The results showed that FXR siRNA specifically knock down FXR in LNCaP cells as confirmed by Western blotting (Figure 5A). Knockdown of FXR attenuates the inhibition of CDCA on SREBP1 (Figure 5A, lane 3). In addition, we analyzed the effect of FXR knockdown on LNCaP cell growth. As shown in Figure 5B, CDCA alone treatment significantly inhibits LNCaP tumor cell growth. However, CDCA treatment fails to inhibit LNCaP cells proliferation when FXR is knocked down. Those data suggest that CDCA decreases SREBP1 levels and inhibits cell proliferation through the FXR dependent pathway.
Figure 5.
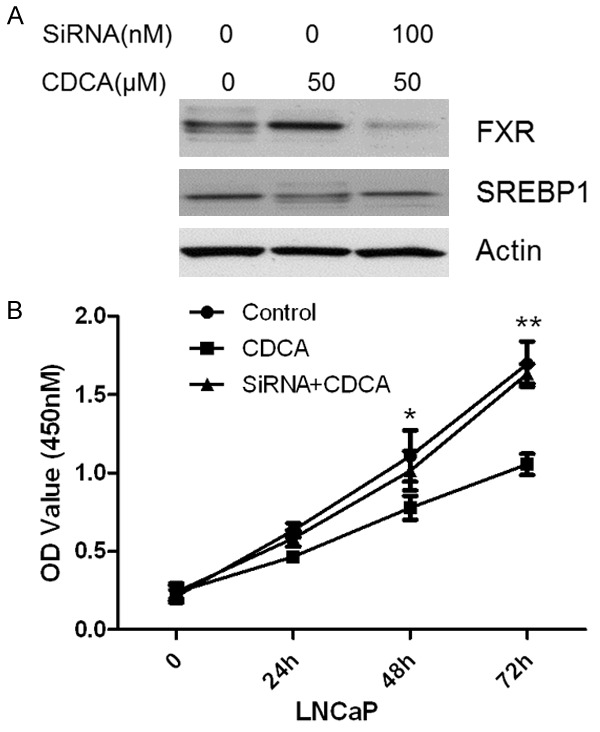
Depletion of FXR on SREBP1 expression and LNCaP cells proliferation induced by CDCA. A. FXR and SREBP1 expression when knockdown FXR. B. Knockdown of FXR failed to inhibit LNCaP cells growth. *P<0.05; **P<0.01.
Discussion
Rapidly proliferating cancer cells require a constant supply of lipids for membrane biogenesis and protein modifications. Several studies have shown that cancer cells either increase their uptake of lipids or activate de novo lipid synthesis to cope with these increased demands [11,12]. Lipid synthesis is especially active in metastatic, castration-resistant patients, which suggests that limiting their availability could provide a therapeutic strategy. In this study, we have observed higher intracellular lipid in LNCaP and DU145 cells instead of RWPE-1 cells, which suggests that lipid synthesis increase in PCa [13].
FXR activation can inhibit the expression of SREBP-1c and its target enzymes such as fatty acid synthase (FAS), stearoyl-coenzyme A desaturase 1 (SCD-1) and acetyl-CoA carboxylase (ACC). It can also prevent excessive fatty acid synthesis and overproduction of triglyceride (TG) [14,15]. However, the function of FXR on lipid metabolism in PCa cells still remains uncertain. Swales, K.E. et al [16] and Glordano, C. et al [17] reported that FXR is expressed in human breast tissues and activation of FXR induces MCF-7 and MDA-MB-468 cell death in vitro. Recent studies also indicated that activation or over-expression of FXR suppresses prostate cancer cell proliferation and upregulates PTEN in LNCaP cells. It suggests that FXR functions as a tumor suppressor in Prostate cancer [14]. Activation of FXR by CDCA reduces intracellular lipid droplets and inhibits cell proliferation in LNCaP and DU145 cells. However, no significant changes were observed on normal prostate epithelial cell following the treatment. These results suggested that the inhibitory effect of CDCA on cell proliferation was specific to PCa cells and the decreased lipid levels in tumor cells may inhibit the PCa cells proliferation.
CDCA has been reported to induce tumor cells apoptosis in vitro and inhibit tumor growth in vito via activation of FXR [18-20]. However, how does CDCA inhibit lipid synthesis is uncertain. As an important transcription factor in lipid metabolism, SREBP1 regulates syntheses of fatty acids, triglycerides and cholesterol. Most enzymes involved in FA and cholesterol synthesis are regulated by SREBPs. Overexpression of SREBP-1 is sufficient to increase tumorigenicity and invasion of PCa [7]. Therefore, inhibition of SREBP-1 is critical for PCa therapy. Our data suggest that FXR activation suppresses SREBP1 expression in LNCaP cells. Meanwhile, we also found that CDCA treatment inhibited FAS, ACC, ACLY mRNA and protein levels as well. These results suggest that SREBP1 is a key regulator for FXR-mediated lipid metabolism inhibition and plays a central role in CDCA-induced PCa cell proliferation inhibition. FXR knockdown attenuated the inhibitory effect on cell proliferation induced by CDCA. It provides further evidence that the activation of FXR inhibits PCa cell growth via the SREBP1 mediated lipogenesis pathway.
In conclusion, our results suggest that FXR ligand CDCA regulates lipid accumulation through SREBP1 mediated lipogenesis pathway in PCa cells. Limiting lipid availability inhibits PCa cell growth, which shows that FXR agonist CDCA could be useful in PCa treatment via targeting lipid lipogenesis.
Acknowledgements
The authors would like to express their gratitude to all the physicians participating in this work. This study was supported by the National Natural Science Foundation of China (81370830, 81000300), and Norman Bethune Program of Jilin University (2012226).
References
- 1.Iranikhah M, Stricker S, Freeman MK. Future of bisphosphonates and denosumab for men with advanced prostate cancer. Cancer Manag Res. 2014;6:217–224. doi: 10.2147/CMAR.S40151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta. 2013;1831:1518–1532. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279:2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 4.Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. FXR, a multipurpose nuclear receptor. Trends Biochem Sci. 2006;31:572–580. doi: 10.1016/j.tibs.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swinnen JV, Heemers H, Deboel L, Foufelle F, Heyns W, Verhoeven G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene. 2000;19:5173–5181. doi: 10.1038/sj.onc.1203889. [DOI] [PubMed] [Google Scholar]
- 7.Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. 2012;10:133–142. doi: 10.1158/1541-7786.MCR-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Tong SJ, Wang X, Qu LX. Farnesoid X receptor inhibits LNcaP cell proliferation via the upregulation of PTEN. Exp Ther Med. 2014;8:1209–1212. doi: 10.3892/etm.2014.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu N, Meng Z, Lou G, Zhou W, Wang X, Zhang Y, Zhang L, Liu X, Yen Y, Lai L, Forman BM, Xu Z, Xu R, Huang W. Hepatocarcinogenesis in FXR-/- mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol Endocrinol. 2012;26:775–785. doi: 10.1210/me.2011-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou HY, Wu HT, Lu FH, Su YC, Hung HC, Wu JS, Yang YC, Wu CL, Chang CJ. Activation of free fatty acid receptor 1 improves hepatic steatosis through a p38-dependent pathway. J Mol Endocrinol. 2014;53:165–174. doi: 10.1530/JME-14-0003. [DOI] [PubMed] [Google Scholar]
- 11.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 12.Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52:585–589. doi: 10.1016/j.plipres.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nambiar DK, Deep G, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1. Oncotarget. 2014;5:10017–10033. doi: 10.18632/oncotarget.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong X, Wang X, Lu Y, Wang E, Zhang Z, Yang J, Zhang H, Li X. Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J Hepatol. 2014;60:847–854. doi: 10.1016/j.jhep.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Li Y, Yang W, Xiao C, Fu S, Deng Q, Ding H, Wang Z, Liu G, Li X. SREBP-1c overexpression induces triglycerides accumulation through increasing lipid synthesis and decreasing lipid oxidation and VLDL assembly in bovine hepatocytes. J Steroid Biochem Mol Biol. 2014;143:174–182. doi: 10.1016/j.jsbmb.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Swales KE, Korbonits M, Carpenter R, Walsh DT, Warner TD, Bishop-Bailey D. The farnesoid X receptor is expressed in breast cancer and regulates apoptosis and aromatase expression. Cancer Res. 2006;66:10120–10126. doi: 10.1158/0008-5472.CAN-06-2399. [DOI] [PubMed] [Google Scholar]
- 17.Giordano C, Catalano S, Panza S, Vizza D, Barone I, Bonofiglio D, Gelsomino L, Rizza P, Fuqua SA, Ando S. Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast cancer cell growth through downregulation of HER2 expression. Oncogene. 2011;30:4129–4140. doi: 10.1038/onc.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai J, Wang H, Shi Y, Dong Y, Zhang Y, Wang J. Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J Hematol Oncol. 2011;4:41. doi: 10.1186/1756-8722-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alasmael N, Mohan R, Meira LB, Swales KE, Plant NJ. Activation of the Farnesoid X-receptor in breast cancer cell lines results in cytotoxicity but not increased migration potential. Cancer Lett. 2016;370:250–259. doi: 10.1016/j.canlet.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Katona BW, Anant S, Covey DF, Stenson WF. Characterization of enantiomeric bile acid-induced apoptosis in colon cancer cell lines. J Biol Chem. 2009;284:3354–3364. doi: 10.1074/jbc.M805804200. [DOI] [PMC free article] [PubMed] [Google Scholar]