

ABSTRACT
The interferon-induced protein with tetratricopeptide repeat 3 (IFIT3 or ISG60) is a host-intrinsic antiviral factor that restricts many instances of DNA and RNA virus replication. Herpes simplex virus 1 (HSV-1), a DNA virus bearing a large genome, can encode many viral proteins to counteract the host immune responses. However, whether IFIT3 plays a role upon HSV-1 infection is little known. In this study, we show for the first time that HSV-1 tegument protein UL41, a viral endoribonuclease, plays an important role in inhibiting the antiviral activity of IFIT3. Here, we demonstrated that ectopically expressed IFIT3 could restrict the replication of vesicular stomatitis virus (VSV) but had little effect on the replication of wild-type (WT) HSV-1. Further study showed that WT HSV-1 infection downregulated the expression of IFIT3, and ectopic expression of UL41, but not the immediate-early protein ICP0, notably reduced the expression of IFIT3. The underlying molecular mechanism was that UL41 diminished the accumulation of IFIT3 mRNA to abrogate its antiviral activity. In addition, our results illustrated that ectopic expression of IFIT3 inhibited the replication of UL41-null mutant virus (R2621), and stable knockdown of IFIT3 facilitated its replication. Taking these findings together, HSV-1 was shown for the first time to evade the antiviral function of IFIT3 via UL41.
IMPORTANCE The tegument protein UL41 of HSV-1 is an endoribonuclease with the substrate specificity of RNase A, which plays an important role in viral infection. Upon HSV-1 infection, interferons are critical cytokines that regulate immune responses against viral infection. Host antiviral responses are significantly boosted or crippled in the presence or absence of IFIT3; however, whether IFIT3 plays a role during HSV-1 infection is still unknown. Our data show for the first time that IFIT3 has little effect on HSV-1 replication, as UL41 decreases the accumulation of IFIT3 mRNA and subverts its antiviral activity. This study identifies IFIT3 as a novel target of the tegument protein UL41 and provides new insight into HSV-1-mediated immune evasion.
INTRODUCTION
In response to viral infection, secretion of type I interferon (IFN-I) is a key step in the host innate immune responses. The antiviral actions of IFN-I are embodied by the products of the IFN-stimulated genes (ISGs) (1, 2). To date, it is estimated that 2,000 human and mouse ISGs have already been identified; however, most of these genes remain uncharacterized (3–5). Among these ISGs, the family of IFN-induced protein with tetratricopeptide repeats (IFIT) has recently gained a great deal of attention (3). Humans have a distinct combination of IFIT family genes, including IFIT1 (ISG56), IFIT2 (ISG54), IFIT3 (ISG60), and IFIT5 (ISG58), which are clustered on human chromosome 10 (6). These IFIT proteins are robustly induced by IFNs (mainly IFN-I), viral infection, and lipopolysaccharide and are characterized by the unique helix-turn-helix motifs, called tetratricopeptide repeats, which mediate a variety of protein-protein and protein-RNA interactions (6–8). In addition, orthologs of the IFIT family are evolutionarily conserved from amphibians to mammals. However, the ortholog of human IFIT5 is absent from mouse (6, 9). Recent studies indicate that IFIT proteins play an important role in antiviral processes and restrict viral replication through altering protein synthesis, binding to viral RNAs, or interacting with structural or nonstructural viral proteins (8, 10, 11). IFIT3 was demonstrated to inhibit the replication of many DNA and RNA viruses, such as hepatitis B virus (HBV), human papillomavirus (HPV), hepatitis C virus (HCV), West Nile virus (WNV), porcine reproductive and respiratory syndrome virus (PRRSV), Dengue virus (DV), vesicular stomatitis virus (VSV), and others (6, 12–15). For example, in HeLa cells, the replication of VSV, whose genomic and leader RNAs carry 5′-ppp, was inhibited by the ectopic expression of IFIT1, IFIT2, and IFIT3, where they form a complex with each other to efficiently recognize 5′-ppp-RNA (12, 16). However, little is known about the function of IFIT3 upon herpes simplex virus 1 (HSV-1) infection.
HSV-1 is a double-stranded linear DNA virus with a large genome encoding over 80 proteins, and it can efficiently spread directly from cell to cell (17–19). HSV-1 tegument protein UL41 plays a vital role in evading IFN responses (20–24). UL41 is an endoribonuclease with the activity of mRNA-specific RNase that triggers rapid degradation of host mRNAs to promote the sequential expression of viral proteins (25–28). Hence, HSV-1 evades host responses to infection and causes a turnover of the host resources to benefit viral macromolecular synthesis via UL41 (29–32). In this study, we demonstrate for the first time that HSV-1 UL41 counteracts IFIT3 antiviral activity through degrading its mRNA.
MATERIALS AND METHODS
Cell culture, viruses, antibodies, and reagents.
Human embryonic kidney 293T (HEK293T), HEK293, and Vero cells were cultured in Dulbecco's modified minimal essential medium (DMEM) (Gibco-BRL) containing 10% fetal bovine serum (FBS) (Gibco-BRL) in a humidified atmosphere containing 5% CO2 at 37°C as described previously (33). HEK293T cells stably expressing short hairpin RNA (shRNA) were maintained in DMEM supplemented with 250 ng/ml puromycin as described previously (34). The wild-type (WT) HSV-1 (F strain) virus, R2621 virus, US11-null mutant virus (ΔUS11-HSV-1), and γ34.5-null mutant virus (Δγ34.5-HSV-1) were propagated in Vero cells, and the titers were determined as described previously (33, 35, 36). Sendai virus (SeV) was propagated as described previously (34). VSV was a gift from Hui Zheng (Institutes of Biology and Medical Sciences, Soochow University). Mouse monoclonal anti-Myc, anti-hemagglutinin (HA), and anti-Flag antibodies (MAbs), rabbit polyclonal anti-IFIT1, anti-IFIT2, and anti-IFIT3, and mouse anti-β-actin antibodies were purchased from Abmart (Shanghai, China), Proteintech (Wuhan, China), and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. Rabbit polyclonal anti-UL46 was made by GL Biochem Ltd. (Shanghai, China). Rabbit polyclonal anti-VP22 was described previously (37). Puromycin was purchased from Gene Operation (Shanghai, China).
Plasmid construction.
All enzymes used for cloning procedures were purchased from Vazyme (Nanjing, China). To construct IFIT3-Myc plasmid, the IFIT3 gene was cloned into the XhoI and HindIII sites of the pCMV-Myc vector (Beyotime, Shanghai, China). Small hairpin RNA specific for IFIT3 (shIFIT3) or scrambled small hairpin RNA (shNC) (NC stands for negative control) was cloned into pSUPER.retro.puro vector (Oligoengine, LA, USA) to yield pSUPER-shIFIT3 and pSUPER-shNC plasmids, respectively, as described in our previous study (38).
Establishment of IFIT3 stably knocked down HEK293T cells.
HEK293T cells were transfected with pSUPER-shIFIT3 (HEK293T-shIFIT3) or pSUPER-shNC plasmid (HEK293T-shNC). At 36 h posttransfection, puromycin was added to cells at a concentration of 1 μg/ml and screened for a week. The stably transfected HEK293T-shNC and HEK293T-shIFIT3 cells were then cultured with puromycin (250 ng/ml).
WB analysis.
Western blot (WB) analysis was performed as previously described (39).
RNA isolation and qRT-PCR.
Total RNA was extracted using TRIzol (Invitrogen, California) according to the manufacturer's manual. Samples were digested with DNase I and subjected to reverse transcription as previously described (40). The cDNA was used as the template for quantitative real-time PCR (qRT-PCR) to test the level of IFIT3 mRNA, and 18S rRNA was used as an internal reference as previously described (34, 40).
Statistical analysis.
Comparison between different groups was analyzed using Student's t test. All differences were considered statistically significant at a P value of <0.05.
RESULTS
Ectopically expressed IFIT3 had no effect on the replication of WT HSV-1.
To explore the role of IFIT3 in WT HSV-1 infection, HEK293T cells were transfected with vector or IFIT3-Myc plasmid. At 8 h posttransfection, cells were infected with WT HSV-1 at a multiplicity of infection (MOI) of 0.5. Cells then were harvested at the indicated time points postinfection and subjected to WB analysis. As shown in Fig. 1A, ectopic expression of IFIT3 did not affect the expression of UL46 and VP22. In addition, viral plaque assay also showed that ectopic expression of IFIT3 failed to inhibit the replication of WT HSV-1 (Fig. 1B).
FIG 1.
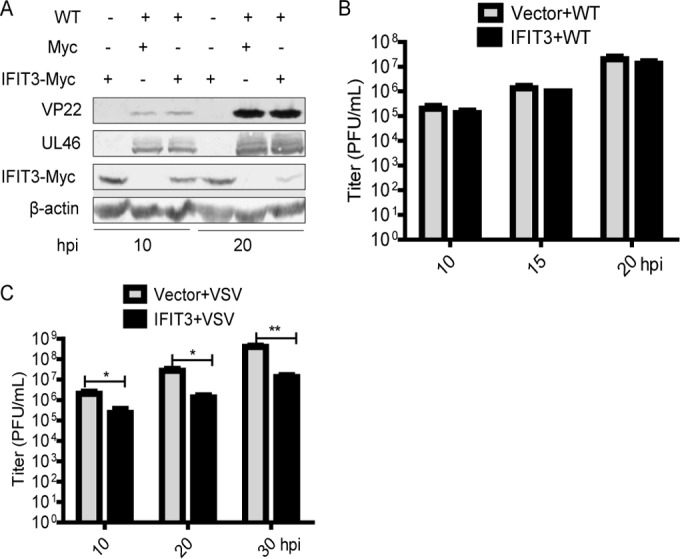
Ectopically expressed IFIT3 had no effect on the replication of WT HSV-1. (A and B) HEK293T cells were transfected with vector or IFIT3-Myc plasmid. At 8 h posttransfection, the cells were infected with WT HSV-1 at an MOI of 0.5, and then cells were harvested at the indicated time points postinfection and subjected to WB analysis with antibodies against UL46, VP22, Myc, or β-actin (A) or viral plaque assay on Vero cells (B). (C) HEK293T cells were transfected with vector or IFIT3-Myc plasmid. At 8 h posttransfection, the cells were infected with VSV at an MOI of 0.2, and then cells were harvested at the indicated time points postinfection and subjected to viral plaque assay on Vero cells. The data represent results from one of the triplicate experiments. Error bars represent mean standard deviations (SD) from three independent experiments. Statistical analysis was performed using Student's t test. *, 0.01< P < 0.05; **, 0.001 < P < 0.01.
To confirm the antiviral activity of IFIT3, viral plaque assay was performed to test the effect of IFIT3 on the replication of VSV. HEK293T cells were transfected with vector or IFIT3-Myc plasmid. At 8 h posttransfection, cells were infected with VSV at an MOI of 0.2. Cells then were harvested at the indicated time points postinfection and subjected to viral plaque assay. As presented in Fig. 1C, ectopic expression of IFIT3 could limit the replication of VSV. Interestingly, we also found that the protein level of IFIT3 was decreased in WT HSV-1-infected cells compared with uninfected cells (Fig. 1A). Taken together, these results showed that ectopically expressed IFIT3 had no effect on the replication of WT HSV-1.
HSV-1 tegument protein UL41 downregulated the expression of IFIT3.
To determine why WT HSV-1 infection could reduce the expression of IFIT3, we hypothesized that at least one of the HSV-1 proteins could abrogate the expression of IFIT3. In our previous and other studies, it has been reported that UL41 specifically degraded both viral and cellular mRNAs containing an AU-rich element (ARE) in its 3′-untranslated regions, such as tetherin, zinc finger antiviral protein, and cig5 (27, 29–32, 41–44). Additionally, ICP0 facilitated degradation of many cellular antiviral proteins, including nuclear factor κB subunit P50 and interferon-inducible protein 16, with the activity of E3 ubiquitin ligase being dependent on the ubiquitin-proteasome pathway (39, 45). In order to verify whether ICP0 or UL41 participated in decreasing the expression of IFIT3, UL41-HA or ICP0-Flag and IFIT3-Myc plasmids were cotransfected into HEK293T cells for 20 h, and then cells were harvested and WB analysis was conducted. As shown in Fig. 2A, HSV-1 tegument protein UL41, not its immediate-early protein, ICP0, downregulated the expression of IFIT3 in a dose-dependent manner.
FIG 2.
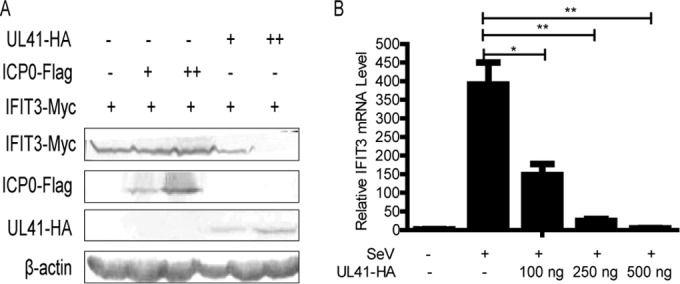
HSV-1 tegument protein UL41 downregulated the expression of IFIT3. (A) HEK293T cells were cotransfected with IFIT3-Myc (800 ng) and UL41-HA or ICP0-Flag plasmid. At 20 h posttransfection, the cells were harvested and subjected to WB analysis with antibodies against Myc, Flag, HA, or β-actin (+, 100 ng; ++, 800 ng). (B) HEK293T cells were transfected with UL41-HA plasmid (100 ng, 250 ng, and 500 ng) for 12 h, and the cells were infected with SeV. At 8 h postinfection, total RNA was extracted for qRT-PCR analysis. The data represent results from one of the triplicate experiments. *, 0.01 < P < 0.05; **, 0.001 < P < 0.01.
UL41 is an endoribonuclease mediating selective degradation of both viral and cellular mRNAs (27, 28, 31). Hence, we speculate that UL41 decreases IFIT3 expression via its RNase activity to degrade IFIT3 mRNA. To confirm this hypothesis, UL41-HA plasmid was transfected into HEK293T cells for 12 h, and cells were infected with SeV. A previous study reported that IFIT3 is not detectable in the absence of stimuli, and we found that SeV infection could significantly induce IFIT3 expression (46). Therefore, in the presence of SeV, cells were harvested at 8 h postinfection and subjected to qRT-PCR analysis. Our data showed that ectopic expression of UL41 downregulated the abundance of IFIT3 mRNA in a dose-dependent manner (Fig. 2B). In conclusion, our results demonstrated that UL41 reduced the expression of IFIT3 via its RNase activity to degrade IFIT3 mRNA.
HSV-1 infection decreased the accumulation of IFIT3 mRNA and reduced the protein expression of IFIT3 via UL41.
To investigate whether IFIT3 mRNA was downregulated during HSV-1 infection, HEK293T cells were either left uninfected or infected with SeV for 8 h and then infected with either WT HSV-1 or R2621 at an MOI of 0.5 for another 16 h. Cells were harvested at the indicated time points and subjected to qRT-PCR (Fig. 3A) and WB analysis (Fig. 3B). As presented in Fig. 3A, we found that in the absence of SeV, infection of WT HSV-1 induced less IFIT3 mRNA (49-fold compared with the mock-infected group) than infection of R2621 (132-fold) (P < 0.001). In addition, in the presence of SeV, we found that coinfection of WT HSV-1 significantly inhibited SeV-induced IFIT3 mRNA levels, while coinfection of R2621 did not. Similarly, WB analysis indicated that compared with R2621, WT HSV-1 significantly inhibited IFIT3 expression (Fig. 3B). Collectively, these results demonstrated that HSV-1 infection decreased the accumulation of IFIT3 mRNA and reduced the protein expression of IFIT3 via UL41.
FIG 3.
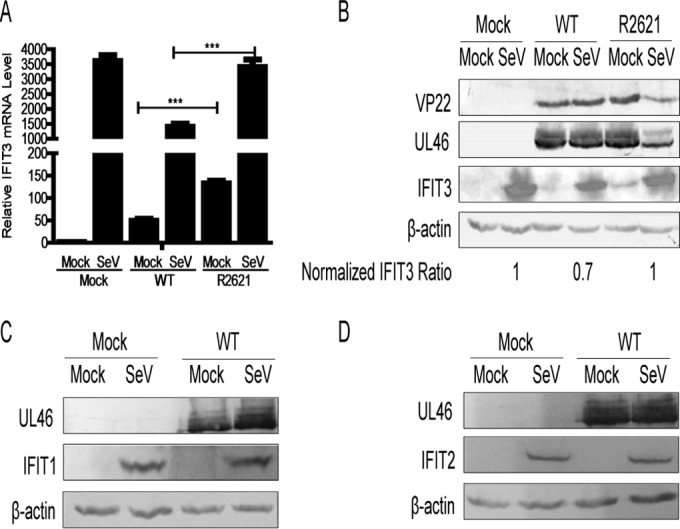
HSV-1 infection decreased the accumulation of IFIT3 mRNA and reduced the protein expression of IFIT3 via UL41. (A) HEK293T cells were infected with SeV for 8 h. At 8 h postinfection, the cells were infected with either WT HSV-1 or R2621 at an MOI of 0.5 for another 16 h. Total RNA then was extracted at the indicated time points for qRT-PCR analysis. (B) HEK293T cells were infected with SeV or HSV-1 as described for panel A, and then the cells were harvested and subjected to WB analysis with antibodies against UL46, VP22, IFIT3, or β-actin. (C and D) HEK293T cells were infected with SeV for 8 h. At 8 h postinfection, the cells were infected with WT HSV-1 at an MOI of 1 for another 16 h, and then cells were harvested at the indicated time points postinfection and subjected to WB analysis with antibodies against UL46, IFIT1, IFIT2, or β-actin. The data represent results from one of the triplicate experiments. ***, P < 0.001.
To demonstrate whether UL41 specifically targets IFIT3, we performed the WB analysis of other IFIT family proteins (IFIT1 and IFIT2). A previous study reported that IFIT1 and IFIT2 are not detectable in the absence of stimuli, and we found that SeV infection could significantly induce the expression of IFIT1 and IFIT2 (47). Therefore, HEK293T cells were infected with SeV. At 8 h postinfection, cells were infected with WT HSV-1 at an MOI of 1 for another 16 h. Cells then were harvested at the indicated time points postinfection and subjected to WB analysis. Our data showed that WT HSV-1 infection failed to affect the expression of IFIT1 and IFIT2 (Fig. 3C and D). Taken together, our data showed that among these IFIT family members, UL41 dampened the antiviral activity of IFIT3 by specifically degrading its mRNA.
The replication of R2621 was inhibited in an IFIT3-dependent manner.
The aforementioned data suggested that HSV-1 evaded the antiviral activity of IFIT3 by UL41 degrading its mRNA, leading us to hypothesize that IFIT3 could restrict R2621 infection. To test this hypothesis, HEK293T cells with ectopic expression of IFIT3 were infected with R2621 and harvested at the indicated time points for WB analysis and viral plaque assay. As shown in Fig. 4A, ectopic expression of IFIT3 could downregulate viral protein UL46 and VP22 levels. Furthermore, viral plaque assay also confirmed that IFIT3 prominently restrained the replication of R2621 (Fig. 4B).
FIG 4.
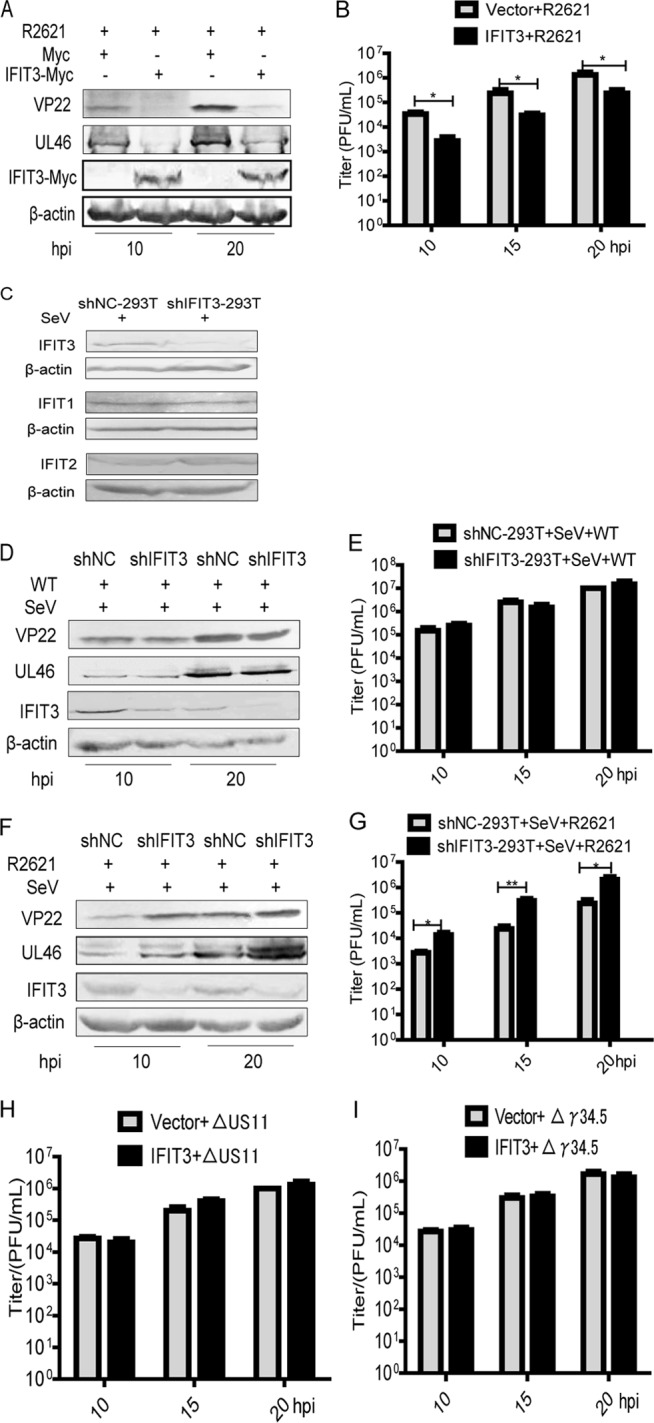
Replication of R2621 was inhibited in an IFIT3-dependent manner. (A and B) HEK293T cells were transfected, infected with R2621 at an MOI of 0.5, and subjected to WB analysis with antibodies against UL46, VP22, Myc, or β-actin (A) or viral plaque assay on Vero cells (B). (C) The stably transfected HEK293T-shNC and HEK293T-shIFIT3 cells were infected with SeV for 20 h. At 20 h postinfection, the cells were harvested and subjected to WB analysis with antibodies against IFIT1, IFIT2, IFIT3, or β-actin. (D, E, F, and G) The stably transfected HEK293T-shNC and HEK293T-shIFIT3 cells were infected with SeV for 8 h. At 8 h postinfection, the cells were infected with WT HSV-1 or R2621 at an MOI of 0.5, harvested at the indicated time points postinfection, and subjected to WB analysis with antibodies against UL46, VP22, IFIT3, or β-actin (D and F) or viral plaque assay on Vero cells (E and G). (H and I) HEK293T cells were transfected and infected with ΔUS11-HSV-1 or Δγ34.5-HSV-1 at an MOI of 0.5 and subjected to viral plaque assay as described for Fig. 1B. The data represent results from one of the triplicate experiments. *, 0.01 < P < 0.05; **, 0.001 < P < 0.01.
To investigate whether endogenous expression of IFIT3 would play an important role in suppression of the replication of R2621, the stably transfected HEK293T-shNC and HEK293T-shIFIT3 cells were constructed to test the effect of IFIT3 on the replication of HSV-1. First, WB analysis was applied to examine the knockdown efficiency of IFIT3 in HEK293T cells (Fig. 4C). As presented in Fig. 4C, we found that compared to HEK293T-shNC cells, the expression of IFIT3 was markedly downregulated in HEK293T-shIFIT3 cells, but the expression of IFIT1 and IFIT2 were not affected under shIFIT3 conditions, indicating that shIFIT3 specifically targeted IFIT3. The stably transfected HEK293T-shNC and HEK293T-shIFIT3 cells then were infected with SeV, and at 8 h postinfection, cells were infected with WT HSV-1 or R2621 at an MOI of 0.5 and harvested at the indicated time points for WB analysis and viral plaque assay. As presented, knockdown of IFIT3 did not affect UL46 and VP22 expression of WT HSV-1 but did increase UL46 and VP22 expression of R2621 (Fig. 4D and F). Similarly, viral plaque assay also demonstrated that knockdown of IFIT3 failed to affect the replication of WT HSV-1 but did facilitate the replication of R2621 (Fig. 4E and G).
To exclude the possibility of the involvement of other late viral proteins in IFIT3 regulation, we chose late protein US11 and γ34.5 as controls. HEK293T cells were transfected with vector or IFIT3-Myc plasmid for 8 h prior to being infected with ΔUS11-HSV-1 or Δγ34.5-HSV-1 at an MOI of 0.5 and harvested at the indicated time points for viral plaque assay. Our results showed that ectopic expression of IFIT3 had no effect on the replication of ΔUS11-HSV-1 or Δγ34.5-HSV-1 (Fig. 4H and I). In short, these results indicated that the replication of R2621 was inhibited in an IFIT3-dependent manner.
DISCUSSION
To control infection by viruses, host cells must recognize invasion and develop a rapid and effective antiviral response (3, 6). In this response, IFN-I, such as IFN-α and IFN-β, are critical to innate immunity against viral infection and the modulation of adaptive immunity through the induction of a wide array of ISGs (2, 48). Over the past decade, it has become clear that IFIT family proteins inhibit viral infection through suppressing translation initiation, binding to uncapped or incompletely capped viral RNA, or sequestering viral proteins or RNA in the cytoplasm (3, 6, 7, 11, 12, 46, 49–51). However, the antiviral activity of individual IFIT family members remains to be defined and corroborated. In this study, we demonstrated for the first time that IFIT3 was a novel target of HSV-1 tegument protein UL41 and revealed a strategy of HSV-1 to evade IFIT3 antiviral activity. Therefore, IFIT3 failed to inhibit the replication of WT HSV-1 but did restrict the replication of R2621. Interestingly, we also found that two other IFIT family proteins, IFIT1 and IFIT2, are not targeted by UL41, as WT HSV-1 infection failed to affect their expression. Therefore, it is worth examining the antiviral activities of other IFIT family proteins upon HSV-1 infection.
In response to IFN-α stimulation, IFIT3 was induced in a variety of tissue cell lines and played a crucial role in IFN-α-mediated innate responses (15, 52). In addition, the subcellular location of IFIT3 affects its function. It has been reported that IFIT3 is mainly located in the cytoplasm and partly located in the mitochondria (3, 7). In the mitochondria, IFIT3 acts as an important adaptor to bridge TNFR-associated factor family member-associated NF-κB activator-binding kinase 1 (TBK1) onto mitochondrial antiviral signaling (MAVS), revealing a new function of IFIT3 in innate immunity (46). In the present study, we found that HSV-1 infection decreased the expression of IFIT3 via its tegument protein, UL41. Whether or not this downregulation of IFIT3 would negatively affect the MAVS-TBK1 signal requires further investigation. It is plausible to consider that HSV-1 efficiently evades a set of host antiviral responses by targeting IFIT3.
HSV-1 is an archetypal member of the alphaherpesvirus subfamily (21). As a double-stranded DNA virus, HSV-1 has evolved multiple mechanisms to evade host innate immunity to establish effective infection. The virion host shutoff protein UL41 is a broad-specificity endoribonuclease that, in vitro, cleaves single-stranded RNA at the 3′ side of U and C residues (28). In vivo, ribosome-associated mRNA is the specific target of UL41 (53). Interacting with the cap-binding initiation factor complex eIF4F, UL41 cleaves mRNA in regions of translation initiation (30, 32, 54). In this study, our results indicated that UL41 could degrade IFIT3 mRNA, which may facilitate the replication of HSV-1.
In summary, we have shown here for the first time that HSV-1 tegument protein UL41 dampened IFIT3 antiviral activity through reducing the accumulation of its mRNA, revealing a strategy by which HSV-1 evades host innate immunity. Findings in this study will help broaden our insight into the mechanisms applied by HSV-1 to dampen host antiviral responses and facilitate its persistence.
ACKNOWLEDGMENTS
We thank Bernard Roizman for R2621 virus and Bin He for Δγ34.5-HSV-1.
Work in the laboratory of C.Z. relevant to this article was supported by grants from the National Natural Science Foundation of China (81371795 and 81571974).
REFERENCES
- 1.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fensterl V, Sen GC. 2009. Interferons and viral infections. Biofactors 35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 3.Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang BX, Fish EN. 2012. The yin and yang of viruses and interferons. Trends Immunol 33:190–197. doi: 10.1016/j.it.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Michal JJ, Zhang L, Ding B, Lunney JK, Liu B, Jiang Z. 2013. Interferon induced IFIT family genes in host antiviral defense. Int J Biol Sci 9:200–208. doi: 10.7150/ijbs.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vladimer GI, Gorna MW, Superti-Furga G. 2014. IFITs: emerging roles as key anti-viral proteins. Front Immunol 5:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbas YM, Pichlmair A, Gorna MW, Superti-Furga G, Nagar B. 2013. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494:60–64. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng C, Zheng Z, Zhang Z, Meng J, Liu Y, Ke X, Hu Q, Wang H. 2015. IFIT5 positively regulates NF-kappaB signaling through synergizing the recruitment of IkappaB kinase (IKK) to TGF-beta-activated kinase 1 (TAK1). Cell Signal 27:2343–2354. doi: 10.1016/j.cellsig.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CUT, Pestova TV. 2013. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp-mRNAs. Nucleic Acids Res 42:3228–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fensterl V, Sen GC. 2015. Interferon-induced Ifit proteins: their role in viral pathogenesis. J Virol 89:2462–2468. doi: 10.1128/JVI.02744-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu YL, Shi SF, Wu WL, Ho LJ, Lai JH. 2013. Protective roles of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) in dengue virus infection of human lung epithelial cells. PLoS One 8:e79518. doi: 10.1371/journal.pone.0079518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Liu J, Bai J, Du Y, Wang X, Liu X, Jiang P. 2013. Poly(I:C) inhibits porcine reproductive and respiratory syndrome virus replication in MARC-145 cells via activation of IFIT3. Antiviral Res 99:197–206. doi: 10.1016/j.antiviral.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Schmeisser H, Mejido J, Balinsky CA, Morrow AN, Clark CR, Zhao T, Zoon KC. 2010. Identification of alpha interferon-induced genes associated with antiviral activity in Daudi cells and characterization of IFIT3 as a novel antiviral gene. J Virol 84:10671–10680. doi: 10.1128/JVI.00818-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiscott J, Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 8:e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajcani J, Durmanova V. 2000. Early expression of herpes simplex virus (HSV) proteins and reactivation of latent infection. Folia Microbiol (Praha) 45:7–28. doi: 10.1007/BF02817445. [DOI] [PubMed] [Google Scholar]
- 18.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res 143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 20.Suazo PA, Ibanez FJ, Retamal-Diaz AR, Paz-Fiblas MV, Bueno SM, Kalergis AM, Gonzalez PA. 2015. Evasion of early antiviral responses by herpes simplex viruses. Mediators Inflamm 2015:593757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Read GS, Frenkel N. 1983. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J Virol 46:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol 78:1063–1068. doi: 10.1128/JVI.78.3.1063-1068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strelow LI, Leib DA. 1995. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J Virol 69:6779–6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotter CR, Kim WK, Nguyen ML, Yount JS, Lopez CB, Blaho JA, Moran TM. 2011. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J Virol 85:12662–12672. doi: 10.1128/JVI.05557-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Svobodova S, Bell S, Crump CM. 2012. Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J Virol 86:473–483. doi: 10.1128/JVI.05981-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pasieka TJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA. 2008. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82:5527–5535. doi: 10.1128/JVI.02047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Everly DN Jr, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J Virol 76:8560–8571. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80:9341–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schek N, Bachenheimer SL. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J Virol 55:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng P, Everly DN Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J Virol 75:10272–10280. doi: 10.1128/JVI.75.21.10272-10280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esclatine A, Taddeo B, Roizman B. 2004. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc Natl Acad Sci U S A 101:18165–18170. doi: 10.1073/pnas.0408272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Read GS. 2013. Virus-encoded endonucleases: expected and novel functions. Wiley Interdiscip Rev RNA 4:693–708. [DOI] [PubMed] [Google Scholar]
- 33.Xing J, Wang S, Lin F, Pan W, Hu CD, Zheng C. 2011. Comprehensive characterization of interaction complexes of herpes simplex virus type 1 ICP22, UL3, UL4, and UL20.5. J Virol 85:1881–1886. doi: 10.1128/JVI.01730-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Wang S, Zhu H, Zheng C. 2011. Cloning of the herpes simplex virus type 1 genome as a novel luciferase-tagged infectious bacterial artificial chromosome. Arch Virol 156:2267–2272. doi: 10.1007/s00705-011-1094-9. [DOI] [PubMed] [Google Scholar]
- 36.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 37.Li M, Wang S, Xing J, Zheng CH. 2011. Antiserum to the recombinant truncated VP22 protein of herpes simplex virus type 1 that also recognizes full-length VP22. Acta Virol 55:69–73. doi: 10.4149/av_2011_01_69. [DOI] [PubMed] [Google Scholar]
- 38.Zhang D, Su C, Zheng C. 2016. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J Virol 90:5824–5829. doi: 10.1128/JVI.00186-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Wang K, Wang S, Zheng C. 2013. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J Virol 87:12935–12948. doi: 10.1128/JVI.01952-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL. 2011. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol 85:11079–11089. doi: 10.1128/JVI.05098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zenner HL, Mauricio R, Banting G, Crump CM. 2013. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J Virol 87:13115–13123. doi: 10.1128/JVI.02167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su C, Zhang J, Zheng C. 2015. Herpes simplex virus 1 UL41 protein abrogates the antiviral activity of hZAP by degrading its mRNA. Virol J 12:203. doi: 10.1186/s12985-015-0433-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esclatine A, Taddeo B, Evans L, Roizman B. 2004. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc Natl Acad Sci U S A 101:3603–3608. doi: 10.1073/pnas.0400354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen G, Wang K, Wang S, Cai M, Li ML, Zheng C. 2014. Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J Virol 88:12163–12166. doi: 10.1128/JVI.01380-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson KE, Chikoti L, Chandran B. 2013. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87:5005–5018. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu XY, Chen W, Wei B, Shan YF, Wang C. 2011. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol 187:2559–2568. doi: 10.4049/jimmunol.1100963. [DOI] [PubMed] [Google Scholar]
- 47.Terenzi F, Hui DJ, Merrick WC, Sen GC. 2006. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J Biol Chem 281:34064–34071. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- 48.Takaoka A, Yanai H. 2006. Interferon signalling network in innate defence. Cell Microbiol 8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 49.Saikia P, Fensterl V, Sen GC. 2010. The inhibitory action of P56 on select functions of E1 mediates interferon's effect on human papillomavirus DNA replication. J Virol 84:13036–13039. doi: 10.1128/JVI.01194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reich NC. 2013. A death-promoting role for ISG54/IFIT2. J Interferon Cytokine Res 33:199–205. doi: 10.1089/jir.2012.0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang B, Liu X, Chen W, Chen L. 2013. IFIT5 potentiates anti-viral response through enhancing innate immune signaling pathways. Acta Biochim Biophys Sin (Shanghai) 45:867–874. doi: 10.1093/abbs/gmt088. [DOI] [PubMed] [Google Scholar]
- 52.Huang X, Yang N, Ou X, Li D, Wang Z, Xie Q, Chen Y, Lin H, Yin G, Wen F. 2008. Sequential activation of protein kinase C delta and JNK is required for interferon-alpha-induced expression of IFIT4. Cell Signal 20:112–119. doi: 10.1016/j.cellsig.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 53.Shiflett LA, Read GS. 2013. mRNA decay during herpes simplex virus (HSV) infections: mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J Virol 87:94–109. doi: 10.1128/JVI.01557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Page HG, Read GS. 2010. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J Virol 84:6886–6890. doi: 10.1128/JVI.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]