

Abstract
Introduction
Xeroderma pigmentosum is a rare autosomal recessive genetic disease. This disease predisposes patients to early-onset skin cancers, particularly squamous cell carcinoma. Here, we report 3 pediatric cases, including 2 deaths.
Observation
The subjects included 2 boys and 1 girl with skin type VI. All subjects were from consanguineous marriages, and the average age was 7.6 years. The patients all had ulcerative budding tumor lesions in the cephalic region, and the mean disease duration was 18 months. In all 3 cases, the diagnosis of xeroderma pigmentosum was made before the poikilodermal appearance of sun-exposed areas and photophobia. Neurological-type mental retardation was noted in 1 case. Histology confirmed squamous cell carcinoma in all 3 cases. The evolutions were marked by the death of 2 children (cases 1 and 3). In one case, the outcome was favorable following cancer excision and subsequent chemotherapy with adjuvant radiotherapy.
Conclusion
Squamous cell carcinoma is a serious complication related to xeroderma pigmentosum in Sub-Saharan Africa. Prevention is based on the early diagnosis of xeroderma pigmentosum, black skin photoprotection, screening and early treatment of lesions, and genetic counseling.
Keywords: Xeroderma pigmentosum, Squamous cell carcinoma, Children
Introduction
Xeroderma pigmentosum (XP) is a precancerous genodermatosis elicited by DNA repair defects. XP is a rare condition that is inherited as an autosomal recessive trait. XP is clinically characterized by extreme skin photosensitivity, pigmentary anomalies of skin that is exposed to sunlight, and ophthalmic and neurological damage [1, 2]. The prevalence of XP is relatively high in the Middle East and Japan. [1] In Africa, a few cases have been previously reported [3, 4]. In this region, the more intense sun exposure predisposes people to early onset of squamous cell carcinoma despite pigmentosa protection [1, 5]. The occurrence of these cancers is related to a defect in DNA repair. We report 3 cases of very advanced early-onset squamous cell carcinomas among black children with XP.
Results
Case 1
A 7-year-old phototype VI boy was admitted for a burgeoning swelling of the lower lip lasting for 3 years. The examination revealed consanguinity of first-degree relatives. A dermatological examination revealed an ulcerative budding swelling of the lower lip (Fig 1) accompanied by macro-poly tumor lymph nodes under the chin and under the angulo-maxillary glands. The diagnosis of XP was made based on the poikilodermal appearance of the sun-exposed areas. Ulcerative crusted lesions on the scalp were also noted. Moreover, ophthalmological examination revealed photophobia with decreased visual acuity. Examinations of the anterior segment and fundus were normal. Mental retardation was present. A pathological examination of a fragment of the tumor confirmed squamous cell carcinoma. The child was stage T3N1M0. The evolution was marked by the death of the child in a Pediatric Oncology Department after 6 months of hospice care. Genetic counseling was provided to the parents.
Fig. 1.
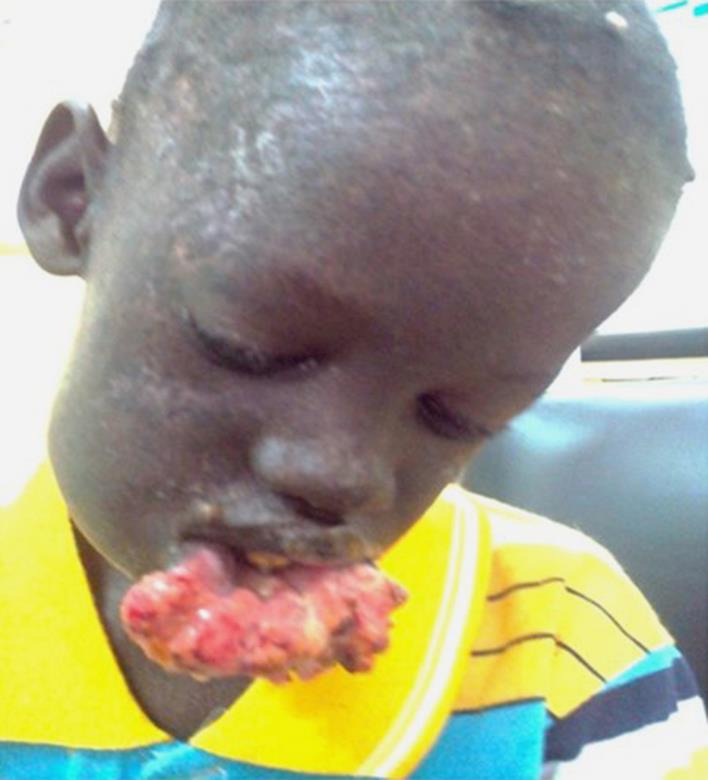
Ulcerative budding swelling of the lower lip lasting for 3 years in a 7-year-old child (case 1, Dakar, Senegal).
Case 2
An 8-year-old phototype VI boy who was also born from first-degree blood relatives was admitted for an ulceration of the scalp that evolved over 1 year (Fig 2). This ulceration was 4 cm in diameter and had smooth edges, a clean red surface, bled on contact and was located in the right temporal-parietal region. A tumor located behind the right ear revealed lymphadenopathy. The child exhibited pigmentation disorders in areas that were exposed to the sun, and the condition was eased by keeping these areas covered. He also exhibited photophobia. Ophthalmological examination revealed corneal scarring and a cataract in the left eye. His psychomotor development was normal. The diagnosis of squamous cell carcinoma was confirmed upon pathological examination. The child was classified as stage T2N1M0. X-rays of the skull and chest were normal. The child underwent a complete surgical resection, and clinical margins greater than 6 mm were achieved. Histological examination of the removed tissue was performed. He also received single adjuvant chemoradiation therapy (radiotherapy and cisplatin) 2 weeks after the intervention. Healing was achieved after 3 months. The outcome was favorable with a decline after 1 year. Wound healing was complete, and there was no lymphadenopathy. Genetic counseling was provided to the parents. Surveillance was set for twice per year for 5 years. The patient was lost to follow-up at the end of 1 year.
Fig. 2.

Swelling, ulcerative budding and crusty lesion in the right temporal region developed on the poikilodermal skin of the scalp and face in an 8-year-old child (case 2, Abidjan, Côte d'Ivoire).
Case 3
An 8-year-old, phototype VI girl was admitted for multiple ulcerative budding tumors evolving over 12 months (Fig 3). She came from a family with 4 children and parental consanguinity in the 2nd degree. She and her sister were followed up for 4 years for XP. Dermatological examination revealed countless ulcerative budding tumor lesions of variable size (1–6 cm) on the midface region and scalp with extension into the left eye. Furthermore, there were dyschromic diffuse blotches, macro-poly-tumoral lymphadenopathies in the left maxillary area and an impaired general condition. Ophthalmological examination revealed a purulent lesion in the right eye, and the left eye was inaccessible due to eyelid tumors. Neurological examination was normal. The diagnosis of squamous cell carcinoma was histologically confirmed (Fig 4). Radiological examinations (skull and thorax) were normal. The child was stage T2N1M0. In a multidisciplinary meeting (pediatric oncology, ophthalmology, and dermatology), the decision for neoadjuvant chemotherapy treatment for tumor mass reduction followed by oncological resection surgery was made. The evolution was marked by the death of the child in the Pediatric Oncology Department after her first chemotherapy treatment. Genetic counseling was provided to the parents.
Fig. 3.

Multiple ulcerative budding tumors in the midface region with an extension into the left eye and other tumors of the scalp in an 8-year-old girl (case 3, Dakar, Senegal).
Fig. 4.

Hyperplasia of the squamous epithelium formed from atypical cells with hyperkeratosis and dermal infiltration of tumor cells (magnification, ×40).
Discussion
We report 3 cases of squamous cell carcinoma in phototype VI, black African children with XP [6]. The patients were also remarkable for their advanced stages, which were linked to long delays in their diagnoses and severe prognoses. The limitation of our series is the absence of a genetic study.
XP is rare in black Africans. A few cases have been reported [3, 4]. XP is far more common in the Middle East due to consanguineous marriages [1]. Consanguinity was found in our 3 observations, and this is common in West Africa. In black subjects, squamous cell carcinomas most often occur on genodermatoses, and among these squamous cell carcinomas, we distinguish albinism, verruciform epidermodysplasia, and XP. The peculiarity observed in our patients was the appearance of multiple de novo carcinomas (3 clinical cases) in addition to the time at onset of the carcinomas and the poor prognoses. Cutaneous neoplasms are found in at least 45% of XP cases. The average age at onset of these tumors is 8 years according to Kraemer and Lee [1]. This closely matched the average age in our series. The clinical diagnosis of XP in its classic form in light-skinned individuals is easy. In our resource-limited countries, the population generally first uses traditional medicine. In addition to this problem, the inaccessibility of health facilities and particularly the scarcity of qualified specialists result in delays to diagnosis. The latter factor can also be attributed to the limited number of dermatology specialists and the low socioeconomic level. The low socioeconomic level was responsible for the delay of treatment in clinical case 3, which had a considerable influence on the prognosis.
In our series, the occurrence of the carcinomas was early, and a similar situation occurred in the study of Beogo et al. [5]. The occurrence of squamous cell carcinoma is primarily linked to ultraviolet light exposure and strongly increased by the genetic abnormality XP [7, 8]. The frequencies of skin cancers in this region (i.e., squamous cell carcinoma, 3%; basal cell carcinoma, 2%; and melanoma, 1%) are 4,800-fold greater than in a control group from the United States [1]. Cancers occur on the most exposed areas, including the face, head, and neck in 97% of cases. Squamous cell carcinomas cause metastases in 4% of cases [1]. The staging was not completed in our patients due to the lack of resources.
In our series, the children had eye problems. Corneal abnormalities are found in 17% of cases and lead to blindness in 12% of patients [1]. Corneal abnormalities are also a marker of the absence of picture protection as determined in a Comoros XP population. In this population, Cartault et al. [9] found a new mutation.
The early deaths in our series were due to late diagnoses. The clinical management of XP includes total avoidance of ultraviolet light and early excision of skin lesions with monitoring of vitamin D status. For neurological and ophthalmological manifestations, multidisciplinary support is needed. In all cases, it is necessary to increase the awareness of the population and increase staff training regarding these rare diseases with prognoses that may be improved if the condition is detected and managed early.
Conclusion
Squamous cell carcinoma is a serious complication of the evolution of XP. The peculiarities of our observations lie in the early occurrences in Blacks, diagnostic delays, and severe prognoses of the patients. Better detection and better prevention are needed.
Statement of Ethics
The authors have no ethical conflict to disclose.
Disclosure Statement
The authors declare no conflict of interest.
References
- 1.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, neurologic, abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 2.Pandey A, Kuldeep K, Koul A, Tyagi M, Singh P, Sharma P, Dimri D. Xeroderma pigmentosa with ocular association: case report. Case Rep Clin Med. 2013;2:466–469. [Google Scholar]
- 3.Dieng MT, Niang SO, Dangou JM, Ndiaye B. Xeroderma pigmentosum: about 6 cases observed in Dakar. Bull Cancer. 2001;88:199–202. [PubMed] [Google Scholar]
- 4.Chidzonga MM, Mahomva L, Makunike-Mutasa R, Masanganise R. Xeroderma pigmentosum. A retrospective case series in Zimbabwe. J Oral Maxillofac Surg. 2009;67:22–31. doi: 10.1016/j.joms.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 5.Beogo R, Andonaba JB, Boulétreau P, Traore Sawadogo H, Traore A. Xeroderma pigmentosum revealed by multiple squamous cell carcinoma of the face in a child. Rev Stomatol Maxillofacial Surg. 2012;113:50–52. doi: 10.1016/j.stomax.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Pathak MA. In memory of Thomas Bernhard Fitzpatrick. J Invest Dermatol. 2004;122:20–21. doi: 10.1046/j.1523-1747.2003.22248.x. [DOI] [PubMed] [Google Scholar]
- 7.Copeland NE, Hanka CW, Michalak JA. The molecular basis of xeroderma pigmentosum. Dermatol Surg. 1997;23:447–453. doi: 10.1111/j.1524-4725.1997.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 8.Nikola AB, JB Natalie, Katie AA, Katherine JB, Rodney JU. Xeroderma pigmentosum complementation groups using gene expression profiling after-UV-light exposure. Int J Mol Sci. 2015;16:15985–15996. doi: 10.3390/ijms160715985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cartault F, C Nava, Malbrunot AC, et al. A new XPC gene splicing mutation has led to the highest worldwide prevalence of xeroderma pigmentosum in black Mahori patients. DNA Repair (Amst) 2011;10:577–585. doi: 10.1016/j.dnarep.2011.03.005. [DOI] [PubMed] [Google Scholar]