

Abstract
Background
The gastrointestinal tract is populated by a complex and vast microbial network, with a composition that reflects the relationships of the symbiosis, co-metabolism, and co-evolution of these microorganisms with their host. The mechanism that underlies such interactions between the genetics of the host and gut microbiota remains elusive.
Results
To understand how genetic variation of the host shapes the gut microbiota and interacts with it to affect the metabolic phenotype of the host, we compared the abundance of microbial taxa and their functional performance between two lines of chickens (fat and lean) that had undergone long-term divergent selection for abdominal fat pad weight, which resulted in a 4.5-fold increase in the fat line compared to the lean line. Our analysis revealed that the proportions of Fusobacteria and Proteobacteria differed significantly between the two lines (8 vs. 18% and 33 vs. 24%, respectively) at the phylum level. Eight bacterial genera and 11 species were also substantially influenced by the host genotype. Differences between the two lines in the frequency of host alleles at loci that influence accumulation of abdominal fat were associated with differences in the abundance and composition of the gut microbiota. Moreover, microbial genome functional analysis showed that the gut microbiota was involved in pathways that are associated with fat metabolism such as lipid and glycan biosynthesis, as well as amino acid and energy metabolism. Interestingly, citrate cycle and peroxisome proliferator activated receptor (PPAR) signaling pathways that play important roles in lipid storage and metabolism were more prevalent in the fat line than in the lean line.
Conclusions
Our study demonstrates that long-term divergent selection not only alters the composition of the gut microbiota, but also influences its functional performance by enriching its relative abundance in microbial taxa. These results support the hypothesis that the host and gut microbiota interact at the genetic level and that these interactions result in their co-evolution.
Electronic supplementary material
The online version of this article (doi:10.1186/s12711-016-0270-5) contains supplementary material, which is available to authorized users.
Background
The development of sequencing technologies for application in metagenomics has increased our capacity to investigate the composition and dynamics of the microbial communities that harbor diverse habitats [1]. The gastrointestinal tract is populated by a complicated and vast microbial network that influences the health and development of the host organism in numerous aspects [2, 3]. The gut microbial composition can be viewed as a polygenic trait, that not only produces essential products and forms a barrier against pathogens, but also has multiple functions in physiology, metabolism, immunity, development, and behavior of the host [4–6]. The gut microbiota causes the suppression of the circulating lipoprotein lipase inhibitor that results in increased lipoprotein lipase activity, which in turn results in a significant increase in body fat deposition in the host [7]. Suppression of the expression of these genes by direct action of the gut microbiota on the villi epithelia also causes increased lipoprotein lipase activity, which leads to increased triglyceride uptake and peripheral fat storage [8]. These findings are in agreement with previous studies in other chicken populations selected for high or low body fat [9, 10] and show that the gut microbiota affects energy uptake from the diet and energy storage in the host [7]. In our previous studies, in order to quantify the influence of genetic variation of the host on the structure of the gut microbiota, the abundance of gut microbiota was considered as a quantitative trait of the host, and we calculated the heritability of abundance of specific microorganisms in the gut microbiota. A few bacterial families of the microbiota had a moderate heritability, which indicated that the host genetics has an effect on the composition of the gut microbiota. Concurrently, we calculated the genetic correlations between specific microorganisms in the gut microbiota to examine if the genetics of the host is involved in the interactions between microorganisms in the gut microbiota. Significant genetic correlations between microorganisms in the gut microbiota were observed. Further analysis showed that such genetic correlations can be altered by genetic variation of the host. These results imply the importance of the host genetic background on the interactions between the microorganisms in the gut microbiota [11, 12]. However, the interactional mechanism between gut microbiota and genetic variation of the host genome has remained obscure. Until now, most studies focused on microbial taxa instead of microbial functional performance to understand the interactions between host genetics and gut microbiota.
Many factors influence the mechanism of the interactions between the host and the gut microbiota [13, 14]. Thus, choosing a model organism that is maintained in a controlled environment should enhance our understanding of the relationships between gut microbiota and host genetic factors. The chicken, which bridges the evolutionary gap between mammals and reptiles, serves as an important experimental model organism for the extant avian species due to the characteristics of its less complex gut microbiota and minimal maternal effect. Here, we analyzed and compared the function and classification of gut microbiota from two divergently selected lines of chickens, i.e. a fat line and a lean line. These lines originated from a single commercial grandsire line and underwent long-term (15 generations) divergent selection for abdominal fat percentage (AFP) and plasma very-low-density lipoprotein (VLDL) concentration. At 7 weeks of age, the mean adipocyte diameter in the fat line was 1.3 times wider than in the lean line, and the number of fat cells was 2.4 times larger in the fat line than in the lean line [15]. The long-term divergent selection also resulted in a 4.5-fold increase in abdominal fat pad weight in the fat line [16]. A total of 230 genes were found to be differentially expressed in the lean and fat lines; these genes are mainly related to signal transduction, tumorigenesis, immunity, and lipid and energy metabolism [17]. The two lines carry two main haplotypes with completely opposite single nucleotide polymorphism (SNP) alleles and a recombinant haplotype with nearly equal frequency in the 0.73-Mb PC1/PCSK1 region of the Z chromosome. Genome-wide association analysis revealed that nearly all regions with evidence of selection signatures had SNP effects on abdominal fat weight and percentage [18].
Methods
Animals and samples collection
Two chicken lines (fat and lean lines) that were divergently selected for abdominal fat content (AFP) and plasma very-low-density lipoprotein (VLDL) were used in this study. Throughout all generations, they were maintained at the same location and reared on the same diets. Fecal samples were collected at 35 weeks of age from 29 fat line males, 26 lean line males, 27 fat line females, and 27 lean line females, for a total of 109 individuals from the 15th generation. The fecal samples were stored at −80 °C after collection. Animals were cared for in accordance with the Institute for Laboratory Animal Research (ILAR) guide for Care and Use of Laboratory Animals at Shanghai Jiao Tong University, China.
Gut microbial 16S rDNA sequencing
Fecal microbial genomic DNA extraction and 16S rDNA amplification and sequencing were performed as previously reported in [11]. A QIAmp DNA Stool Mini Kit (Qiagen, cat#51504) was used for microbial genomic DNA extraction. Extracted DNA was measured using a nanodrop spectrophotometer (Thermo Fisher Scientific) to assess DNA quantity and quality. The V4 hypervariable region of the 16S rDNA gene was PCR-amplified from microbiota genomic DNA using sample-specific sequence barcode fusion primers (forward 5′AYTGGGYDTAAAGNG 3′, reverse 5′ TACNVGGGTATCTAATCC 3′). PCR reactions and PCR product purification were performed as previously reported in [11]. Purified PCR products from the 109 samples were combined at equal concentrations and used to construct a metagenomic library using Illumina TruSeq sample preparation kit (Illumina, USA) according to the manufacturer’s protocol. Sequencing was carried out by the Shanghai Personal Biotechnology Limited Company (Shanghai, P. R. China) using an Illumina MiSeq (Illumina, USA) sequencing platform. Sequence reads were quality-checked and removed based on the following criteria: reads that (1) contained ambiguous bases, (2) had an average phred score lower than 25, (3) contained a homopolymer run that exceeded 6, (4) contained mismatches in the primers, and (5) had sequence lengths that were outside the limits of 200 and 1000 bp. The filtered sequences with an overlap longer than 10 bp between Read 1 and Read 2 and without mismatches were assembled according to their overlapping sequences. Reads that could not be assembled were discarded. The barcode and sequencing primers were trimmed from each sequence.
Analysis of classification and abundance
Based on the V4 region of the 16S rDNA sequence that passed the quality criteria, 2,301,532 amplicons were used for this study, with an average of 21,115 amplicons for each sample (ranging from 12,137 to 30,067) [see Additional file 1: Table S1]. The average sequenced amplicon length was 225 bp. Following filtering, each sample’s trimmed and filtered sequences were submitted to Metagenome Rapid Annotation using Subsystem Technology (MG-RAST) [19] and compared to the Ribosomal database project databases (RDP) [20] using the best hit classification option to classify the abundance count of each taxon. The metagenome sequences used in this paper are publicly available from MG-RAST under the project name “fatandleanchicken”. Data were generated at the species level, using cutoffs for the parameter classification at 8 for maximum e-value, 98% for minimum percentage identity, and 120 bp for minimum alignment length. A total of 37,590 taxa on the phylum, class, order, family, genus and species levels were annotated [see Additional file 2: Table S2]. Taxa that were present in at least 28 samples were considered as commonly existing classifications; 51 genera and 109 species met that criterion and their abundance counts were used for further analysis.
The taxon abundance counts were log2 transformed and normalized by subtracting the mean of all transformed values and dividing by the standard deviation of all log-transformed values for the given sample. After this procedure, the abundance profiles for all samples exhibited a mean of 0 and a standard deviation of 1. In order to detect if host genetic factors influence gut microbiota, T test was performed between fat and lean lines for specific microbes using Microsoft Excel, with adjustment of p values by Benjamini Hochberg FDR (FDR < 0.05) [21]. Alpha-diversity analysis was performed in mothur 1.31.2 [22] with the alpha-diversity.py script to calculate the index of chao1 and Shannon.
Sequencing of the whole microbial genome
Microbial genomic DNA of three females from each fat and lean line was used to construct whole microbial genome sequencing libraries with insert sizes of 300 and 400 bp. Each library was sequenced by high-throughput sequencing at 2 × 100 bp using the Illumina HiSeq 2000 (Illumina, USA). Eighty percent of the whole microbial genome sequence data with paired-end Illumina sequences were accounted for across all samples. A data cleaning process was applied to all samples. Low-quality reads, and low-compositional-complexity reads were removed. An average of 37.9 million reads per sample were used in the analysis. The DNA sequences are publicly available in MG-RAST under the project name “Hiseqchicken-six”.
Annotation of microbial function
Quality-filtered reads were submitted to MG-RAST and compared to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [23] using the ‘all annotation’ option for functional annotation with a maximum e-value cutoff of 1e-5, a minimum percent identity cutoff of 90%, and a minimum alignment length cutoff of 20 amino acids. Functional pathways, which had a relative abundance that was greater than 0.1% and for at least two samples, were chosen for further analysis. The relative abundance of each functional pathway was normalized within each sample. Clustering analysis was performed by Cluster 3.0 and Java Treeview [24] and differential analysis was evaluated by STAMP v2.0 [25] between fat and lean lines for abundance of each functional pathway by applying the two-side Welch’s t-test [26] and Benjamini-Hochberg FDR correction (FDR < 0.05).
Results
Diversity of gut microbial composition between the fat and lean lines
16S rDNA amplicon sequencing was used to analyze the microbial diversity and abundance in the gut microbiota of the fat and lean chicken lines. Alpha diversity results suggested that the richness and diversity of gut microbiota were influenced by the long-term divergent selection [see Additional file 3: Figure S1]. Four major phyla dominated the chicken gut bacterial community; Firmicutes was the most predominant phylum, followed by Proteobacteria, Fusobacteria, and Actinobacteria (Fig. 1a). Consistent with previous studies related to avian microbial diversity, Firmicutes and Proteobacteria were the main ubiquitous members in the gut microbiota, but more Fusobacteria were classified compared with several other avian gut microbial studies [27–29]. The gut microbial composition differed between the fat and lean lines. The percentage of Fusobacteria was significantly lower in the fat line (8%) than in the lean line (18%). Conversely, the percentage of Proteobacteria was 33% in the fat line and approximately 24% in the lean line (Fig. 1a). At the genus level, Gallibacterium, which belongs to the phylum of Proteobacteria and comprises the Gallibacterium anatis species, was the most abundant genus in chicken gut microbiota (Fig. 1c). The genus Fusobacterium, belonging to the phylum of Fusobacteria, was in lower proportion in the fat line (9%) than in the lean line (20%) (Fig. 1b). The analysis showed that one (Proteobacteria) of the eight phyla [see Additional file 4: Table S3], eight of the 52 genera (Table 1) and 11 species [see Additional file 5: Table S4] were significantly influenced by the genetics of the host.
Fig. 1.

Aggregate microbiota composition at different levels in the fat and lean lines. a Phylum level, b genus level, c species level. Only major taxonomic groups are shown
Table 1.
Comparison of bacterial genus abundance in the gut microbiota between the fat and lean lines
| Phylum | Genus | Relative fold changea
Fat versus lean line |
p value (*p < 0.05, **p < 0.01) |
|---|---|---|---|
| Actinobacteria | Rothia | 1.28 | 0.004** |
| Micrococcus | 1.20 | 0.009** | |
| Bacteroidetes | Bacteroides | −1.08 | 0.019* |
| Proteobacteria | Gallibacterium | 1.25 | 0.011* |
| Tenericutes | Acholeplasma | −1.19 | 0.03* |
| Firmicutes | Aerococcus | 1.14 | 0.037* |
| Pectinatus | −1.27 | 0.003** | |
| Selenomonas | 1.17 | 0.04* |
a+ fat/lean; − lean/fat
The effect of host genetics on gene functional enrichment of gut microbiota
In order to investigate the influence of host genetic variation on the functional performance of the microbiota, we sequenced the whole gut microbial genome using three biological replicates from each line. Based on the associated KEGG orthologous group markers, we compared predicted microbial functions between the fat and lean lines and detected that amino acid metabolism, energy metabolism, lipid metabolism, and cell motility were nearly twofold more enriched in the lean line than in the fat line (Fig. 2). Pathways that were more enriched in the fat line included translation, signal transduction mechanisms, metabolism of terpenoids and polyketides, protein folding and degradation, biosynthesis of secondary metabolites, and cancers (Fig. 2). These results are consistent with the previous findings of a study on obese rats [30].
Fig. 2.
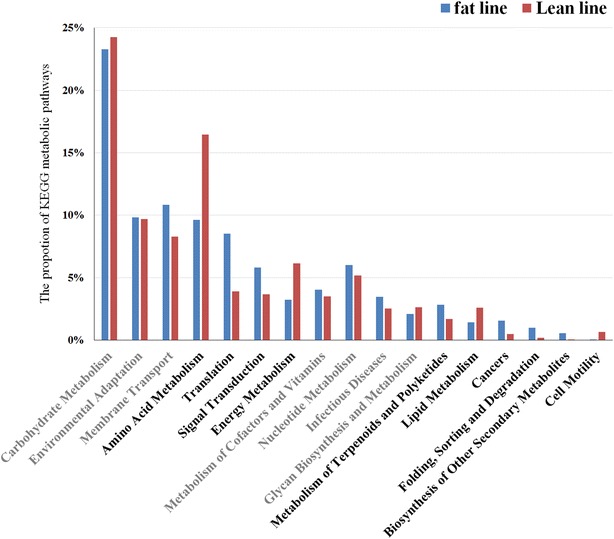
Distribution of KEGG metabolic pathways in the fat and lean lines. Profile bar plots show the relative proportion of each metabolic pathway. The pathways labeled in black were differentially expressed (fold change >1.5)
Based on the analysis of whole microbial genome sequencing data, we observed that enriched markers were frequently involved in the functional pathways of inositol phosphate metabolism, antigen processing and presentation, and phosphonate and phosphinate metabolism in the lean line. In contrast, the gut microbiota of the fat line showed enrichment in citrate cycle, other types of o-glycan biosynthesis, peroxisome proliferator activated receptor (PPAR) signaling pathway, carbon fixation in photosynthetic organisms, ribosomes, and cell adhesion molecules (Fig. 3). A hierarchy cluster heatmap was generated to visualize the distribution of microbial functions in the fat and lean lines (Fig. 4). The microbial functions were also matched to the microbial metabolic pathway results from the study on obese mice [31]. The heatmap results suggested that aminotransferase, arginine decarboxylase, cytochrome o ubiquinol oxidase subunit III, and 1-phosphatidylinositol-3-phosphate 5-kinase, which are involved in amino acid metabolism, energy metabolism, and carbohydrate metabolism respectively, were more abundant in the lean line than in the fat line (Fig. 4) and [see Additional file 6: Table S5]. Compared to the lean line, the gut microbiota in the fat line had a higher functional performance related to bacitracin transport system permease protein, citrate (pro-3s)-lyase ligase, and ribonucleoside-diphosphate reductase alpha chain, which are respectively involved in immune system diseases, signal transduction and nucleotide metabolism (Fig. 4) and [see Additional file 6: Table S5].
Fig. 3.
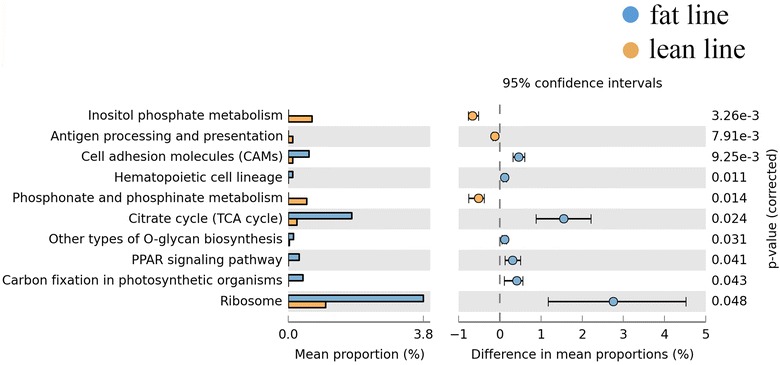
Significant differences in microbial metabolism pathways between the fat and lean lines
Fig. 4.

Heatmap of microbial function pathways in the fat and lean lines. Colors reflect relative abundance from low (green) to high (red); detailed categories for each gene are in Table S5 [see Additional file 6: Table S5]
Discussion
Previous studies reported that the genetics of the host can influence the abundance and composition of gut microbiota. In this study, significant differences in 11 microbial species were observed between the fat and lean lines [see Additional file 5: Table S4]. Among these species, Pectinatus frisingensis, Lactobacillus salivarius, and Micrococcus sp. SMCC ZAT352 were found to be associated with adipogenesis. For example, P. frisingensis synthesizes lipopolysaccharide with polymeric O-specific chains that are related to host obesity [32], L. salivarius can modify the fecal microbiota, which in turn affects metabolic pathways in obese chickens and humans [33, 34], and Micrococcus is involved in lipolytic activity, which shows a positive correlation with fatty acid biosynthesis [35]. Although 16S rDNA amplicon sequencing was the primary method used to analyze microbial diversity, we also used the computation tool PICRUSTs [36] to predict microbial community functions [see Additional file 7]. Functional prediction results revealed that signal transduction mechanisms and fatty acid biosynthesis were more abundant in the fat line than in the lean line and this was consistent with the results of the microbial composition of the gut microbiota [see Additional file 5: Table S4 and Additional file 8: Figure S2].
Several studies have shown that multiple transcription factors and signaling pathways are involved in the regulation of adipogenesis [37–39]. PPAR plays an important role in adipogenesis, adipocyte gene expression, and fat cell differentiation, which promote lipid storage and metabolism [40, 41]. Moreover, the PPAR signaling axis is also a potential target for the modulation of adipogenesis [42]. Interestingly, our whole microbial genome sequencing results suggested that, compared to the lean line, the PPAR signal pathway of gut microbiota in the fat line had a significantly higher functional performance (Fig. 4). This suggests the possibility that the PPAR signal pathway may also be involved in lipid storage. The citrate cycle is another key metabolic pathway that unifies carbohydrate, lipid, and protein metabolism. A significant correlation between citrate synthase level and obesity, together with a decreased activity of this enzyme in the mitochondria of human omental adipose tissue, were reported in obese humans [43]. Previous studies showed that citrate synthase activity was suppressed in obese mice, resulting in excessive carbon flow into the citrate cycle prompting energy storage [44]. Gut microbiota appears to play a key role in the development and progression of obesity, together with changes in citrate synthase activity [45, 46]. In this study, analysis of the results of whole microbial genome sequencing suggested that enrichment in the microbial function that relates to citrate cycle was significantly different between the fat and lean lines (Fig. 4). We have reasons to believe that microbes undertake many metabolic tasks, and that functional interactions between host genetic factors and gut microbiota are inevitable.
Conclusions
We found that long-term divergent selection for abdominal fat has considerable influence on the abundance and composition of gut microbiota by altering the frequencies of obesity-related alleles. Furthermore, whole microbial genome sequencing results revealed that functional activities of the microbiota, such as those related to the citrate cycle and PPAR signaling pathway, differed significantly between the fat and lean lines and were affected by the gut microbiota and by differences in frequencies of host alleles. Our results provide further evidence for the hypothesis that host genetic factors interact and co-evolve with gut microbiota.
Authors’ contributions
JD, YZ and HM wrote the manuscript. JD and LZ performed statistical analysis and prepared the necessary scripts. JD, HZ, HL and HM conceived and designed the experimental procedure and supervised the study. LW, WZ, ZZ, LL, YW and CH participated in sample collection, DNA extraction, and data processing. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Christa F. Honaker for editing the manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Funding
This study was supported by the National Science Foundation of China (Grant No. 31572384) and the National High Technology Research and Development Program of China (Grant No. 2011AA100901).
Additional files
Additional file 1: Table S1. Summary of sequencing data for all samples.
Additional file 2: Table S2. Numbers of taxa at each level.
Additional file 3: Figure S1. Alpha diversity of bacteria in the gut microbiota of fat and lean lines. Boxes indicate the IQR (75th to 25th of the data). The median value is shown as a line within the box and the mean value as a star. Whiskers extend to the most extreme value within 1.5 * IQR. Outliers are shown as crosses. Higher chao1 suggests greater richness of microbes. Higher Shannon suggests greater diversity of microbes.
Additional file 4: Table S3. Comparison of microorganisms in the gut microbiota at the phylum level between the fat and lean lines. This table lists the relative fold change and p value of the differences in microorganisms at the phylum level between the fat and lean lines.
Additional file 5: Table S4. Comparison of microorganisms in the gut microbiota at the species level between the fat and lean lines. This table lists the relative fold change and p value of the differences in microorganisms in the gut microbiota at the species level between the fat and lean lines.
Additional file 6: Table S5. Corresponding metabolic pathways of the significant functions in the heatmap (related to Fig. 4). This table lists the corresponding metabolic pathways that are in Fig. 4.
Additional file 7. Prediction of microbial functions. This file describes the method for functional prediction based on 16S rDNA sequencing data.
Additional file 8: Figure S2. Predicted functional pathways in fat and lean lines based on 16S rDNA sequencing data.
Footnotes
Jinmei Ding, Lele Zhao and Lifeng Wang contributed equally to this work
Contributor Information
Jinmei Ding, Email: dingjinmei@sjtu.edu.cn.
Lele Zhao, Email: lelezhao2011@163.com.
Lifeng Wang, Email: jmd721@163.com.
Wenjing Zhao, Email: zhaodalv@qq.com.
Zhengxiao Zhai, Email: zhaizx@hotmail.com.
Li Leng, Email: lengli1981@163.com.
Yuxiang Wang, Email: wyx2000@neau.edu.cn.
Chuan He, Email: hehc2005@sina.com.
Yan Zhang, Email: yanzhang@personalbio.cn.
Heping Zhang, Email: hepingdd@vip.sina.com.
Hui Li, Email: lihui@neau.edu.cn.
He Meng, Email: menghe@sjtu.edu.cn.
References
- 1.Waldor MK, Tyson G, Borenstein E, Ochman H, Moeller A, Finlay BB, et al. Where next for microbiome research? PLoS Biol. 2015;13:e1002050. doi: 10.1371/journal.pbio.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley RE, Backhed F, Turnbaugh PJ, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 6.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf G. Gut microbiota: a factor in energy regulation. Nutr Rev. 2006;64:47–50. doi: 10.1111/j.1753-4887.2006.tb00173.x. [DOI] [PubMed] [Google Scholar]
- 9.Simon J, Leclercq B. Longitudinal study of adiposity in chickens selected for high or low abdominal fat content: further evidence of a glucose-insulin imbalance in the fat line. J Nutr. 1982;112:1961–1973. doi: 10.1093/jn/112.10.1961. [DOI] [PubMed] [Google Scholar]
- 10.Hermier D, Quignard-Boulangé A, Dugail I, Guy G, Salichon MR, Brigant L, et al. Evidence of enhanced storage capacity in adipose tissue of genetically fat chickens. J Nutr. 1989;119:1369–1375. doi: 10.1093/jn/119.10.1369. [DOI] [PubMed] [Google Scholar]
- 11.Zhao LL, Wang G, Siegel P, He C, Wang H, Zhao WJ, et al. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci Rep. 2013;3:1163. doi: 10.1038/srep01163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng H, Zhang Y, Zhao LL, Zhao WJ, He C, Honaker CF, et al. Body weight selection affects quantitative genetic correlated responses in gut microbiota. PLoS One. 2014;9:e89862. doi: 10.1371/journal.pone.0089862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota. Proc Natl Acad Sci USA. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo L, Sun B, Shang Z, Leng L, Wang Y, Wang N, et al. Comparison of adipose tissue cellularity in chicken lines divergently selected for fatness. Poult Sci. 2011;90:2024–2034. doi: 10.3382/ps.2010-00863. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Wang SZ, Wang ZP, Da Y, Wang N, Hu XX, et al. A genome-wide scan of selective sweeps in two broiler chicken lines divergently. BMC Genomics. 2012;13:704. doi: 10.1186/1471-2164-13-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang HB, Li H, Wang QG, Zhang XY, Wang SZ, Wang YX, et al. Profiling of chicken adipose tissue gene expression by genome array. BMC Genomics. 2007;8:193. doi: 10.1186/1471-2164-8-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Hu XX, Wang ZP, Zhang YD, Wang SZ, Wang N, et al. Selection signature analysis implicates the PC1/PCSK1 region for chicken abdominal fat content. PLoS One. 2012;7:e40736. doi: 10.1371/journal.pone.0040736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 22.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 25.Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26:715–721. doi: 10.1093/bioinformatics/btq041. [DOI] [PubMed] [Google Scholar]
- 26.Welch BL. The generalisation of student’s problems when several different population variances are involved. Biometrika. 1947;34:28–35. doi: 10.1093/biomet/34.1-2.28. [DOI] [PubMed] [Google Scholar]
- 27.Lu J, Domingo JS. Turkey fecal microbial community structure and functional gene diversity revealed by 16S rRNA gene and metagenomic sequences. J Microbiol. 2008;46:469–477. doi: 10.1007/s12275-008-0117-z. [DOI] [PubMed] [Google Scholar]
- 28.Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One. 2011;6:e27949. doi: 10.1371/journal.pone.0027949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waite DW, Taylor MW. Characterizing the avian gut microbiota: membership, driving influences, and potential function. Front Microbiol. 2014;5:223. doi: 10.3389/fmicb.2014.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An Y, Xu W, Li H, Lei H, Zhang L, Hao F, et al. High-fat diet induces dynamic metabolic alterations in multiple biological matrices of rats. J Proteome Res. 2013;12:3755–3768. doi: 10.1021/pr400398b. [DOI] [PubMed] [Google Scholar]
- 31.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 32.Vinogradov E, Li J, Sadovskaya I, Jabbouri S, Helander I. The structure of the carbohydrate backbone of the lipopolysaccharide of pectinatus frisingensis strain VTT E-79104. Carbohydr Res. 2004;339:1637–1642. doi: 10.1016/j.carres.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Guban J, Korver DR, Allison GE, Tannock GW. Relationship of dietary antimicrobial drug administration with broiler performance, decreased population levels of Lactobacillus salivarius, and reduced bile salt deconjugation in the ileum of broiler chickens. Poult Sci. 2006;85:2186–2194. doi: 10.1093/ps/85.12.2186. [DOI] [PubMed] [Google Scholar]
- 34.Larsen N, Vogensen FK, Gobel RJ, Michaelsen KF, Forssten SD, Lahtinen SJ, et al. Effect of Lactobacillus salivarius Ls-33 on fecal microbiota in obese adolescents. Clin Nutr. 2013;32:935–940. doi: 10.1016/j.clnu.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Coppola R, Iorizzo M, Saotta R, Sorrentino E, Grazia L. Characterization of micrococci and staphylococci isolated from soppressata molisana, a southern Italy fermented sausage. Food Microbiol. 1997;14:47–53. doi: 10.1006/fmic.1996.0062. [DOI] [Google Scholar]
- 36.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi H, Wang Q, Wang Y, Leng L, Zhang Q, Shang Z, et al. Adipocyte fatty acid-binding protein: an important gene related to lipid metabolism in chicken adipocytes. Comp Biochem Physiol B Biochem Mol Biol. 2010;157:357–363. doi: 10.1016/j.cbpb.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Di LJ, Noguchi CT. AMPK is involved in mediation of erythropoietin influence on metabolic activity and reactive oxygen species production in white adipocytes. Int J Biochem Cell Biol. 2014;54:1–9. doi: 10.1016/j.biocel.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Di LJ. Wnt/β-catenin mediates AICAR effect to increase GATA3 expression and inhibit adipogenesis. J Biol Chem. 2015;290:29759. doi: 10.1074/jbc.A115.641332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Investig. 1997;99:2416–2422. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 42.Jiang X, Huang L, Xing D. Photoactivation of Dok1/ERK/PPAR gamma signaling axis inhibits excessive lipolysis in insulin-resistant adipocytes. Cell Signal. 2015;27:1265–1275. doi: 10.1016/j.cellsig.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Christe M, Hirzel E, Lindinger A, Kern B, von Flüe M, Peterli R, et al. Obesity affects mitochondrial citrate synthase in human omental adipose tissue. ISRN Obes. 2013;2013:826027. doi: 10.1155/2013/826027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cummins TD, Holden CR, Sansbury BE, Gibb AA, Shah J, Zafar N, et al. Metabolic remodeling of white adipose tissue in obesity. Am J Physiol Endocrinol Metab. 2014;307:E262–E277. doi: 10.1152/ajpendo.00271.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta. 2013;10:1518–1532. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferramosca A, Conte A, Zara V. Krill oil ameliorates mitochondrial dysfunctions in rats treated with high-fat diet. Biomed Res Int. 2015;2015:645984. doi: 10.1155/2015/645984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.