

Abstract
Hydroxyurea is a well-established inhibitor of ribonucleotide reductase that has a long history of scientific interest and clinical use for the treatment of neoplastic and non-neoplastic diseases. It is currently the staple drug for the management of sickle cell anemia and chronic myeloproliferative disorders. Due to its reversible inhibitory effect on DNA replication in various organisms, hydroxyurea is also commonly used in laboratories for cell cycle synchronization or generating replication stress. However, incubation with high concentrations or prolonged treatment with low doses of hydroxyurea can result in cell death and the DNA damage generated at arrested replication forks is generally believed to be the direct cause. Recent studies in multiple model organisms have shown that oxidative stress and several other mechanisms may contribute to the majority of the cytotoxic effect of hydroxyurea. This review aims to summarize the progress in our understanding of the cell-killing mechanisms of hydroxyurea, which may provide new insights towards the improvement of chemotherapies that employ this agent.
Keywords: hydroxyurea, ribonucleotide reductase, oxidative stress, cytokinesis arrest, DNA replication checkpoint, cell cycle
1. Introduction
Hydroxyurea (HU, also called hydroxycarbamide, see Figure 1) is a non-alkylating antineoplastic and antiviral agent that has been used for a variety of conditions in the disciplines of hematology, oncology, infectious disease and dermatology. It was first synthesized over a century ago in 1869 [1], but it was not until ~60 years later in 1928 that the biological effects of this simple antimetabolite compound on blood cells in rabbits were reported [2]. A large-scale drug screen carried out in the 1960s showed that it has anti-tumor activities, which revived interest in HU as a potential antineoplastic drug [3,4]. Subsequent studies showed that it could be used to treat several types of solid tumors and myeloproliferative disorders. The therapeutic spectrum for HU was also expanded to include various infectious diseases such as the human immunodeficiency virus [5,6,7,8,9]. Some earlier studies also reported that HU could be successfully used for the treatment of psoriasis, particularly in cases that have not responded to other treatment [10,11]. Although newer and more efficient agents have replaced HU in certain instances, as an established, reliable and well-tolerable small molecule drug for multiple neoplastic and non-neoplastic diseases, it is still being used in clinics. Currently, it serves as the staple drug for the treatment of sickle cell anemia and chronic myeloproliferative disorders [12,13] and is listed as an “essential medicine” by the World Health Organization [14].
Figure 1.
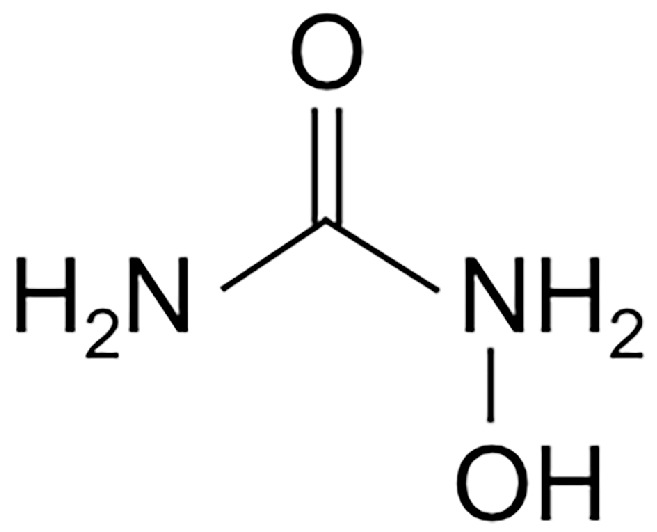
Hydroxyurea (HU).
HU is an inhibitor of DNA synthesis in many organisms and in cell culture systems [15,16]. As a result, HU is mainly active in the S-phase of the cell cycle and because of the easy reversibility of its action, HU has been commonly used in laboratories as a synchronizing agent in cell cultures. HU has been shown to induce chromosome damage in various organisms and is also cytotoxic depending on the concentration that is used, the duration of exposure, and the sensitivity of the cell lines [17]. HU can also cross the placenta and is teratogenic in animals [17,18]. Thus, the DNA damage such as the strand breaks caused by inhibition of DNA synthesis is generally believed to be responsible for its cytotoxicity, the anti-neoplastic activity and the teratogenic effects. However, the reversible effect of HU on DNA replication suggests that it is a cytostatic agent, and, in addition to the DNA damage, its cytotoxic effects may involve a more complex mechanism. Furthermore, the genetic backgrounds in most of the mammalian cell lines that were used in earlier studies are unknown, and thus the previously reported cytotoxic effects and the underlying mechanisms need to be reconsidered more carefully. Recent studies in several model organisms with defined genetic backgrounds showed that HU also generates oxidative stress and induces cytokinesis arrest in certain mutant cells [19,20,21,22,23,24], which likely contributes to the majority of the cell-killing and thus the therapeutic effects. The benefits of HU for the treatment of sickle cell anemia are likely the increased production of fetal hemoglobin via nitric oxide production [25] and the decreased adherence of red blood cells to vascular endothelial cells [26]. The effectiveness for management of refractory psoriasis is likely due to its inhibitory effect on epithelial proliferation, which restores the patients’ thickened epidermis to a more normal appearance [10]. Interestingly, endogenous HU has been found in the plasma and various tissues of many animal groups [27], including humans [28], which is likely produced by arginase from the intermediate of nitric oxide synthesis pathway hydroxyarginine. Because the concentrations of endogenous HU vary by as much as 25 folds between tissues, and the concentrations in certain types of tissues are high enough to be effective against bacterial or viral infections, HU could also act as a natural defense agent. However, the exact function of the endogenous HU remains largely unknown. In the following, we will briefly review the action of HU on its primary target ribonucleotide reductase (RNR) and then summarize the recent research progress on the cell-killing mechanisms of this clinically important drug.
2. Inhibition of RNR and Other Potential Metalloenzymes
RNR is the well-established primary cellular target of HU [29,30]. This enzyme catalyzes the reduction of ribonucleoside diphosphates to their corresponding deoxyribonucleotides as the precursors for DNA replication and repair. RNRs are unique enzymes in that they all require a protein thiyl radical for catalysis. There are three classes of RNRs, which employ different mechanisms for the generation of the protein thiyl radical. Class I RNRs exist in mammals, plants, yeasts and prokaryotes. They contain two dissociable dimeric subunits termed R1 and R2 and require oxygen for the generation of a stable tyrosyl radical by a di-iron center in the smaller R2 subunit. During catalysis, the tyrosyl radical is continuously shuttled to a cysteine residue in the larger R1 subunit and generates the thiyl protein radical required for activation of the substrate [31]. Computer modeling showed that this path of radical transfer is ~35 Å long in class I reductases [32]. In class Ia RNRs, the redox-active cysteines of thioredoxin or glutaredoxin are the electron donors [15,30,33]. In addition to the catalytic site, the R1 subunit also contains allosteric sites for the regulation of RNR activity and specificity. Due to the allosteric regulation, all RNRs can provide an appropriate balance of the four deoxyribonucleotide triphosphate (dNTP) precursors for DNA synthesis [34]. Because of the essential function in DNA replication and repair, RNR is also highly regulated during the cell cycle and in response to DNA damage or perturbed DNA replication via multiple mechanisms [35,36,37,38].
HU inhibits RNR by directly reducing the diferric tyrosyl radical center in the smaller R2 subunit via one-electron transfer from the drug [16,29,39,40]. Since urea does not have such an effect [29], the –NH2-OH moiety of HU is the minimal structural requirement for the inhibitory effect. This conclusion is also supported by structure activity relationship studies [41,42,43]. Because the free radical catalysis mechanism is conserved among different RNRs from prokaryotes to higher eukaryotes, including mammals, HU has been proved to be active in many organisms. Free radicals are generally very reactive and short-lived. Therefore, few proteins utilize free radical chemistry. RNRs are remarkable in that they accomplish the catalysis through a complex radical storage and a long-range radical transfer mechanism. The tyrosyl radical in the R2 subunit is relatively stable. For example, the radical in Escherichia coli R2 can last for days at room temperature, although the same radical in mouse R2 needs to be continuously regenerated [44,45]. One explanation for the stability is that the tyrosyl radical is buried deep inside the protein. The three-dimensional structure of E. coli R2 protein showed that the radical is located more than 10Å from the closest surface within a hydrophobic pocket, an environment that is absolutely required for radical storage [46,47]. Because the crystal structure of a tetrameric RNR holoenzyme containing both R1 and R2 subunits has not been solved yet, the exact mechanism by which HU scavenges the tyrosyl radical and thus inhibits RNRs remains unclear. Since HU is a relatively small and simple molecule, it may penetrate into the R2 protein via small channels and directly access the tyrosyl radical [43,48]. Alternatively, HU scavenges the radical from the surface of RNR via a long-range electron transfer [44,48]. Since several bulkier and structurally unrelated compounds such as guanazole, pyrazoloimidazole (IMPY) and resveratrol [49,50] can also scavenge the tyrosyl radical, it is more likely that the radical is quenched via the long-range electron transfer mechanism. Kinetic studies of the HU scavenging reaction using purified E. coli R2 also support this mechanism [40,51]. Since the regulatory state of RNRs affects the radical stability and the radical in an active RNR holoenzyme is less stable in the presence of HU [48,51], HU may also exploit alternative sites along the electron-transfer path between the tyrosyl radical and the catalytic site on R1 through either direct or indirect access [48].
In addition to RNR, it has been reported that HU can target catalase in plant cells in vivo (see Table 1) [52]. HU can also suppress several other metalloenzymes in vitro such as carbonic anhydrase and matrix metalloproteases [53,54,55,56]. Because suppression of these metal enzymes occurs only in the presence of high concentrations of HU, whether HU targets these enzymes in vivo, particularly in the mutant cells with defects that can synergize with this HU effect, remains to be seen (see below).
Table 1.
List of potentially new targets of hydroxyurea (HU) that have been discovered recently.
| Potential Targets | Discovery Methods | Organisms | Biological Functions | Ref. |
|---|---|---|---|---|
| Catalase | Genetics | A. thaliana | Decomposition of H2O2 | [52] |
| Carbonic anhydrase | in vitro | ? | Interconversion of CO2 and H2O to H2CO3 | [53] |
| Matrix metalloproteinases | in vitro | ? | Cleavage of the peptide bond | [54] |
| Unknown yet | Genetics | S. pombe | Cytokinesis | [21] |
3. S Phase Arrest, DNA Damage and the Checkpoint Response
Because RNR catalyzes the rate-limiting step in the biosynthesis of all four precursors for DNA replication, its activity is tightly regulated during the cell cycle, which generates a periodic fluctuation of the dNTP concentration in proliferating cells. As mentioned above, the enzyme’s allosteric specificity regulation controls the balanced concentrations of dNTPs. In mammalian cells, RNR activity in the G0/G1 phase is suppressed by transcriptional repression of the R2 gene and by anaphase-promoting complex Cdh1-dependent degradation of the R2 subunit in the M phase [57,58]. The enzyme activity and R1 and R2 mRNAs reach maximal levels during S phase [59,60,61]. The R1 subunit has a long half-life of ~18–24 h, and its protein levels are relatively constant and in excess throughout the cell cycle. The R2 protein has a shorter half-life of ~3–4 h and is specifically expressed during the S phase [60,61]. In addition to the conserved transcriptional repression mechanism, the RNR activity is also controlled by a small inhibitor protein in yeasts (Sml1 in Saccharomyces cerevisiae or Spd1 in Schizosaccharomyces pombe), that binds to RNR in the G1 phase [62,63]. The small inhibitor proteins are degraded upon entry into S phase or in response to DNA damage. Another regulation of RNR is achieved by differential cellular localization of its subunits during the cell cycle and after DNA damage or S phase arrest, and this regulation mechanism appears to be conserved among eukaryotic organisms [63,64,65,66,67]. In bacteria, the transcriptional regulation of RNR activity also plays a critical role during the cell division or under various growth conditions [68,69].
In the presence of HU, proliferating cells are arrested in S phase due to the decreased levels of dNTPs, which slows the DNA polymerase movement at replication forks. In eukaryotes, slowed forks activate the replication checkpoint, a highly conserved intracellular signaling pathway that is crucial for the maintenance of genome stability under replication stress [70,71]. The activated checkpoint stimulates RNR activity by increasing the production of R2, removing the small inhibitor proteins, and regulating the subcellular localization of R2. The activated checkpoint also delays mitosis, suppresses the firing of late origins, and stabilizes the slowed replication forks against collapse so that normal DNA synthesis can properly resume when the HU effect diminishes [72,73,74]. Without the checkpoint protection, the HU-treated forks are unstable and may undergo catastrophic collapse. Collapsed forks generate strand breaks and oxidative stress [22,75], which is generally believed to be the direct cause of cell death. Since the activated checkpoint delays cell division, mitotic catastrophe of the HU-treated cells lacking a functional checkpoint is likely another cause of the cell death [76,77]. Therefore, checkpoint mutants are highly sensitive to HU. However, cells with an intact checkpoint response are relatively insensitive to HU and the HU-induced S phase arrest is generally reversible in wild type cells after the drug removal [72,78].
Due to the reversible S phase arrest, HU is generally considered to be cytostatic, particularly to non-cycling cells [7,78,79,80]. However, earlier studies showed that at high concentrations or with prolonged exposure at lower doses, HU is cytotoxic to various mammalian cells such as Chinese hamster cells, mouse lymphoma cells, Ehrlich ascites tumor cells, and human lymphocytes [79,81,82,83,84], although HeLa and A549 lung carcinoma cells appear to be less sensitive [85,86]. Cytotoxicity after HU administration has also been found in rat and mouse proliferating tissues and embryos [87,88,89,90]. At high concentrations (more than 10 mM), HU is also cytotoxic to E. coli. Earlier studies showed that the cytotoxic effect of HU in both mammalian cells and E. coli appears to be linked to the accumulation of DNA strand breaks in HU-treated cells [91,92] or caused directly by reactive intermediates of HU that are generated in prolonged incubation [93,94,95,96]. A more recent report showed that, in vitro, HU can directly cause Cu(II)-mediated DNA damage particularly at thymine and cytosine residues, probably via the formation of H2O2 and nitric oxide [97]. However, whether HU induces DNA damage by itself or via its reactive derivatives in vivo remains unknown. Furthermore, since the checkpoint and the recently found sterol or heme biosynthesis mutants in S. pombe are highly sensitive to HU (see below) and the genetic backgrounds of the cell lines used in the earlier studies are unknown, the linkage between the DNA damage and the cell killing effect of HU may need to be reconsidered with caution.
4. Accumulation of Reactive Oxygen Species (ROS)
ROS is a collective term used to describe ions and free radicals containing derivatives of molecular oxygen that are more reactive than oxygen itself. The ROS formed inside living cells commonly includes superoxide anion, hydrogen peroxide, and hydroxyl radical [98]. The normal process of respiration in mitochondria is a major source of endogenous ROS. Production of ROS is enhanced when mitochondrial function is perturbed or when the cells are under stress conditions. Accumulation of large amounts of ROS, particularly the deleterious hydroxyl radical, causes extensive oxidation of macromolecules, which directly contributes to cell killing.
To explain the mechanisms of rapid cell killing in the S phase and the teratogenic effect of HU, DeSesso hypothesized in 1979 that HU may exert its cytotoxic effects through radical chain reactions initiated by its hydroxylamine group, and predicted that antioxidants should ameliorate the cytotoxic and teratogenic effects of HU [18]. Subsequent studies by his group and other labs showed that radical scavengers substantially ameliorated the cytotoxic and teratogenic effects of HU [83,99,100]. These earlier studies suggest that accumulation of ROS might be involved in the cell-killing process of HU. More recent studies in E. coli using systems-level analyses have revealed the genomic and physiological effects of HU treatment that lead to cell death [19,20]. It was found that during the initial stage of HU treatment, several cell survival responses are activated, including upregulation of the SOS response, downregulation of cell division inhibition, and induction of the synthesis of RNR and the primosome components at the forks. As the HU treatment continues, the toxin modules MazF and RelE are activated, which trigger membrane stress and a cascade of events that eventually lead to the production of highly reactive hydroxyl radicals [101]. Production of hydroxyl radical is exacerbated by increased iron uptake, which promotes hydroxyl radical formation via Fenton chemistry [102]. An accumulation of harmful amounts of ROS is believed to contribute to the majority of HU-mediated cell death in E. coli [20,98]. Consistent with this notion, addition of the hydroxyl radical scavenger thiourea to the medium, suppresses HU sensitivity, and depletion of AphC, a component of the major scavenger enzyme of endogenous H2O2 alkyl hydroperoxide reductase [103], enhances HU sensitivity. Furthermore, deletion of genes involved in respiration and energy production, which decreases endogenous ROS production, confers resistance to HU [20]. Interestingly, elevated ROS levels and the resulting oxidation of guanine nucleotide pool has been shown to be a common mechanism that underlies cell death induced by all three major classes of bactericidal antibiotics [104,105].
Wild type yeasts such as S. cerevisiae and S. pombe are relatively insensitive to HU. However, recent studies suggest that HU treatment may generate ROS in both species. In addition to the DNA damage and environmental stress responses, HU treatment activates the Yap and Aft regulons in S. cerevisiae that function in redox and iron homeostasis respectively [24,106,107]. As a result, depletion of Yap1 moderately sensitizes the cells to HU, suggesting that ROS may be increased at an intermediate level or redundant factors exist in S. cerevisiae [24,106,107,108]. The Yap1 homologous protein in S. pombe is Pap1. Similar to that in S. cerevisiae, depletion of Pap1 also moderately sensitized S. pombe to HU [109], suggesting that HU treatment may generate oxidative stress in various eukaryotic organisms. Interestingly, overexpression or increased nuclear accumulation of Pap1 also confers the resistance on S. pombe to various other agents such as staurosporine [110], caffeine [111], and berefeldin A [112] and to DNA damage in checkpoint deficient mutants [113]. Scavenging the tyrosyl radical in RNR may also generate the hydroperoxy radical form of HU [17], which diffuses away and directly or indirectly modifies Yap1, leading to its accumulation inside the nucleus and transcriptional activation of genes involved in the redox response [106,114]. The activated Aft regulon promotes iron uptake, which may exacerbate the oxidative stress via Fenton reaction [19,115]. Consistent with these possibilities, overexpression of Yap1 can suppress the HU sensitivity caused by mutations in iron binding proteins such as Apd1 [108]. Apd1 is a thioredoxin-like ferredoxin protein. Mutation of the iron binding pocket or loss of Apd1 moderately sensitizes the cells to HU and the sensitivity can be rescued by antioxidant N-acetyl-cysteine [108].
The ROS generated by HU treatment can also alter the functions of proteins that contain iron–sulfur centers. For example, Dre2-Tah18 protein complex functions in cytosolic iron–sulfur protein biogenesis [116,117] and RNR metallocofactor assembly [118,119]. Mutation in Tah18 sensitizes S. cerevisiae to chronic treatment with HU [118,120] and overexpression of Yap1 can suppress the HU sensitivity caused by the Tah18 mutation [23]. Similar to that in S. cerevisiae, E. coli cells devoid of YfaE protein, which contains an iron–sulfur cluster and is required for the diferric tyrosyl radical cofactor maintenance of RNR, are also sensitive to HU [20,121].
Together, these studies show that HU may kill the cells by affecting the iron–sulfur clusters in proteins that function in the maintenance of the diferric tyrosyl radical center in RNRs or other cellular processes. Without a proper maintenance of diferric tyrosyl radical center in RNRs, the radical may leak into the cytoplasm and generate superoxide [20]. Interestingly, because iron–sulfur centers are sensitive to oxidative agents [122,123,124] and several eukaryotic replication proteins such as primase and Pol3 are known to contain iron–sulfur clusters [116,125,126], it is possible that oxidative stress generated by HU may directly suppress DNA replication. Although this mechanism of HU on DNA replication needs further investigation, it may provide an explanation to the replication arrest in the presence of basal dNTP levels that have been observed in HU-treated cells [23]. A recent study showed that HU could also trigger the accumulation of ROS in plant cells [127], suggesting that it is likely that this cell-killing mechanism of HU is highly conserved.
We have recently found in S. pombe that, similar to a previous study [22], the levels of ROS are only slightly increased in HU-treated wild type cells. However, in a hem13 mutant, in which the heme level is low due to the hypomorphic mutation of the enzyme coproporphyrinogen III oxidase in the heme biosynthesis pathway, the levels of ROS as well as protein carbonylation, an indicator of the oxidation of various macromolecules, were significantly increased in HU-treated cells (our unpublished data). Unlike the checkpoint mutants that usually die within 2 to 3 h or one cell cycle time in HU, the hem13 mutant is highly sensitive only to chronic treatment of HU. Similar chronic HU sensitivity was also observed in S. cerevisiae lacking Sod1, the enzyme that catalyzes the decomposition of superoxide [128], which suggests that the cell killing caused by HU-induced oxidative stress is a slow process. Furthermore, the HU sensitivity of the hem13 mutant can be suppressed by culturing the cells under anaerobic conditions, which inhibits aerobiosis and thus decreases the production of endogenous ROS. Like the S. cerevisiae cells lacking Sod1, increased RNR activity cannot rescue the HU sensitivity of the hem13 mutant, which is consistent with the notion that the hem13 mutant is killed by a mechanism that is unrelated to dNTP depletion.
5. Cytokinesis Arrest and the Potentially Unidentified Cellular Target(s) of HU
While screening for new mutants in S. pombe that are sensitive to replication stress, we identified a new hypomorphic mutation, erg11-1, that dramatically sensitizes the cells to chronic, but not acute treatment with HU [21] (Figure 2). The gene product of erg11 is the enzyme sterol-14α-demethylase, which is required for ergosterol biosynthesis and a major target of antifungal agents. We found that, unlike wild type cells that are arrested in S phase, HU arrests the mutant cells mainly in cytokinesis. The HU-induced cytokinesis arrest is relatively stable and occurs at low doses of HU, which likely explains the remarkable HU sensitivity. HU hypersensitivity has also been observed in several erg mutants in S. cerevisiae, including erg10-1, which encodes the first enzyme in the ergosterol biosynthesis pathway acetolacetyl-CoA thiolase [129], and erg3 that encodes C-5 sterol desaturase [130]. Although the underlying mechanism of the HU sensitivity in these S. cerevisiae mutants remains to be determined, HU may suppress cell division in the presence of sterol deficiency in diverse eukaryotic organisms.
Figure 2.

HU induces cytokinesis arrest in Schizosaccharomyces pombe erg11-1 mutant. (A) unlike wild type (WT) cells that are arrested in S phase, HU arrests erg11-1 cells in G2/M phase. Cell cycle progression of the wild type and erg11-1 mutant cells cultured in YE6S medium containing 15 mM HU was monitored during the course of incubation at the indicated time points by flow cytometry. Dashed lines indicate the cells with a 2C DNA content. Since most of the S. pombe cell cycle time is at G2 phase, the majority of the logarithmically growing cells (Log) have a 2C DNA content; (B) wild type S. pombe, the checkpoint mutant rad3∆ lacking the sensor protein kinase Rad3 (ortholog of human ATR and Saccharomyces cerevisiae Mec1), and erg11-1 cells were treated with 15 mM HU for 3 h at 30 °C in YE6S medium and then stained with propidium iodide (PI) for genomic DNA and Blankophor for cell wall and the septum. The stained cells were examined under a fluorescent microscope. Arrowheads indicate cells with the “cell untimely torn” or cut phenotype in rad3∆ cells, a strong indicator of aberrant mitosis in HU-treated checkpoint deficient mutants [77]. (This figure is adapted from the reference [21] with permission from The Genetics Society of America).
Since the cytokinesis arrest occurs at HU concentrations much lower (1–3 mM) than that required for replication arrest (more than 6 mM) in S. pombe, it is possible that, in addition to RNR, HU may have a secondary target(s) involved in cell division that can be unmasked by sterol deficiency and become druggable to HU (Figure 3). As mentioned above, HU has been reported to inhibit catalase and several metalloproteases. It is possible that sterol deficiency may synergize with HU in suppressing the secondary target(s) and thus arrest the cells in cytokinesis. In support of this hypothesis, various combinations of HU and sterol synthesis inhibitors have shown synergistic antifungal effects (our unpublished data). Since almost all of HU-treated erg11-1 S. pombe contains two nuclei and a brightly stained septum that is well positioned in the middle of the cells, the arrest may be caused by a defect in the late stage of cytokinesis. Several S. pombe mutants have been reported that show the similar late stage cytokinesis arrest [131]. Genetic studies by crossing the erg11-1 mutant with cytokinesis mutants such as byr4, cdc16, dma1 and nuc2, may pinpoint the exact step where the arrest occurs and thus the identification of the target(s) of HU [132,133,134,135]. Cytokinesis is the last step of the cell division cycle that is crucial for cell proliferation. It has been extensively exploited for the development of anti-neoplastic chemotherapeutics [136,137]. Identification of the secondary target(s) of HU in cytokinesis may therefore help to develop new therapeutics for the treatment of cancers or infectious diseases.
Figure 3.

The cell-killing mechanisms of HU. HU inhibits its primary cellular target ribonucleotide reductase (RNR), which decreases the deoxyribonucleotide triphosphate (dNTP) levels and slows the movement of DNA polymerases at the forks (red cross). Slowed forks activate the DNA replication checkpoint. Activated checkpoint stimulates RNR to increase the dNTP production for DNA synthesis and fork recovery. Activated checkpoint can also suppress mitosis to prevent aberrant cell division (not shown). Without a functional checkpoint, slowed forks collapse and thus generate DNA damage, which leads to cell inviability. Recent studies suggest that, in addition to RNR, HU may have a secondary target(s) (red question mark) such as the metal enzymes and the matrix proteases that have been reported recently [52,53,54]. Suppression of the secondary target(s) may arrest the cells in cytokinesis or generate oxidative stress, which also leads to cell lethality. In Escherichia coli, oxidative stress is the common mechanism underlying the cell killing process of all three major classes of bactericidal antibiotics [105]. It has been shown that fork collapse generates oxidative stress in yeast [22]. Whether the HU-induced cytokinesis arrest also generates oxidative stress in eukaryotes remains to be investigated.
6. Conclusions
This review has focused upon the mechanisms by which HU exerts its cytotoxic effects. Clearly our knowledge is far from complete. For example, how are ROS generated in HU-treated cells? Fork collapse can clearly generate ROS [19,20,22]. However, the exact mechanism by which fork collapse causes ROS accumulation remains to be determined. Because some of the HU hypersensitive yeast mutants are killed at drug concentrations significantly lower than that required for slowing down the fork progression, ROS have to be accumulated via a different mechanism. In addition, the cytokinesis arrest observed in the S. pombe erg11-1 mutant is clearly caused by a previously unknown mechanism [21] and is consistent with the existence of the secondary unknown target(s) in eukaryotic organisms [52,53,54,55] (Figure 3). Since HU has been used for the treatment of various cancers and infectious diseases, identification of such targets and characterization of the new cell-killing mechanisms of HU, particularly in the non-proliferating cells, may provide new strategies for improving the HU-based chemotherapies [45,56].
Acknowledgments
We would like to thank Michael Kemp and anonymous reviewers for critical reading and suggestions. The research of the authors’ laboratory is supported by an NIH RO1 grant GM110132 and the start-up fund provided by Wright State University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Dresler W.F.C., Stein R. Ueber Den Hydroxylharnstoff. Eur. J. Org. Chem. 1869;150:242–252. doi: 10.1002/jlac.18691500212. [DOI] [Google Scholar]
- 2.Rosenthal F., Wislicki L., Kollek L. Über die Beziehungen von Schwersten Blutgiften zu Abbauprodukten des Eiweisses. Klin. Wochenschr. 1928;7:972–977. doi: 10.1007/BF01716922. [DOI] [Google Scholar]
- 3.Stock C.C., Clarke D.A., Philips F.S., Barclay R.K., Myron S.A. Sarcoma 180 Screening Data. Cancer Res. 1960;20 Pt 2:193–381. [PubMed] [Google Scholar]
- 4.Stearns B., Losee K.A., Bernstein J. Hydroxyurea. A New Type of Potential Antitumor Agent. J. Med. Chem. 1963;6:201. doi: 10.1021/jm00338a026. [DOI] [PubMed] [Google Scholar]
- 5.Rosenkranz H.S., Gutter B., Becker Y. Studies on the Developmental Cycle of Chlamydia Trachomatis: Selective Inhibition by Hydroxyurea. J. Bacteriol. 1973;115:682–690. doi: 10.1128/jb.115.2.682-690.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feiner R.R., Coward J.E., Rosenkranz H.S. Effect of Hydroxyurea on Staphylococcus Epidermidis and Micrococcus Lysodeikticus: Thickening of the Cell Wall. Antimicrob. Agents Chemother. 1973;3:432–435. doi: 10.1128/AAC.3.3.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gale G.R., Kendall S.M., Mclain H.H., Dubois S. Effect of Hydroxyurea on Pseudomonas Aeruginosa. Cancer Res. 1964;24:1012–1020. [PubMed] [Google Scholar]
- 8.Nozaki A., Numata K., Morimoto M., Kondo M., Sugimori K., Morita S., Miyajima E., Ikeda M., Kato N., Maeda S., et al. Hydroxyurea Suppresses Hcv Replication in Humans: A Phase I Trial of Oral Hydroxyurea in Chronic Hepatitis C Patients. Antivir. Ther. 2010;15:1179–1183. doi: 10.3851/IMP1668. [DOI] [PubMed] [Google Scholar]
- 9.Lori F., Malykh A., Cara A., Sun D., Weinstein J.N., Lisziewicz J., Gallo R.C. Hydroxyurea as an Inhibitor of Human Immunodeficiency Virus-Type 1 Replication. Science. 1994;266:801–805. doi: 10.1126/science.7973634. [DOI] [PubMed] [Google Scholar]
- 10.Leavell U.W., Jr., Yarbro J.W. Hydroxyurea. A New Treatment for Psoriasis. Arch. Dermatol. 1970;102:144–150. doi: 10.1001/archderm.1970.04000080016003. [DOI] [PubMed] [Google Scholar]
- 11.Yarbro J.W. Hydroxyurea in the Treatment of Refractory Psoriasis. Lancet. 1969;2:846–847. doi: 10.1016/S0140-6736(69)92297-1. [DOI] [PubMed] [Google Scholar]
- 12.Donehower R.C. An Overview of the Clinical Experience with Hydroxyurea. Semin. Oncol. 1992;19:11–19. [PubMed] [Google Scholar]
- 13.Spivak J.L., Hasselbalch H. Hydroxycarbamide: A User’s Guide for Chronic Myeloproliferative Disorders. Expert Rev. Anti-Cancer Ther. 2011;11:403–414. doi: 10.1586/era.11.10. [DOI] [PubMed] [Google Scholar]
- 14.19th WHO Model List of Essential Medicines (April 2015) http://www.who.int/medicines/publications/essentialmedicines/EML2015_8-May-15.pdf.
- 15.Reichard P., Ehrenberg A. Ribonucleotide Reductase—A Radical Enzyme. Science. 1983;221:514–519. doi: 10.1126/science.6306767. [DOI] [PubMed] [Google Scholar]
- 16.Akerblom L., Ehrenberg A., Graslund A., Lankinen H., Reichard P., Thelander L. Overproduction of the Free Radical of Ribonucleotide Reductase in Hydroxyurea-Resistant Mouse Fibroblast 3t6 Cells. Proc. Natl. Acad. Sci. USA. 1981;78:2159–2163. doi: 10.1073/pnas.78.4.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timson J. Hydroxyurea. Mutat. Res. 1975;32:115–132. doi: 10.1016/0165-1110(75)90002-0. [DOI] [PubMed] [Google Scholar]
- 18.Desesso J.M. Cell Death and Free Radicals: A Mechanism for Hydroxyurea Teratogenesis. Med. Hypotheses. 1979;5:937–951. doi: 10.1016/0306-9877(79)90043-4. [DOI] [PubMed] [Google Scholar]
- 19.Davies B.W., Kohanski M.A., Simmons L.A., Winkler J.A., Collins J.J., Walker G.C. Hydroxyurea Induces Hydroxyl Radical-Mediated Cell Death in Escherichia coli. Mol. Cell. 2009;36:845–860. doi: 10.1016/j.molcel.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayashiki T., Mori H. Genome-Wide Screening with Hydroxyurea Reveals a Link between Nonessential Ribosomal Proteins and Reactive Oxygen Species Production. J. Bacteriol. 2013;195:1226–1235. doi: 10.1128/JB.02145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y.J., Singh A., Alter G.M. Hydroxyurea Induces Cytokinesis Arrest in Cells Expressing a Mutated Sterol-14alpha-Demethylase in the Ergosterol Biosynthesis Pathway. Genetics. 2016 doi: 10.1534/genetics.116.191536. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchetti M.A., Weinberger M., Murakami Y., Burhans W.C., Huberman J.A. Production of Reactive Oxygen Species in Response to Replication Stress and Inappropriate Mitosis in Fission Yeast. J. Cell Sci. 2006;119:124–131. doi: 10.1242/jcs.02703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang M.E., Facca C., Fatmi Z., Baille D., Benakli S., Vernis L. DNA Replication Inhibitor Hydroxyurea Alters Fe-S Centers by Producing Reactive Oxygen Species in Vivo. Sci. Rep. 2016;6:29361. doi: 10.1038/srep29361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubacq C., Chevalier A., Courbeyrette R., Petat C., Gidrol X., Mann C. Role of the Iron Mobilization and Oxidative Stress Regulons in the Genomic Response of Yeast to Hydroxyurea. Mol. Genet. Genom. 2006;275:114–124. doi: 10.1007/s00438-005-0077-5. [DOI] [PubMed] [Google Scholar]
- 25.Cokic V.P., Smith R.D., Beleslin-Cokic B.B., Njoroge J.M., Miller J.L., Gladwin M.T., Schechter A.N. Hydroxyurea Induces Fetal Hemoglobin by the Nitric Oxide-Dependent Activation of Soluble Guanylyl Cyclase. J. Clin. Investig. 2003;111:231–239. doi: 10.1172/JCI200316672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adragna N.C., Fonseca P., Lauf P.K. Hydroxyurea Affects Cell Morphology, Cation Transport, and Red Blood Cell Adhesion in Cultured Vascular Endothelial Cells. Blood. 1994;83:553–560. [PubMed] [Google Scholar]
- 27.Fraser D.I., Liu K.T., Reid B.J., Hawkins E., Sevier A., Pyle M., Robinson J.W., Ouellette P.H., Ballantyne J.S. Widespread Natural Occurrence of Hydroxyurea in Animals. PLoS ONE. 2015;10:E0142890. doi: 10.1371/journal.pone.0142890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kettani T., Cotton F., Gulbis B., Ferster A., Kumps A. Plasma Hydroxyurea Determined by Gas Chromatography-Mass Spectrometry. J. Chromatogr. B. 2009;877:446–450. doi: 10.1016/j.jchromb.2008.12.048. [DOI] [PubMed] [Google Scholar]
- 29.Krakoff I.H., Brown N.C., Reichard P. Inhibition of Ribonucleoside Diphosphate Reductase by Hydroxyurea. Cancer Res. 1968;28:1559–1565. [PubMed] [Google Scholar]
- 30.Nordlund P., Reichard P. Ribonucleotide Reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 31.Stubbe J., Van Der Donk W.A. Ribonucleotide Reductases: Radical Enzymes with Suicidal Tendencies. Chem. Biol. 1995;2:793–801. doi: 10.1016/1074-5521(95)90084-5. [DOI] [PubMed] [Google Scholar]
- 32.Uhlin U., Eklund H. Structure of Ribonucleotide Reductase Protein R1. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 33.Fontecave M. Ribonucleotide Reductases and Radical Reactions. Cell. Mol. Life Sci. 1998;54:684–695. doi: 10.1007/s000180050195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimanyi C.M., Chen P.Y., Kang G., Funk M.A., Drennan C.L. Molecular Basis for Allosteric Specificity Regulation in Class Ia Ribonucleotide Reductase From Escherichia coli. Elife. 2016;5:E07141. doi: 10.7554/eLife.07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou C., Elia A.E., Naylor M.L., Dephoure N., Ballif B.A., Goel G., Xu Q., Ng A., Chou D.M., Xavier R.J., et al. Profiling DNA Damage-Induced Phosphorylation in Budding Yeast Reveals Diverse Signaling Networks. Proc. Natl. Acad. Sci. USA. 2016;113:E3667–E3675. doi: 10.1073/pnas.1602827113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis N.A., Zhou C., Elia A.E., Murray J.M., Carr A.M., Elledge S.J., Rhind N. Identification of S-Phase DNA Damage-Response Targets in Fission Yeast Reveals Conservation of Damage-Response Networks. Proc. Natl. Acad. Sci. USA. 2016;113:E3676–E3685. doi: 10.1073/pnas.1525620113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chabes A., Georgieva B., Domkin V., Zhao X., Rothstein R., Thelander L. Survival of DNA Damage in Yeast Directly Depends on Increased Dntp Levels Allowed by Relaxed Feedback Inhibition of Ribonucleotide Reductase. Cell. 2003;112:391–401. doi: 10.1016/S0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 38.Elledge S.J., Zhou Z., Allen J.B. Ribonucleotide Reductase: Regulation, Regulation, Regulation. Trends Biochem. Sci. 1992;17:119–123. doi: 10.1016/0968-0004(92)90249-9. [DOI] [PubMed] [Google Scholar]
- 39.Nyholm S., Thelander L., Graslund A. Reduction and Loss of the Iron Center in the Reaction of The Small Subunit of Mouse Ribonucleotide Reductase with Hydroxyurea. Biochemistry. 1993;32:11569–11574. doi: 10.1021/bi00094a013. [DOI] [PubMed] [Google Scholar]
- 40.Lassmann G., Thelander L., Graslund A. Epr Stopped-Flow Studies of the Reaction of the Tyrosyl Radical of Protein R2 from Ribonucleotide Reductase with Hydroxyurea. Biochem. Biophys. Res. Commun. 1992;188:879–887. doi: 10.1016/0006-291X(92)91138-G. [DOI] [PubMed] [Google Scholar]
- 41.Shao J., Zhou B., Zhu L., Bilio A.J., Su L., Yuan Y.C., Ren S., Lien E.J., Shih J., Yen Y. Determination of the Potency and Subunit-Selectivity of Ribonucleotide Reductase Inhibitors with a Recombinant-Holoenzyme-Based in Vitro Assay. Biochem. Pharmacol. 2005;69:627–634. doi: 10.1016/j.bcp.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 42.Zhu L., Zhou B., Chen X., Jiang H., Shao J., Yen Y. Inhibitory Mechanisms of Heterocyclic Carboxaldehyde Thiosemicabazones for Two Forms of Human Ribonucleotide Reductase. Biochem. Pharmacol. 2009;78:1178–1185. doi: 10.1016/j.bcp.2009.06.103. [DOI] [PubMed] [Google Scholar]
- 43.Larsen I.K., Sjoberg B.M., Thelander L. Characterization of the Active Site of Ribonucleotide Reductase of Escherichia coli, Bacteriophage T4 and Mammalian Cells by Inhibition Studies with Hydroxyurea Analogues. Eur. J. Biochem. 1982;125:75–81. doi: 10.1111/j.1432-1033.1982.tb06653.x. [DOI] [PubMed] [Google Scholar]
- 44.Gerez C., Elleingand E., Kauppi B., Eklund H., Fontecave M. Reactivity of the Tyrosyl Radical of Escherichia coli Ribonucleotide Reductase—Control by the Protein. Eur. J. Biochem. 1997;249:401–407. doi: 10.1111/j.1432-1033.1997.t01-2-00401.x. [DOI] [PubMed] [Google Scholar]
- 45.Shao J., Zhou B., Chu B., Yen Y. Ribonucleotide Reductase Inhibitors and Future Drug Design. Curr. Cancer Drug Targets. 2006;6:409–431. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 46.Nordlund P., Sjoberg B.M., Eklund H. Three-Dimensional Structure of the Free Radical Protein of Ribonucleotide Reductase. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 47.Ormo M., Regnstrom K., Wang Z., Que L., Jr., Sahlin M., Sjoberg B.M. Residues Important for Radical Stability in Ribonucleotide Reductase from Escherichia coli. J. Biol. Chem. 1995;270:6570–6576. doi: 10.1074/jbc.270.12.6570. [DOI] [PubMed] [Google Scholar]
- 48.Sneeden J.L., Loeb L.A. Mutations in the R2 Subunit of Ribonucleotide Reductase That Confer Resistance to Hydroxyurea. J. Biol. Chem. 2004;279:40723–40728. doi: 10.1074/jbc.M402699200. [DOI] [PubMed] [Google Scholar]
- 49.Wheeler G.P., Bowdon B.J., Adamson D.J., Vail M.H. Comparison of the Effects of Several Inhibitors of the Synthesis of Nucleic Acids Upon the Viability and Progression through the Cell Cycle of Cultured H. Ep. No. 2 Cells. Cancer Res. 1972;32:2661–2669. [PubMed] [Google Scholar]
- 50.Fontecave M., Lepoivre M., Elleingand E., Gerez C., Guittet O. Resveratrol, a Remarkable Inhibitor of Ribonucleotide Reductase. FEBS Lett. 1998;421:277–279. doi: 10.1016/S0014-5793(97)01572-X. [DOI] [PubMed] [Google Scholar]
- 51.Karlsson M., Sahlin M., Sjoberg B.M. Escherichia coli Ribonucleotide Reductase. Radical Susceptibility to Hydroxyurea Is Dependent on the Regulatory State of the Enzyme. J. Biol. Chem. 1992;267:12622–12626. [PubMed] [Google Scholar]
- 52.Juul T., Malolepszy A., Dybkaer K., Kidmose R., Rasmussen J.T., Ersen G.R., Johnsen H.E., Jorgensen J.E., Andersen S.U. The in Vivo Toxicity of Hydroxyurea Depends on Its Direct Target Catalase. J. Biol. Chem. 2010;285:21411–21415. doi: 10.1074/jbc.M110.103564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scozzafava A., Supuran C.T. Hydroxyurea Is a Carbonic Anhydrase Inhibitor. Bioorg. Med. Chem. 2003;11:2241–2246. doi: 10.1016/S0968-0896(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 54.Campestre C., Agamennone M., Tortorella P., Preziuso S., Biasone A., Gavuzzo E., Pochetti G., Mazza F., Hiller O., Tschesche H., et al. N-Hydroxyurea as Zinc Binding Group in Matrix Metalloproteinase Inhibition: Mode of Binding in a Complex with Mmp-8. Bioorg. Med. Chem. Lett. 2006;16:20–24. doi: 10.1016/j.bmcl.2005.09.057. [DOI] [PubMed] [Google Scholar]
- 55.Temperini C., Innocenti A., Scozzafava A., Supuran C.T. N-Hydroxyurea—A Versatile Zinc Binding Function in the Design of Metalloenzyme Inhibitors. Bioorg. Med. Chem. Lett. 2006;16:4316–4320. doi: 10.1016/j.bmcl.2006.05.068. [DOI] [PubMed] [Google Scholar]
- 56.Day J.A., Cohen S.M. Investigating the Selectivity of Metalloenzyme Inhibitors. J. Med. Chem. 2013;56:7997–8007. doi: 10.1021/jm401053m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chabes A.L., Pfleger C.M., Kirschner M.W., Thelander L. Mouse Ribonucleotide Reductase R2 Protein: A New Target for Anaphase-Promoting Complex-Cdh1-Mediated Proteolysis. Proc. Natl. Acad. Sci. USA. 2003;100:3925–3929. doi: 10.1073/pnas.0330774100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chabes A.L., Bjorklund S., Thelander L. S Phase-Specific Transcription of the Mouse Ribonucleotide Reductase R2 Gene Requires Both a Proximal Repressive E2f-Binding Site and an Upstream Promoter Activating Region. J. Biol. Chem. 2004;279:10796–10807. doi: 10.1074/jbc.M312482200. [DOI] [PubMed] [Google Scholar]
- 59.Bjorklund S., Skog S., Tribukait B., Thelander L. S-Phase-Specific Expression of Mammalian Ribonucleotide Reductase R1 and R2 Subunit Mrnas. Biochemistry. 1990;29:5452–5458. doi: 10.1021/bi00475a007. [DOI] [PubMed] [Google Scholar]
- 60.Engstrom Y., Eriksson S., Jildevik I., Skog S., Thelander L., Tribukait B. Cell Cycle-Dependent Expression of Mammalian Ribonucleotide Reductase. Differential Regulation of the Two Subunits. J. Biol. Chem. 1985;260:9114–9116. [PubMed] [Google Scholar]
- 61.Eriksson S., Graslund A., Skog S., Thelander L., Tribukait B. Cell Cycle-Dependent Regulation of Mammalian Ribonucleotide Reductase. The S Phase-Correlated Increase in Subunit M2 Is Regulated by de Novo Protein Synthesis. J. Biol. Chem. 1984;259:11695–11700. [PubMed] [Google Scholar]
- 62.Zhao X., Muller E.G., Rothstein R. A Suppressor of Two Essential Checkpoint Genes Identifies a Novel Protein That Negatively Affects Dntp Pools. Mol. Cell. 1998;2:329–340. doi: 10.1016/S1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 63.Nestoras K., Mohammed A.H., Schreurs A.S., Fleck O., Watson A.T., Poitelea M., O’shea C., Chahwan C., Holmberg C., Kragelund B.B., et al. Regulation of Ribonucleotide Reductase by Spd1 Involves Multiple Mechanisms. Genes Dev. 2010;24:1145–1159. doi: 10.1101/gad.561910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hakansson P., Dahl L., Chilkova O., Domkin V., Thelander L. The Schizosaccharomyces pombe Replication Inhibitor Spd1 Regulates Ribonucleotide Reductase Activity and Dntps by Binding to the Large Cdc22 Subunit. J. Biol. Chem. 2006;281:1778–1783. doi: 10.1074/jbc.M511716200. [DOI] [PubMed] [Google Scholar]
- 65.Yao R., Zhang Z., An X., Bucci B., Perlstein D.L., Stubbe J., Huang M. Subcellular Localization of Yeast Ribonucleotide Reductase Regulated by the DNA Replication and Damage Checkpoint Pathways. Proc. Natl. Acad. Sci. USA. 2003;100:6628–6633. doi: 10.1073/pnas.1131932100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee Y.D., Wang J., Stubbe J., Elledge S.J. Dif1 Is a DNA-Damage-Regulated Facilitator of Nuclear Import for Ribonucleotide Reductase. Mol. Cell. 2008;32:70–80. doi: 10.1016/j.molcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee Y.D., Elledge S.J. Control of Ribonucleotide Reductase Localization through an Anchoring Mechanism Involving Wtm1. Genes Dev. 2006;20:334–344. doi: 10.1101/gad.1380506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mckethan B.L., Spiro S. Cooperative and Allosterically Controlled Nucleotide Binding Regulates the DNA Binding Activity of Nrdr. Mol. Microbiol. 2013;90:278–289. doi: 10.1111/mmi.12364. [DOI] [PubMed] [Google Scholar]
- 69.Herrick J., Sclavi B. Ribonucleotide Reductase snd the Regulation of DNA Replication: An Old Story and an Ancient Heritage. Mol. Microbiol. 2007;63:22–34. doi: 10.1111/j.1365-2958.2006.05493.x. [DOI] [PubMed] [Google Scholar]
- 70.Ciccia A., Elledge S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Furuya K., Carr A.M. DNA Checkpoints in Fission Yeast. J. Cell Sci. 2003;116:3847–3848. doi: 10.1242/jcs.00790. [DOI] [PubMed] [Google Scholar]
- 72.Alvino G.M., Collingwood D., Murphy J.M., Delrow J., Brewer B.J., Raghuraman M.K. Replication in Hydroxyurea: It’s a Matter of Time. Mol. Cell. Biol. 2007;27:6396–6406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zegerman P., Diffley J.F. Checkpoint-Dependent Inhibition of DNA Replication Initiation by Sld3 and Dbf4 Phosphorylation. Nature. 2010;467:474–478. doi: 10.1038/nature09373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lopez-Mosqueda J., Maas N.L., Jonsson Z.O., Defazio-Eli L.G., Wohlschlegel J., Toczyski D.P. Damage-Induced Phosphorylation of Sld3 Is Important to Block Late Origin Firing. Nature. 2010;467:479–483. doi: 10.1038/nature09377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carr A.M., Lambert S. Replication Stress-Induced Genome Instability: The Dark Side of Replication Maintenance by Homologous Recombination. J. Mol. Biol. 2013;425:4733–4744. doi: 10.1016/j.jmb.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 76.Hirano T., Funahashi S., Uemura T., Yanagida M. Isolation and Characterization of Schizosaccharomyces pombe Cut Mutants That Block Nuclear Division But Not Cytokinesis. EMBO J. 1986;5:2973–2979. doi: 10.1002/j.1460-2075.1986.tb04594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saka Y., Yanagida M. Fission Yeast Cut5+, Required for S Phase Onset and M Phase Restraint, Is Identical to the Radiation-Damage Repair Gene Rad4+ Cell. 1993;74:383–393. doi: 10.1016/0092-8674(93)90428-S. [DOI] [PubMed] [Google Scholar]
- 78.Sinclair W.K. Hydroxyurea: Differential Lethal Effects on Cultured Mammalian Cells during the Cell Cycle. Science. 1965;150:1729–1731. doi: 10.1126/science.150.3704.1729. [DOI] [PubMed] [Google Scholar]
- 79.Sinclair W.K. Hydroxyurea: Effects on Chinese Hamster Cells Grown in Culture. Cancer Res. 1967;27:297–308. [PubMed] [Google Scholar]
- 80.Barranco S.C., Novak J.K. Survival Responses of Dividing and Nondividing Mammalian Cells after Treatment with Hydroxyurea, Arabinosylcytosine, or Adriamycin. Cancer Res. 1974;34:1616–1618. [PubMed] [Google Scholar]
- 81.Timson J. Hydroxyurea: Comparison of Cytotoxic and Antimitotic Activities against Human Lymphocytes in Vitro. Br. J. Cancer. 1969;23:337–339. doi: 10.1038/bjc.1969.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu C.K., Sinclair W.K. Cytological Effects on Chinese Hamster Cells of Synchronizing Concentrations of Hydroxyurea. J. Cell. Physiol. 1968;72:39–42. doi: 10.1002/jcp.1040720107. [DOI] [PubMed] [Google Scholar]
- 83.Przybyszewski W.M., Malec J. Protection against Hydroxyurea-Induced Cytotoxic Effects in L5178y Cells by Free Radical Scavengers. Cancer Lett. 1982;17:223–228. doi: 10.1016/0304-3835(82)90036-2. [DOI] [PubMed] [Google Scholar]
- 84.Li J.C., Kaminskas E. Progressive Formation of DNA Lesions in Cultured Ehrlich Ascites Tumor Cells Treated with Hydroxyurea. Cancer Res. 1987;47:2755–2758. [PubMed] [Google Scholar]
- 85.Veale D., Cantwell B.M., Kerr N., Upfold A., Harris A.L. Phase 1 Study of High-Dose Hydroxyurea in Lung Cancer. Cancer Chemother. Pharmacol. 1988;21:53–56. doi: 10.1007/BF00262739. [DOI] [PubMed] [Google Scholar]
- 86.Mohler W.C. Cytotoxicity of Hydroxyurea (Nsc-32065) Reversible by Pyrimidine Deoxyribosides in a Mammalian Cell Line Grown in Vitro. Cancer Chemother. Rep. 1964;34:1–6. [PubMed] [Google Scholar]
- 87.Philips F.S., Sternberg S.S., Schwartz H.S., Cronin A.P., Sodergren J.E., Vidal P.M. Hydroxyurea. I. Acute Cell Death in Proliferating Tissues in Rats. Cancer Res. 1967;27:61–75. [PubMed] [Google Scholar]
- 88.Farber E., Baserga R. Differential Effects of Hydroxyurea on Survival of Proliferating Cells In Vivo. Cancer Res. 1969;29:136–139. [PubMed] [Google Scholar]
- 89.Scott W.J., Ritter E.J., Wilson J.G. DNA Synthesis Inhibition and Cell Death Associated with Hydroxyurea Teratogenesis in Rat Embryos. Dev. Biol. 1971;26:306–315. doi: 10.1016/0012-1606(71)90129-1. [DOI] [PubMed] [Google Scholar]
- 90.Hennings H., Devik F. Comparison of Cytotoxicity of Hydroxyurea in Normal and Rapidly Proliferating Epidermis and Small Intestine in Mice. Cancer Res. 1971;31:277–282. [PubMed] [Google Scholar]
- 91.Coyle M.B., Strauss B. Cell Killing and the Accumulation of Breaks in the DNA of Hep-2 Cells Incubated in the Presence of Hydroxyurea. Cancer Res. 1970;30:2314–2319. [PubMed] [Google Scholar]
- 92.Walker I.G., Yatscoff R.W., Sridhar R. Hydroxyurea: Induction of Breaks in Template Strands of Replicating DNA. Biochem. Biophys. Res. Commun. 1977;77:403–408. doi: 10.1016/S0006-291X(77)80212-X. [DOI] [PubMed] [Google Scholar]
- 93.Massafi K., Carr H.S., Rosenkranz H.S. Hydroxyurea and Cell Death. Isr. J. Med. Sci. 1972;8:559–561. [PubMed] [Google Scholar]
- 94.Sinha N.K., Snustad D.P. Mechanism of Inhibition of Deoxyribonucleic acid Synthesis in Escherichia coli by Hydroxyurea. J. Bacteriol. 1972;112:1321–1324. doi: 10.1128/jb.112.3.1321-1334.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jacobs S.J., Rosenkranz H.S. Detection of a Reactive Intermediate in the Reaction between DNA and Hydroxyurea. Cancer Res. 1970;30:1084–1094. [PubMed] [Google Scholar]
- 96.Rosenkranz H.S., Rosenkranz S. Degradation of DNA by Carbamoyloxyurea—An Oxidation Product of Hydroxyurea. Biochim. Biophys. Acta. 1969;195:266–267. doi: 10.1016/0005-2787(69)90628-5. [DOI] [PubMed] [Google Scholar]
- 97.Sakano K., Oikawa S., Hasegawa K., Kawanishi S. Hydroxyurea Induces Site-Specific DNA Damage via Formation of Hydrogen Peroxide and Nitric Oxide. Jpn. J. Cancer Res. 2001;92:1166–1174. doi: 10.1111/j.1349-7006.2001.tb02136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Imlay J.A. Pathways of Oxidative Damage. Annu. Rev. Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 99.Przybyszewski W.M., Kasperczyk J. [Radical Mechanism of Hydroxyurea Side Toxicity] Postepy Hig. Med. Dosw. 2006;60:516–526. [PubMed] [Google Scholar]
- 100.Desesso J.M. Amelioration of Teratogenesis. I. Modification of Hydroxyurea-Induced Teratogenesis by the Antioxidant Propyl Gallate. Teratology. 1981;24:19–35. doi: 10.1002/tera.1420240104. [DOI] [PubMed] [Google Scholar]
- 101.Godoy V.G., Jarosz D.F., Walker F.L., Simmons L.A., Walker G.C. Y-Family DNA Polymerases Respond to DNA Damage-Independent Inhibition of Replication Fork Progression. EMBO J. 2006;25:868–879. doi: 10.1038/sj.emboj.7600986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Imlay J.A., Chin S.M., Linn S. Toxic DNA Damage by Hydrogen Peroxide through the Fenton Reaction in Vivo and in Vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 103.Seaver L.C., Imlay J.A. Alkyl Hydroperoxide Reductase Is the Primary Scavenger of Endogenous Hydrogen Peroxide in Escherichia coli. J. Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Foti J.J., Devadoss B., Winkler J.A., Collins J.J., Walker G.C. Oxidation of the Guanine Nucleotide Pool Underlies Cell Death by Bactericidal Antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kohanski M.A., Dwyer D.J., Hayete B., Lawrence C.A., Collins J.J. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 106.Rowe L.A., Degtyareva N., Doetsch P.W. DNA Damage-Induced Reactive Oxygen Species (Ros) Stress Response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008;45:1167–1177. doi: 10.1016/j.freeradbiomed.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Delaunay A., Pflieger D., Barrault M.B., Vinh J., Toledano M.B. A Thiol Peroxidase Is an H2O2 Receptor and Redox-Transducer in Gene Activation. Cell. 2002;111:471–481. doi: 10.1016/S0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 108.Tang H.M., Pan K., Kong K.Y., Hu L., Chan L.C., Siu K.L., Sun H., Wong C.M., Jin D.Y. Loss of Apd1 in Yeast Confers Hydroxyurea Sensitivity Suppressed by Yap1p Transcription Factor. Sci. Rep. 2015;5:7897. doi: 10.1038/srep07897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Han T.X., Xu X.Y., Zhang M.J., Peng X., Du L.L. Global Fitness Profiling of Fission Yeast Deletion Strains by Barcode Sequencing. Genome Biol. 2010;11:R60. doi: 10.1186/gb-2010-11-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Toda T., Shimanuki M., Yanagida M. Fission Yeast Genes That Confer Resistance to Staurosporine Encode an Ap-1-Like Transcription Factor and a Protein Kinase Related to the Mammalian Erk1/Map2 and Budding Yeast Fus3 and Kss1 Kinases. Genes Dev. 1991;5:60–73. doi: 10.1101/gad.5.1.60. [DOI] [PubMed] [Google Scholar]
- 111.Benko Z., Fenyvesvolgyi C., Pesti M., Sipiczki M. The Transcription Factor Pap1/Caf3 Plays a Central Role in the Determination of Caffeine Resistance in Schizosaccharomyces pombe. Mol. Genet. Genom. 2004;271:161–170. doi: 10.1007/s00438-003-0967-3. [DOI] [PubMed] [Google Scholar]
- 112.Turi T.G., Webster P., Rose J.K. Brefeldin a Sensitivity and Resistance in Schizosaccharomyces pombe. Isolation of Multiple Genes Conferring Resistance. J. Biol. Chem. 1994;269:24229–24236. [PubMed] [Google Scholar]
- 113.Belfield C., Queenan C., Rao H., Kitamura K., Walworth N.C. The Oxidative Stress Responsive Transcription Factor Pap1 Confers DNA Damage Resistance on Checkpoint-Deficient Fission Yeast Cells. PLoS ONE. 2014;9:E89936. doi: 10.1371/journal.pone.0089936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boronat S., Domenech A., Paulo E., Calvo I.A., Garcia-Santamarina S., Garcia P., Encinar Del Dedo J., Barcons A., Serrano E., Carmona M., et al. Thiol-Based H2O2 Signalling in Microbial Systems. Redox Biol. 2014;2:395–399. doi: 10.1016/j.redox.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Imlay J.A., Linn S. DNA Damage and Oxygen Radical Toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 116.Netz D.J., Stith C.M., Stumpfig M., Kopf G., Vogel D., Genau H.M., Stodola J.L., Lill R., Burgers P.M., Pierik A.J. Eukaryotic DNA Polymerases Require an Iron-Sulfur Cluster for the Formation of Active Complexes. Nat. Chem. Biol. 2012;8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang Y., Lyver E.R., Nakamaru-Ogiso E., Yoon H., Amutha B., Lee D.W., Bi E., Ohnishi T., Daldal F., Pain D., et al. Dre2, a Conserved Eukaryotic Fe/S Cluster Protein, Functions in Cytosolic Fe/S Protein Biogenesis. Mol. Cell. Biol. 2008;28:5569–5582. doi: 10.1128/MCB.00642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Y., Li H., Zhang C., An X., Liu L., Stubbe J., Huang M. Conserved Electron Donor Complex Dre2-Tah18 Is Required for Ribonucleotide Reductase Metallocofactor Assembly and DNA Synthesis. Proc. Natl. Acad. Sci. USA. 2014;111:E1695–E1704. doi: 10.1073/pnas.1405204111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Y., Liu L., Wu X., An X., Stubbe J., Huang M. Investigation of in Vivo Diferric Tyrosyl Radical Formation in Saccharomyces cerevisiae Rnr2 Protein: Requirement of Rnr4 and Contribution of Grx3/4 and Dre2 Proteins. J. Biol. Chem. 2011;286:41499–41509. doi: 10.1074/jbc.M111.294074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vernis L., Facca C., Delagoutte E., Soler N., Chanet R., Guiard B., Faye G., Baldacci G. A Newly Identified Essential Complex, Dre2-Tah18, Controls Mitochondria Integrity and Cell Death after Oxidative Stress in Yeast. PLoS ONE. 2009;4:E4376. doi: 10.1371/journal.pone.0004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu C.H., Jiang W., Krebs C., Stubbe J. Yfae, a Ferredoxin Involved in Diferric-Tyrosyl Radical Maintenance in Escherichia coli Ribonucleotide Reductase. Biochemistry. 2007;46:11577–11588. doi: 10.1021/bi7012454. [DOI] [PubMed] [Google Scholar]
- 122.Jang S., Imlay J.A. Hydrogen Peroxide Inactivates the Escherichia coli Isc Iron-Sulphur Assembly System, and Oxyr Induces the Suf System to Compensate. Mol. Microbiol. 2010;78:1448–1467. doi: 10.1111/j.1365-2958.2010.07418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jang S., Imlay J.A. Micromolar Intracellular Hydrogen Peroxide Disrupts Metabolism by Damaging Iron-Sulfur Enzymes. J. Biol. Chem. 2007;282:929–937. doi: 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Flint D.H., Tuminello J.F., Emptage M.H. The Inactivation of Fe-S Cluster Containing Hydro-Lyases by Superoxide. J. Biol. Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 125.Klinge S., Hirst J., Maman J.D., Krude T., Pellegrini L. An Iron-Sulfur Domain of the Eukaryotic Primase Is Essential for RNA Primer Synthesis. Nat. Struct. Mol. Biol. 2007;14:875–877. doi: 10.1038/nsmb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weiner B.E., Huang H., Dattilo B.M., Nilges M.J., Fanning E., Chazin W.J. An Iron-Sulfur Cluster in the C-Terminal Domain of the P58 Subunit of Human DNA Primase. J. Biol. Chem. 2007;282:33444–33451. doi: 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- 127.Yi D., Alvim Kamei C.L., Cools T., Vanderauwera S., Takahashi N., Okushima Y., Eekhout T., Yoshiyama K.O., Larkin J., Van Den Daele H., et al. The Arabidopsis Siamese-Related Cyclin-Dependent Kinase Inhibitors Smr5 and Smr7 Regulate the DNA Damage Checkpoint in Response to Reactive Oxygen Species. Plant Cell. 2014;26:296–309. doi: 10.1105/tpc.113.118943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Carter C.D., Kitchen L.E., Au W.C., Babic C.M., Basrai M.A. Loss of Sod1 and Lys7 Sensitizes Saccharomyces cerevisiae to Hydroxyurea and DNA Damage Agents and Downregulates Mec1 Pathway Effectors. Mol. Cell. Biol. 2005;25:10273–10285. doi: 10.1128/MCB.25.23.10273-10285.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mcculley A., Haarer B., Viggiano S., Karchin J., Feng W. Chemical Suppression of Defects in Mitotic Spindle Assembly, Redox Control, and Sterol Biosynthesis by Hydroxyurea. Genes Genomes Genet. 2014;4:39–48. doi: 10.1534/g3.113.009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yun Y., Yin D., Dawood D.H., Liu X., Chen Y., Ma Z. Functional Characterization of Fgerg3 and Fgerg5 Associated with Ergosterol Biosynthesis, Vegetative Differentiation and Virulence of Fusarium Graminearum. Fungal Genet. Biol. 2014;68:60–70. doi: 10.1016/j.fgb.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 131.Gould K.L., Simanis V. The Control of Septum Formation in Fission Yeast. Genes Dev. 1997;11:2939–2951. doi: 10.1101/gad.11.22.2939. [DOI] [PubMed] [Google Scholar]
- 132.Lai L.A., Morabito L., Holloway S.L. A Novel Yeast Mutant That Is Defective in Regulation of the Anaphase-Promoting Complex by the Spindle Damage Checkpoint. Mol. Genet. Genom. 2003;270:156–164. doi: 10.1007/s00438-003-0912-5. [DOI] [PubMed] [Google Scholar]
- 133.Gruneberg U., Nigg E.A. Regulation of Cell Division: Stop the Sin! Trends Cell Biol. 2003;13:159–162. doi: 10.1016/S0962-8924(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 134.Yanagida M. Gene Products Required for Chromosome Separation. J. Cell Sci. Suppl. 1989;12:213–229. doi: 10.1242/jcs.1989.Supplement_12.18. [DOI] [PubMed] [Google Scholar]
- 135.Jwa M., Song K. Byr4, a Dosage-Dependent Regulator of Cytokinesis in S. Pombe, Interacts with a Possible Small Gtpase Pathway Including Spg1 and Cdc16. Mol. Cells. 1998;8:240–245. [PubMed] [Google Scholar]
- 136.Lee I.J., Coffman V.C., Wu J.Q. Contractile-Ring Assembly in Fission Yeast Cytokinesis: Recent Advances and New Perspectives. Cytoskeleton. 2012;69:751–763. doi: 10.1002/cm.21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bathe M., Chang F. Cytokinesis and the Contractile Ring in Fission Yeast: Towards a Systems-Level Understanding. Trends Microbiol. 2010;18:38–45. doi: 10.1016/j.tim.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]