

Abstract
Geneticists have long sought the ability to manipulate vertebrate genomes by directly altering the information encoded in specific genes. The recently discovered CRISPR-Cas9 endonuclease has the ability to bind single loci within vertebrate genomes and generate double-strand breaks at those sites. These double stranded breaks induce an endogenous double-strand break repair response that results in small insertions or deletions at the targeted site. Alternatively, a template can be supplied in which case homology-directed repair results in generation of engineered alleles at the break site. These changes alter the function of the targeted gene facilitating the analysis of gene function. This tool has been widely adopted in the zebrafish model; we discuss the development of this system in the zebrafish and how it can be manipulated to facilitate genome engineering.
Keywords: CRISPR-Cas9, Zebrafish, Genome editing, Knockout, Knock-in, genetic screen
The CRISPR-Cas System
The CRISPR-Cas9 system is a diverse family of endonucleases found in archaeal and bacterial genomes that have been adapted for use in engineering metazoan genomes [1,2]. The best studied CRISPR-Cas9 system is from the bacteria S. pyogenes and this is the system that has been used for most zebrafish genome engineering work. The endonuclease consists of a large protein encoded by the cas9 gene and two small RNA molecules, tracRNA and crRNA, which form a complex (see Glossary). The small RNAs are encoded by the tracr gene and cleavage products of the CRISPR repeat locus, respectively [3-7]. The complex between the Cas9 protein, the crRNA, and the tracRNA is referred to as the Cas9 holoendonuclease. The Cas9 protein has a domain that binds to a short sequence of DNA, referred to as the Protospacer Adjacent Motif (PAM), but most of the DNA-binding specificity comes from basepairing between the 20 nucleotide “spacer” domain of the crRNA:tracrRNA duplex and the target DNA strand. Successful basepairing of the spacer domain of the crRNA to the genomic DNA target activates two endonuclease domains in Cas9, which each cleave a single strand of the DNA target. Because the spacer provides 20 nucleotides of specificity to the holoendonuclease in addition to the two nucleotides of specificity provided by the PAM, the holoendonuclease has sufficient specificity to uniquely cleave DNA at a single site in a metazoan organism. The generation of a double strand break (DSB) in genomic DNA is toxic and is rapidly repaired by an endogenous DSB repair system (Box 1). One repair mechanism, non-homologous end joining results in small insertions or deletions at the site of the DSB. The resulting alleles are generally knock-out alleles as they are predicted to ablate gene function. A second repair mechanism, homology directed repair, uses a template, which can be supplied by the geneticist or can be the sister chromosome, to generate alleles of a specified sequence. This second approach is considerably more difficult than the first.
Box 1. Double-stranded DNA break repair and mutagenesis.
Double stranded DNA breaks (DSBs) can result from physical stress on chromosomes or nuclei, ionizing radiation, mitosis, or exogenously added nucleases like the Cas9-holoenzyme. Cells have multiple mechanisms to identify and repair DSBs. DSB repair falls into two general categories which vary in their fidelity: homology-directed repair (HDR) and non-homologous repair (NHR) which includes non-homologous end joining (NHEJ) or alternative end joining (alt-NHEJ) repair (Figure I) [74,75]. HDR uses the sister chromosome as the preferred template to repair the DSB and this templated repair therefore has high fidelity. In contrast, NHR processes vary in mechanism, but involve exonuclease resection, fill-in, and ligation of the broken ends. This process is error prone, generally resulting in the creation of a small insertion or deletion (indel) at the cleavage site. The persistent activity of an exogenous site-specific nucleases (for example Cas9) favors NHR since HDR regenerates the target that is a substrate for subsequent nuclease cleavage. If HDR occurs, it will result in repair to the sister chromosome that is itself cleavable. A futile cycle of cleavage and repair to the wildtype may occur until an error in the repair process results in a change to the target. Experimentally, what can be observed are the non-faithful repair events mediated by NHR pathways, though it is possible that faithful repair has occurred prior to the non-faithful event which locks the target in an uncleavable state. HDR in the presence of a user generated template lacking the cleavage site will convert the wt allele to the templated allele.
Evidence supports NHR as the observed DSB repair pathway induced by Cas9 generated DSBs. Ligation of the broken DNA ends through the canonical, ligase IV-dependent, NHEJ (C-NHEJ) process has been observed [76]. The alternative NHEJ (alt-NHEJ) relying on ligase III has also been suggested to mediate repair following Cas9 cleavage [77]. Moreover, the DNA polymerase polq, essential for alt-NHEJ, is required for repairing DSB generated either by CRISPR-Cas9 or by ionizing radiation through the 1000-cell stage, though this requirement is not stringent by the dome stage [78]. polq mutants injected with highly active holo-Cas9 directed to any of 3 targets generate indels at greatly reduced frequency. This result strongly implicates alt-NHEJ as the dominant response in most CRISPR-Cas9 mutagenesis experiments.
Since the seminal work that used CRISPR-Cas9 in genome engineering in vitro [8] and in cultured cells in 2013 [9-11], the system has been engineered to modify gene function, gene expression, and even to mark nucleic acids fluorescently. One of the first modifications of this system was to fuse the crRNA:tracRNA duplex into a single guide RNA (sgRNA) that is fully functional [8]. This sgRNA architecture has been used in most subsequent CRISPR-Cas9 applications. The Cas9 protein was also modified by the addition of nuclear localization sequences in order to target the endonuclease activity to the nucleus in eukaryotes. This review focuses on the use of CRISPR-Cas9 in the zebrafish model and the ways that CRISPR-Cas9 are allowing geneticists to explore biological processes that were technically inaccessible prior to the development of these tools. The first section describes the use of CRISPR-Cas9 to direct mutations to a single gene, the second section discusses the use of transgenically expressed CRISPR-Cas9, and the third section describes how these approaches can be expanded to target multiple genes which is likely to open new fields to genetic study.
Targeted mutagenesis in zebrafish
Although several endonucleases with specificity for a single site in a vertebrate genome have been developed for directed mutagenesis experiments, the RNA-directed endonuclease activity of Cas9 was immediately recognized as a game-changing advance in directed mutagenesis experiments (Box 2). The endonuclease generated DSBs repair can be manipulated so that gene function is lost (targeted gene knock-out) or so that new function is added to a gene (targeted knock-in).
Box 2. Limitations of Zinc Finger Nucleases and TALE Nucleases.
Before the discovery of the CRISPR-Cas9 system, Zinc Finger Nucleases (ZFNs) and Transcription Activator-Llike Effector Nucleases (TALENs) were used in zebrafish to edit the genome [54-57]. Like CRISPR-Cas9, ZFN and TALEN generate site-specific double-stranded breaks (DSBs) in vivo. It has long been realized that repair of a DSB would likely be sufficient to introduce a mutation at that site [58,59]. Each of these endonucleases is modular with target recognition provided by one or more DNA-binding modules and DNA cleavage provided by endonuclease modules. The development of these reagents as genetic tools was driven by the ability to manipulate the binding specificity of the DNA binding modules so that they recognize at least 20 nucleotides of genomic DNA.
Zinc-finger proteins (ZF) were identified as promising candidates for target-specific binding as each ZF domain is stable, folds independently, and recognizes 3 nucleotides. These modular DNA-binding domains can be linked together into a single ZF protein [60-63]. Two engineered ZF proteins recognizing adjacent stretches of genomic DNA on opposite strands in a head-to-head orientation are each fused to a FokI endonuclease domain making the ZF nuclease (ZFN, Figure I). Dimerization, mediated by the FokI domains, is required for endonuclease activity [64-66]. While identification of ZFN proteins that are specific for a single genomic target have been successful for many targets, the work required to identify these proteins is considerable. Because the specificity of each zinc finger is context dependent [67], successful generation of in vivo functional ZFNs, even by highly experienced groups, is not assured. The number of available ZF modules also limits the number of target sites in the genome [56].
TALENs employ modular single nucleotide DNA-binding domains derived from Transcription Activator-Like Effector (TALE) domains that are not context-dependent [68-71]. Like ZFNs they are composed of two TALE proteins that bind adjacent genomic sequences and are linked through dimerization of the FokI endonuclease domains. The TALE proteins have far fewer restrictions on the choice of sequence, requiring a single T at the 5′ end of each binding site [68,69], however they cannot distinguish T from methylated C in the major groove [72,73].
ZFN and TALEN have several disadvantages compared to the CRISPR/Cas9 system. First, ZFN and TALEN require the de novo design and synthesis of two new proteins for each target rather than the de novo design and synthesis of a single short RNA which requires simply ordering a single DNA oligo. Second, even with a DNA-binding domain that has demonstrated DNA-binding activity, there is no guarantee that the proteins will be mutagenic in vivo. Third, the FokI endonuclease used in both ZFN and TALEN is sensitive to DNA methylation while Cas9 is not [15].
Targeted knock-out
Generating knock-out alleles in zebrafish using CRISPR-Cas9 is rapid and not difficult. The basic procedures used are outlined here. Zebrafish lines carrying homozygous CRISPR-cas9 mutant alleles can be obtained in just two generations or less (Key Figure, Figure 1). Selection of the sgRNA target sequence is guided by heuristic rules developed from analysis of the cutting efficiency of different sgRNA molecules in vivo (Box 3). Multiple databases (Box 4) are available online showing all available guides in the genomes of commonly used model systems [15-21]. Following purchase of a DNA oligomer template of the sgRNA, in vitro synthesis of the sgRNA, and its purification can be performed with commercially available reagents using standard molecular biology techniques [12-14]. The sgRNAs are injected directly into the zebrafish zygote either with in vitro synthesized mRNA encoding a nuclear localized Cas9 or commercially available Cas9 protein with a nuclear localization sequence [12,22-25]. Low fidelity DSB repair (Box 1) occurs at each target of each diploid cell independently resulting in the generation of distinct alleles. Sequence analysis of the targets in the injected animals show that many alleles are generated in each animal. The injection of preformed Cas9 protein-sgRNA holoendonuclease into the 1-cell stage embryo also results in highly mosaic F0 animals [12], suggesting that translation of the injected cas9 mRNA is not the limiting step in generating endonuclease activity. Injection of the zygote or early blastula stage embryo thus generates highly mosaic zebrafish embryos that grow into mosaic adults. The mosaic adults are either in-crossed to another animal with the same gene targeted or out-crossed to wildtype lines. The offspring of the in-cross will contain transheterozygous offspring which can be analyzed for phenotype, while the out-cross will contain heterozygotes which can be raised and in-crossed to generate homozygous mutants. Germline mutations can be identified in the offspring in both cases using standard molecular biology methods.
Figure 1. Key Figure, Strategy for zebrafish genome engineering with CRISPR-Cas9.
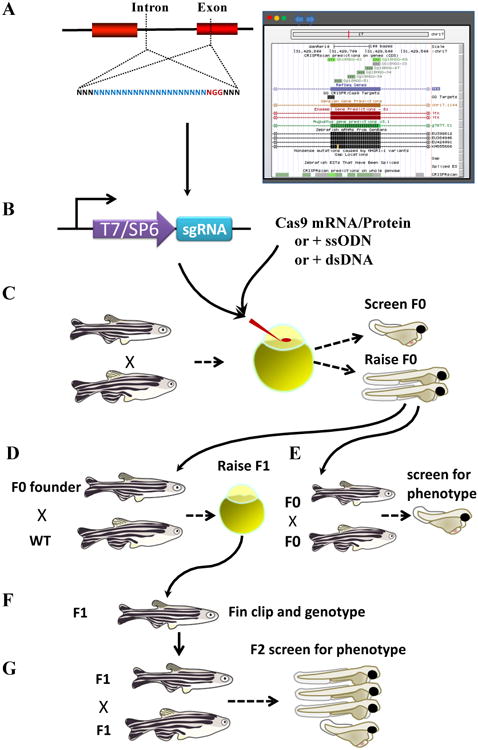
Schematic depiction of the steps in a zebrafish CRISPR experiment. The target is selected (A), the sgRNA is synthesized by in vitro RNA synthesis (B), and injected together with Cas9 protein or cas9 mRNA into the 1-cell stage zebrafish (C). Mutagenesis efficiency is evaluated for each sgRNA using various methods including T7 endonuclease, heteroduplex mobility shift (HMA), and sequencing (D). The phenotype of the F0 animals can also be assessed. Successfully mutagenized F0 animals are raised to adulthood and crossed to WT animals; or crossed to other F0 and the phenotype of the offspring assessed (E). F1 animals are genotyped and sequenced (F) and crossed either to siblings or to independently generated alleles in the same gene (G).
Box 3. Sequence constraints on the binding of the Cas9 holoendonuclease.
Both the sequence requirements of the Cas9 protein and the synthetic requirements of the RNA polymerases that synthesize the sgRNA constrain target selection. The S. pyogenes Cas9 PAM sequence 5′-nGG occurs theoretically every 42 = 16 nucleotides on each strand in the genome. The RNA polymerases that synthesize the sgRNA in vitro require conserved nucleotides at their 5′ ends; for example, T7 RNA polymerase requires 5′-GG which also occurs every 16 nucleotides. Alternatively, SP6 RNA polymerase can initiate with 5′-G[A/G] doubling the number of potential sites [12]. So a simplistic model of target frequency would use the sequence GGN18nGG [23], where N refers to the target sequence and n refers to the non-specific PAM nucleotide. This sequence will occur every 44 = 256 nucleotides per strand in a random genome, or every 128 nucleotides. As the median exon size is 124 nucleotides [79], many exons will have at least one target matching this sequence. The actual frequency of target sites is greater than this as a mismatch at the 5′ end of the target is allowed and targets as short as 18 nucleotides are also cleavable [16,80,81]. This increases the number of possible sites more than 8 fold, allowing most exons to have multiple targets.
Various groups have found experimentally that some targets are cleaved more efficiently than others. Heuristic rules have been derived from systematic assays of cleavage efficiency of very large libraries of sgRNAs [82-84]. In zebrafish, optimal target sites should have 50% G/C content and a G adjacent to the PAM; G-rich and A-depleted sgRNAs are also more stable and efficient [12,13,16]. Also, secondary structure in the target can interfere in target recognition [24]. Some sequences occur in near exact copies in the genome which can result in off-target effects. Off-target sites can be identified using common bioinformatics tools [15] and assessment of potential off-target activity is generally included in the public target databases. More commonly the Cas9 holoenzyme may bind non-productively to sites that imperfectly match the target [80,85-87]. Binding non-productively to many sites may result in reducing the available free Cas9 holoenzyme preventing cleavage at the correct target, a phenomena referred to as ‘distraction’ [16]. Targets with many distraction targets can also be removed from the database further increasing the likelihood of selecting a high efficiency sgRNA [16].
Box 4. CRISPR experimental design and informatics tools.
Perhaps the greatest risk in experimental science is false positive results, in genetic experiments this is most likely to occur with off-target effects. Reagent design is insufficient to prevent errors. Errors can be avoided however through careful experimental design and appropriate controls. There are two principle ways to avoid mistaking off-target events for on-target phenotype. The first is to design and use multiple different sgRNA molecules targeting the same gene; null mutations in the same gene should have identical phenotypes and should not complement each other. The second is to perform a linkage analysis to assure that phenotype and genotype cosegregate. It is relatively trivial to genotype 48 animals from several different crosses, which should provide sufficient mapping resolution so that unlinked off-targets are readily identified.
The single most important design decision is the choice of the target site within the gene of interest. Good CRISPR target sites can be found in most exons of a target gene, so site choice is driven by experimental needs, not the engineering constraints of the endonuclease. Often the 5′ most exon is targeted to generate functionally null alleles; however, caution should be used when choosing a 5′ exon of a predicted ORF, as it may be incorrect. Three targeting approaches are suggested. The first two can be used either in generating germline or F0 mutants while the fourth is recommended only for making germline mutants. First, target sequences that encode a conserved and functionally important component of the gene, for example the DNA binding domain or active site of an enzyme. Thus, even if the alleles generated retain their open reading frame, the protein activity will be greatly reduced or abrogated. Second, use two targeting sgRNAs together to generate multiple mutations and/or a deletion. Third, use HDR to insert an in-frame stop codon in a well characterized exon of the target gene.
Following the choice of exon or exons, several different databases can be consulted for targets that are likely to be effective. Different databases have different strengths and use different scoring algorithms. For example, the CHOPCHOP database has a convenient web interface that also provides genotyping oligo pairs. Both the CHOPCHOP and the CRISPR-Scan database are integrated directly with the UCSC genome browser allowing the user to readily find targets relative to other annotations. We find that this is particularly useful when targeting the NHGRI line [88] as a separate track showing the polymorphisms present in this line and between this line and the standard genome sequence [79] can be displayed. Polymorphisms can affect target efficiency and genotyping assays. Our approach is to choose 3 or 4 different target sites that score highly in more than one program.
CRISPRScan (http://www.crisprscan.org)
Optimized CRISPR Design (http://crispr.mit.edu/)
E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html)
CHOPCHOP (http://chopchop.cbu.uib.no/)
CasBLASTR (http://www.casblastr.org)
CT-Scan (http://gt-scan.braembl.org.au/gt-scan/)
CRISPRz (http://research.nhgri.nih.gov/crisprz)
CRISPRdirect (http://crispr.dbcls.jp/)
ZiFiT Targeter (http://zifit.partners.org/ZiFiT/)
WU-CRISPR (http://crispr.wustl.edu/)
Mutagenesis of an incorrect target, i.e. off-target mutagenesis, may result in attribution of genetic function to the wrong gene. Good experimental design can avoid this error (Box 4). In cell culture systems, off-target DSB events may occur in cells with high-level constitutive expression of cas9 activity and any off-target effects may accumulate with time. Both the cas9 mRNA/sgRNA and Cas9 protein/sgRNA injection techniques used in zebrafish laboratories result in very robust, but transient, expression of the holoendonuclease; transient expression may assist in keeping off-target events to a minimum [12,16,22,24,26]. A second class of off-target events that may occur as a result of a DSB is the formation of a chromosomal rearrangement. The genotyping procedures used to identify mutant alleles, PCR reactions that span the site of the CRISPR-cas9 induced DSB, will not detect chromosomal rearrangements. As a result only small indels, not chromosomal rearrangements, will generate mutant lines. Any rare off target events can be bred out of the targeted line and the phenotype should be confirmed by linkage analysis for any new alleles (Box 4). These procedures are well established practices for all chemically induced mutant alleles as the founder animals are very heavily mutagenized [49,50].
Targeted Knock-in
Targeted integration of an exogenous DNA sequence is desired for a range of applications. Generation of targeted CRISPR-Cas9 alleles of a gene removes that genetic function. In contrast, targeted knock-in adds function to a gene. Knock-in events may change the open reading frame to recapitulate a patient allele, insert a restriction site to assist genotyping, fuse the open reading frame to a fluorescent protein, or insert genetic elements that can be used to later change the locus, for example by the insertion of loxP, I-SceI, or phiC31 integration sites. In mouse, knock-in occurs via homologous recombination (HR) in ES cells in culture [27]. In most cases, quite long regions of homology (“homology arms”), spanning several thousand bp or more are required flanking the sequence to be inserted [28]. These HR events, while rare, can be identified in cell culture systems where many cells can be manipulated and the HR products can be selected for directly. In zebrafish, it is impractical to inject sufficient numbers of blastulas to obtain successful HR events; however, since DSBs can stimulate HR by more than 1000-fold in ES cells [29], the availability of ZFN, TALEN and CRISPR-Cas9 has prompted the development of knock-in methods in zebrafish.
A number of successful TALEN and CRISPR-Cas9 mediated knock-ins in zebrafish have been reported which have incorporated small changes, such as SNPs, stop codons, HA tags, and loxP sites [12,30-33]. Because the molecular mechanism of these knock-ins has not yet been elucidated, they are referred to as homology directed repair (HDR) rather than HR which has a well understood mechanism. The most straightforward approach to use HDR is to use single-stranded DNA oligodeoxynucleotides (ssODNs) as donors. Several studies have used ssODNs with homology arms between 20 and 50 bp to incorporate small changes at the CRISPR-Cas9 induced DSBs. Most integrations are imprecise [12,25,30,34,35] suggesting that the mechanism may not be HR as seen in ES cells. In many cases, precise integration is not necessary, such as intronic insertion of loxP sites or exonic insertion of a cassette containing stop codons in all frames [12,25]. Although the fecundity of zebrafish allows identification of rare individuals with precise integration, improvements in the frequency of HDR are still desirable. One possible solution is to use longer homology arms. Using rescue of the albino (alb) phenotype as a sensitive assay of HDR, it was found that co-injection of donor plasmid DNA with homology arms of at least 986 bp in length flanking the mutant SNP results in precise HDR [36]. Importantly, linearization of the donor DNA in vivo with Cas9 improves the HDR efficiency [36]. Germline transmission frequency still has plenty of room for improvement and generating donor vectors with long homology arms is certainly more difficult than ordering a single oligonucleotide. Mechanistic studies currently underway should provide guidance to improve the ssODN-mediated HDR frequency. For example, a recent study in cell culture indicates that Cas9 remains tightly bound to 3 of the 4 strands at the DSB, leaving the PAM distal non-target strand accessible. Donor ssODN with optimal homology arms complementary to the non-target strand results in higher HDR frequency [37]. Similar design improvements may increase the precision and the frequency of HDR events in zebrafish.
Another commonly used tool in zebrafish is the use of reporter genes. CRISPR-Cas9 directed DSB together with plasmid-based donors with long, short, or no homology arms have been used to knock-in reporter genes. Reporter expression may result from HDR or homology-independent repair events (Figure I). One study used a 200 bp fragment of EGFP coding sequence as “bait” to convert EGFP into Gal4 in existing transgenic lines. Although very efficient (up to 75%) in-frame fusion of Gal4 was found, there were also imprecise insertions. Further analyses indicate that homology is not required for targeted insertion [38]. However, one study showed that homology arms as short as 10 bp can result in precise knock-in at the target site in 60% of somatic integrations. Increase in the length of the homology arms to 40 bp improves the rate of precise integration to 77% of all somatic integrations [39]. The reason for the difference is unknown, but may be locus specific. The prevalence of homology-independent repair led to an alternative approach to generate fusion proteins at the endogenous locus. In this approach, a DSB is generated in the intron upstream of the last coding exon. A construct containing an in-frame fusion of EGFP and a polyadenylation sequence to the last coding exon of the target gene is inserted at the DSB by non-homologous repair [40]. This results in splicing to the fusion exon, marking the protein product with the GFP fusion. Many variants of this procedure are possible.
Figure I. Genomic engineering using ZFN, TALEN, and the CRISPR-Cas9 endonucleases.

The three different endonucleases can be designed to site-specifically recognize a single-site in a vertebrate genome. Both ZFN and TALEN are constructed as dimers recognizing two sites in a head-to-head orientation (indicated in green). Modular ZF or TALE domains are used to recognize the specific locus while the FokI endonuclease domain cuts the target. The FokI dimerization interface has been engineered to only allow formation of heterodimers. Expression of both proteins is required to make a functional endonuclease. CRISPR-Cas9 is a complex between the Cas9 protein and a small RNA molecule, the sgRNA. Only the 5′ end of the sgRNA need be changed to change the specificity of this endonuclease. All three endonucleases generate double stranded DNA breaks (DSB) which are repaired by the endogenous DNA repair machinery. This can occur without a template by NHEJ mechanisms or can be templated by exogenous DNA for example in HDR or can incorporate exogenously supplied DNA in a homology independent process. The exact mechanisms of repair are currently being investigated.
It is often useful to block gene function spatially (in particular cells or tissues) or temporally (only at particular developmental time points). This can be done with conditional alleles. In vertebrates conditional alleles generally use site-specific recombination between two inserted genetic elements flanking an exon. For example using the cre recombinase to invert or excise an exon flanked by two loxP sites, ablating the function of the gene. Although intronic recombinase sites (e.g. loxP) may be “knocked-in” via ssODN-mediated integration [25,30], the difficulty of introducing 2 sites in the same gene in one generation is considerable. Plasmids with an excisable donor containing long homology arms and a selectable marker similar to gene targeting vector in mouse ES cells may be more practical [33], particularly if germline transmission efficiency is improved.
Manipulating genes using transgenic expression of CRISPR-Cas9
The previous section described the use of CRISPR-Cas9 by transient introduction of the Cas9-holoenzyme from Cas9 protein or cas9 mRNA in blastula stage embryos. Transgenic expression of a Cas9-holoenzyme that targets the gene-of-interest in selected tissues or at selected times can be used analogously to conditional alleles. A distinct advantage of conditional mutagenesis by transgenic expression of the Cas9-holoenzyme is that it can be achieved in one generation, regardless of how many genes are targeted. In contrast, spatiotemporal control of gene inactivation using recombinase-dependent conditional alleles requires engineering at the locus and more breeding, especially when targeting multiple genes. Transgenic expression of the Cas9 holoenzyme using Tol2 transposons leverages the ease and efficiency of the Tol2 transgenesis system with the specificity and modularity of the CRISPR-Cas9 system. Two conceptually similar approaches to transgenic expression of the Cas9 holoendonuclease have been reported in zebrafish (Figure 2) [41,42]. In one approach, a single Tol2 transposon was used to express both cas9 and the targeting sgRNA [42]. This approach can successfully ablate the target gene in the desired tissue and requires the generation and injection of only a single transposable element. The second approach uses two different Tol2 transposons, one that expresses a tissue specific or otherwise regulatable cas9 gene. The other Tol2 element expresses one or more sgRNA molecules also under the control of distinct U6 promoters [14]. Animals carrying both transposons will be mutagenized in the cas9 expressing tissue; these animals are readily identified by fluorescent markers encoded within both transposable elements [14]. For example, a cross between a line which carries a Tol2 transposon expressing cas9 from a liver-specific promoter and a line which carries a Tol2 transposon expressing multiple sgRNAs targeting the insulin receptor genes under the control of a ubiquitously expressed U6 snRNA promoter results in liver-specific gene knockout of the insulin receptor genes. This second approach has the advantage that both Tol2 elements are phenotypically silent on their own, allowing stock maintenance even when the mutants fail to thrive or reproduce. While this approach requires the generation of more than one Tol2 element, it is also more flexible as each Tol2 line can be used for multiple experiments. For example, a heat shock inducible, a drug inducible, or a ubiquitously expressed cas9 line can be crossed to multiple different sgRNA expressing Tol2 lines [14]. The use of the improved cas9 with minimal off-target activity may address the concern that off-target events may accumulate with persistent expression of the Cas9-holoenzyme [43,44]. A significant limitation of both of these approaches is that biallelic frameshift mutations occur in only 44.4% of cells (4 in 9 events) with a single sgRNA. This limitation may be overcome by selecting an sgRNA that targets a critical structural or functional domain in the target gene such that even mutations that conserve the ORF will result in loss-of-function. Another approach is to co-express 2 or more sgRNAs targeting the same gene, increasing the potential biallelic frameshift mutations to more than 70% of cells (64 in 81 with 2 sgRNAs). The combination of both approaches is recommended [14]. Other newly developed conditional Cas9 systems including a photoactivatable Cas9 [45] and a 4-hydroxytamoxifen-sensitive Cas9 [46,47] may also allow highly refined expression of the holoendonuclease activity with fine spatial, tissue, or temporal control.
Figure 2. Mutagenesis using transgenic expression of the cas9 holoendonuclease.

Transgenesis in zebrafish is generally mediated through the use of the Tol2 transposon carried by a plasmid that is injected at the 1 cell stage. The Tol2 inverted repeats (TIR) are recognized by the Tol2 transposase, generally supplied as injected mRNA, and transposition of the engineered Tol2 transposon occurs into the genome. In one approach (left panel) the two components of the holoendonuclease are expressed from different Tol2 elements. One Tol2 element expresses cas9 using a desired promoter that can be tissue specific, ubiquitously expressed, or driven by a small molecule. The other Tol2 element expresses one or more sgRNAs directed against the target gene or genes. Offspring of a cross between lines carrying these two Tol2 elements will express the holoendonuclease and have a mutated target. These offspring can be identified by markers, not shown, on each Tol2 transposon. For example, the sgRNA expressing Tol2 might be marked with GFP expressed specifically in the lens, while the cas9 expressing Tol2 might be marked with mCherry expressed in the heart. The “lens green, heart red” embryos also express the holoendonuclease. In a separate approach (right panel), both components of the holoendonuclease are expressed from a single transgene and all carriers will also be mutagenized at the target gene or genes. These animals may also be marked as in the left panel.
Whether conditional CRISPR-Cas9 mutagenesis can replace the Cre/loxP based conditional mutagenesis commonly used in mice remains to be seen. The mutagenesis efficiency of CRISPR-Cas9 is somewhat lower than with Cre/loxP [14]. For conditional inactivation of a single gene, it may be more appropriate to use CRISPR-Cas9 to generate loxP-containing alleles and then use the appropriate Cre lines to achieve gene-disruption in the cells of interest.
Multigenic CRISPR-Cas9 mutagenesis
As eluded to in the previous section, the CRISPR-Cas9 system can be used to target multiple genes in the same animal as multiple sgRNAs can be used simultaneously. The ability to target multiple genes expands the number of tools available to the geneticist. Multigenic targeting experiments can overcome the limitation of functional redundancy associated with genetic analysis as well as to test models of genetic interaction. Multigenic targeting experiments can also be used in so-called forward genetic experiments to identify novel genetic functions or phenotypes. Targeting many genes in a single cross is essential to find the rare genes that affect the function of interest. The following sections describe how multiplex CRISPR-Cas9 mutagenesis is being used to facilitate classical genetic techniques to dissect genetic function.
Multigenic mutagenesis to explore genetic redundancy and genetic interaction
There are two common cases in which being able to make mutations in more than one gene, multigenic mutagenesis, are useful. In the first case, some genes are found in multiple copies (paralogs) with overlapping or redundant function; null alleles of a single paralog may not result in a detectable phenotype. Studying genetic function in these cases requires assessment of phenotype in animals in which more than one paralog is mutant. The second common case in which multiplex mutagenesis is useful is in determination of genetic interactions between genes; this approach can identify genetic suppressors or enhancers thus elucidating genetic pathways. Using a classical breeding approach, it is relatively straightforward to generate animals with mutant alleles in more than one locus, but the number of animals with the desired genotype is reduced by a factor of 2 and the number of generations required to generate the stocks increases with each allele. Multigenic mutagenesis methods in which more than one gene is targeted at once in the same animal allow much more rapid analysis of genetic function in these classes of experiments. Co-injection of cas9 mRNA with five different sgRNAs into one-cell stage zygotes resulted in successful mutagenesis of the five different genes. Mutagenesis rates of 75-99% of all loci in F0 animals were observed. This resulted in multiple loss-of-function phenotypes in the same fish [22]. Consistent with these results, a later study using a similar strategy also showed efficient modification of five loci (four genes) in zebrafish by the CRISPR/Cas9 system [48]. Alternatively, using Tol2 elements expressing cas9 and multiple sgRNAs multiple genes can be targeted in a single animal [14].
Multigenic mutagenesis for genetic screen
Forward genetic screens are performed to identify the genes that are essential for a particular biological process and identify the set of phenotypes that arise (common phenotypic end points) resulting from loss-of-function in these genes. This technology has had profound impacts in cell biology (cell cycle regulatory genes), developmental biology (the genes required to pattern the Drosophila embryo), molecular biology (the genes required for pre-mRNA splicing), and behavior (the genes required for learning and memory in Drosophila). Classically, a forward genetic screen is performed by randomly mutagenizing the genome with a chemical mutagen. The phenotype is assessed in animals that are bred to homozygosity revealing the phenotypic consequence of the mutagenized gene. However, the offspring from more than 10,000 crosses must be phenotypically assessed in a screen performed on chemically mutagenized zebrafish to find the rare mutants that cause detectable phenotype and several generations are required to breed the screened animals [49-52]. Because these screens are very labor intensive, screening has largely been limited to phenotypes that can be very rapidly assessed, for instance embryonic lethality or pigmentation defects. However, many interesting phenotypes are subtle or not readily visible, like myelination, or are quantitative and time consuming to measure, like growth rate. These traits are poorly suited to a labor intensive classical forward screen. Further, in chemical mutagenesis screens, mutations in small genes are under-represented, saturation is difficult to achieve, and uncovering the identity of genes responsible for the phenotypes requires laborious mapping and positional cloning. In theory, a directed-forward screen in which each gene is targeted by sgRNAs could be used to screen for phenotypes as fewer generations are required for a CRISPR-Cas9 based directed forward screen. Because the animal husbandry and labor is reduced, even phenotypes that are subtle or time consuming to measure can be screened. Moreover, because the targets are known, negative results are informative. Since genes that affect particular phenotypes are rare, multigenic mutagenesis increases throughput and efficiency.
At the present time no full genome scale CRISPR-cas9 based forward screens have been reported in zebrafish, but pilot scale studies have demonstrated that these screens can identify genes and new phenotypes. One efficient approach would be to use multiple highly active sgRNAs for each gene to target multiple genes at once and screen for phenotype in F0. In one experiment, sgRNAs targeting 48 genes were pooled, injected, and then screened. This screen identified two genes involved in electrical synapse formation [53]. Alternatively, a phenotypic screen may be performed in F1 fish from founders injected with the same pool of sgRNA, as was done in another study in which 162 sgRNAs targeting 83 genes were injected in pools of 10 [13]. Because the mutagenesis is reproducible using the sequence of the sgRNA, stocking of animals or cryopreservation of sperm is not required and screening can be performed continuously against the whole genome [53].
Concluding Remarks and Future Perspectives
In just three years, CRISPR-Cas9 has revolutionized genome engineering. Various CRISPR-Cas9 based methods allow functional genome engineering to manipulate the genome for in many organisms including zebrafish. The CRISPR-Cas9 system has been successfully applied in zebrafish, including knockout, high-throughput mutagenesis screens, multiplex knockout, conditional knockout and knock-in. Many other interesting new developments in the CRISPR-Cas9 system may soon find their successful application in zebrafish.
Outstanding Questions.
The CRISPR-Cas9 system allows researchers to engineer the genome to control gene function. The set of tools that can be used at the endogenous locus is large; we have seen only a few of those tools implemented in zebrafish thus far. Tools under development include:
Spatiotemporal control of gene function by integration of conditional gene-trap cassettes.
Monitoring gene expression and protein localization through the integration of a reporter protein at the endogenous locus.
Control of gene expression by targeted integration of exogenous regulatory sequences to control gene transcription or mRNA stability.
Targeted integration of transgenes into a common locus that allows consistent and reproducible expression of transgenes, similar to the ROSA26 locus in mouse [89].
Development of targeted whole-genome mutagenesis screen methods.
Development of methods to analyze the non-coding transcriptome.
Development of these systems requires further refinement of the CRISPR-Cas9 methods. These refinements include:
Continued development of improved informatics tools that reliably predict optimal targets.
Development of more efficient homologous directed repair to efficiently perform gene conversion to an engineered template DNA.
Development of more efficient and inexpensive genotyping methods which work on both large and small changes to the genome.
Engineering the PAM-interacting domain and development of other class II CRISPR systems to expand the targetable genome.
Mutants of Cas9 that lack nuclease activity can be used for site-specific DNA and RNA binding. These tools have great potential in regulating gene expression both transcriptionally and post-transcriptionally. Additionally, they can be used to characterize localization of non-coding RNA and mRNA in vivo.
Development of non-catalytic systems to track RNA in zebrafish in vivo.
Development of regulators of mRNA stability and/or translation.
Trends Box.
□ CRISPR-Cas9 is an RNA directed endonuclease that can generate double stranded breaks in the zebrafish genome.
□ CRISPR-Cas9 mutagenesis can be used to make conditional alleles to study tissue-specific or stage-specific gene function.
□ Multigenic mutagenesis using CRISPR-Cas9 can be used to identify and study genetic redundancy and to establish epistatic relationships between genes.
□ CRISPR-Cas9 can be used for whole genome forward genetic screens. This opens biological processes that have been genetically intractable to exploration.
□ Elucidation of the precise mechanism by which the CRISPR-Cas9 double stranded break is repaired will likely expand the ability to perform gene conversion through homology dependent and homology independent means.
Acknowledgments
We apologize that owing to space limitations we have been unable to cite many important zebrafish CRISPR-Cas9 studies. This work was supported by the Vanderbilt Diabetes Research and Training Centers and DK088686 (WC), by the American Diabetes Association grant 1-13-BS-027 (WC), and by the National Natural Science Foundation of China (81670709 to ML and 41506164 to LZ), an R21 UDN grant (1R21GM110311 to PPM and WC), and is supported by a National Institute of Food and Agriculture, U.S. Department of Agriculture award (2015-67015-23488 to PPM and WC).
Glossary
- Cas9
The gene that encodes the protein component of the Cas9 holoendonuclease. It encodes a large multi-domain protein that binds the crRNA-tracrRNA duplex and the target DNA strand. Two endonuclease domains cleave each strand of the target DNA.
- Clustered regularly interspaced short palindromic repeats (CRISPR)
Bacterial and archaeal loci in which the same DNA sequence is repeated many times. Each repeat is separated by a short apparently random DNA sequence called the interspacer or spacer. Informatics experiments demonstrated that these spacer sequences matched the sequence of known bacteriophages and that the repeats contain near palindromes. These features of the locus give rise to the acronym.
- Conditional Alleles
Conditional alleles are mutations in a gene in which the mutant phenotype is only expressed in a particular condition. For example, temperature sensitive alleles are non-functional at elevated temperatures, but wildtype at standard temperatures. In vertebrate systems the genetic locus is engineered so an exon of the gene is flanked by recombinase binding sites. Regulated expression of a recombinase results in inversion (flipping) or deletion of the targeted locus. Controlling the expression of the recombinase allows the geneticist to control where in the organism or when in development the phenotype is expressed.
- CRISPR RNA (crRNA)
The CRISPR locus is transcribed into pre-crRNA, which is then cleaved at the end of each repeat to form a crRNA. This results in an RNA molecule in which one half is encoded by the spacer and the other is encoded by the repeat.
- Double-strand Break (DSB) Repair
Environmental damage, DNA metabolism, and nucleases can result in scission of both strands of the DNA. Left unrepaired, this event is highly toxic to cells and there are robust systems in cells to repair the damage. These repair systems can restore the original DNA sequence, as in HDR, or result in altered DNA sequence, as in NHEJ.
- Homology-directed repair (HDR)
A high fidelity DSB repair pathway that relies on a homologous DNA template for repair. In vivo, the sister chromosome is generally used as the homologous template. In genetic engineering systems, HDR is used to produce precise base changes, insertions, or deletions by introducing an engineered donor template to substitute for the sister chromosome.
- Non-homologous end joining (NHEJ)
A non-templated DSB repair pathway that causes insertion/deletion (indel) mutations. NHEJ pathways include both the canonical ligase IV-dependent NHEJ (C-NHEJ) process and the ligase I/III alternative NHEJ (alt-NHEJ) process.
- Protospacer Adjacent Motif (PAM)
PAM are short DNA sequences which are recognized by the Cas9 protein PAM-interacting (PI) domain. Following binding of Cas9 to the PAM, Cas9 unwinds the genomic DNA facilitating access of the spacer domain of crRNA target strand to the target DNA strand.
- single guide RNA (sgRNA)
sgRNA is an engineered chimeric RNA that mimics the natural S. pyogenes crRNA:tracrRNA duplex. It fuses the 5′ end of the target binding crRNA to the 3′ end of the tracrRNA which is tightly bound by Cas9.
- Tol2 transgenesis system
Tol2 transgenesis system is based on Tol2 transposon, a mobile genetic element discovered in medaka that also functions in zebrafish [90] and several other species [90,91]. It consists of a plasmid containing a transgene flanked by Tol2 terminal inverted repeats (TIR) and a plasmid that encodes Tol2 transposase. Tol2 transposase recognizes the Tol2 terminal inverted repeats (TIR) and excises the element from the plasmid and inserts it into the host genome. Most commonly the transgene is assemble with the open source Tol2Kit [92]. Co-injection of the transgenic plasmid and the transposase mRNA results in efficient transgenesis.
- transactivating CRISPR RNA (tracrRNA)
tracrRNA is a noncoding RNA gene adjacent to the CRISPR locus. The tracrRNA product is an RNA that forms a stable double-stranded RNA duplex with the repeat region of the crRNA. tracrRNA acts as an adapter or handle allowing each crRNA to be stably bound by the Cas9 protein via the tracrRNA.
- Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALEN)
Both ZFN and TALEN are engineered protein nucleases that create DSBs at specific genomic sites. Both systems require significant time and skill in order to generate the site-specific nuclease than the CRISPR-Cas9 system.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van der Oost J, et al. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Wright AV, et al. Biology and Applications of CRISPR Systems: Harnessing Nature's Toolbox for Genome Engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 3.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Brouns SJ, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen R, et al. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 7.Makarova KS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mali P, et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinek M, et al. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagnon JA, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLOS ONE. 2014;9:e98186. doi: 10.1371/journal.pone.0098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varshney GK, et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015;25:1030–1042. doi: 10.1101/gr.186379.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin L, et al. Generation of Targeted Mutations in Zebrafish Using the CRISPR/Cas System. Methods Mol Biol. 2015;1332:205–217. doi: 10.1007/978-1-4939-2917-7_16. [DOI] [PubMed] [Google Scholar]
- 15.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreno-Mateos MA, et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heigwer F, et al. E-CRISP: fast CRISPR target site identification. Nat Methods. 2014;11:122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- 18.Prykhozhij SV, et al. CRISPR MultiTargeter: A Web Tool to Find Common and Unique CRISPR Single Guide RNA Targets in a Set of Similar Sequences. PLOS ONE. 2015;10:e0119372. doi: 10.1371/journal.pone.0119372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montague TG, et al. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–7. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varshney GK, et al. CRISPRz: a database of zebrafish validated sgRNAs. Nucleic Acids Res. 2016;44:D822–6. doi: 10.1093/nar/gkv998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Speir ML, et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44:D717–25. doi: 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jao LE, et al. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci USA. 2013;110:13904–13909. doi: 10.1073/pnas.1308335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang WY, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thyme SB, et al. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat Commun. 2016;7:11750. doi: 10.1038/ncomms11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang N, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465–472. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sollu C, et al. Autonomous zinc-finger nuclease pairs for targeted chromosomal deletion. Nucleic Acids Res. 2010;38:8269–8276. doi: 10.1093/nar/gkq720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nature Reviews Genetics. 2005;6:507–512. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- 28.Mansour SL, et al. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 29.Donoho G, et al. Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol Cell Biol. 1998;18:4070–4078. doi: 10.1128/mcb.18.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bedell VM, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zu Y, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nature Methods. 2013;10:329–331. doi: 10.1038/nmeth.2374. [DOI] [PubMed] [Google Scholar]
- 32.Shin J, et al. Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development. 2014;141:3807–3818. doi: 10.1242/dev.108019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoshijima K, et al. Precise Editing of the Zebrafish Genome Made Simple and Efficient. Dev Cell. 2016;36:654–667. doi: 10.1016/j.devcel.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hruscha A, et al. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development. 2013;140:4982–4987. doi: 10.1242/dev.099085. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong GAB, et al. Homology Directed Knockin of Point Mutations in the Zebrafish tardbp and fus Genes in ALS Using the CRISPR/Cas9 System. PLOS ONE. 2016;11:e0150188. doi: 10.1371/journal.pone.0150188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irion U, et al. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development. 2014;141:4827–4830. doi: 10.1242/dev.115584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auer TO, et al. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014;24:142–153. doi: 10.1101/gr.161638.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hisano Y, et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci Rep. 2015;5:8841. doi: 10.1038/srep08841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, et al. Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res. 2015;25:634–637. doi: 10.1038/cr.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin L, et al. Multiplex Conditional Mutagenesis Using Transgenic Expression of Cas9 and sgRNAs. Genetics. 2015;200:431–441. doi: 10.1534/genetics.115.176917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ablain J, et al. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell. 2015;32:756–764. doi: 10.1016/j.devcel.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleinstiver BP, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nihongaki Y, et al. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol. 2015;33:755–760. doi: 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]
- 46.Davis KM, et al. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oakes BL, et al. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol. 2016;34:646–651. doi: 10.1038/nbt.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ota S, et al. Multiple genome modifications by the CRISPR/Cas9 system in zebrafish. Genes Cells. 2014;19:555–564. doi: 10.1111/gtc.12154. [DOI] [PubMed] [Google Scholar]
- 49.Haffter P, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Driever W, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- 51.Muto A, et al. Forward genetic analysis of visual behavior in zebrafish. PLoS Genet. 2005;1:e66. doi: 10.1371/journal.pgen.0010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amsterdam A, et al. Identification of 315 genes essential for early zebrafish development. PNAS. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shah AN, et al. Rapid reverse genetic screening using CRISPR in zebrafish. Nat Methods. 2015;12:535–540. doi: 10.1038/nmeth.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doyon Y, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nature Biotechnology. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meng X, et al. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nature Biotechnology. 2008;26:695–701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sander JD, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat Methods. 2011;8:67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang P, et al. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- 58.Smih F, et al. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995;23:5012–5019. doi: 10.1093/nar/23.24.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rouet P, et al. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Segal DJ, et al. Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5“-GNN-3” DNA target sequences. PNAS. 1999;96:2758–2763. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rebar EJ, Pabo CO. Zinc finger phage: affinity selection of fingers with new DNA-binding specificities. Science. 1994;263:671–673. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 62.Isalan M, et al. A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nature Biotechnology. 2001;19:656–660. doi: 10.1038/90264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wolfe SA, et al. DNA Recognition by Cys2His2 Zinc Finger Proteins. Annual Rev Biophys. 2003;29:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 64.Li L, et al. Functional domains in Fok I restriction endonuclease. PNAS. 1992;89:4275–4279. doi: 10.1073/pnas.89.10.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim YG, et al. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. PNAS. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller JC, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nature Biotechnology. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 67.Ramirez CL, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–375. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moscou MJ, Bogdanove AJ. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science. 2009;326:1501–1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 69.Boch J, et al. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 70.Christian M, et al. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li T, et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011;39:359–372. doi: 10.1093/nar/gkq704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deng D, et al. Recognition of methylated DNA by TAL effectors. Cell Res. 2012;22:1502–1504. doi: 10.1038/cr.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valton J, et al. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J Biol Chem. 2012;287:38427–38432. doi: 10.1074/jbc.C112.408864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chapman JR, et al. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 76.Maruyama T, et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He MD, et al. Efficient ligase 3-dependent microhomology-mediated end joining repair of DNA double-strand breaks in zebrafish embryos. Mutat Res. 2015;780:86–96. doi: 10.1016/j.mrfmmm.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 78.Thyme SB, Schier AF. Polq-Mediated End Joining Is Essential for Surviving DNA Double-Strand Breaks during Early Zebrafish Development. Cell Rep. 2016;15:707–714. doi: 10.1016/j.celrep.2016.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Howe K, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dahlman JE, et al. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol. 2015;33:1159–1161. doi: 10.1038/nbt.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu Y, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang T, et al. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chari R, et al. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12:823–826. doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sternberg SH, et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuscu C, et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 87.Wu X, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.LaFave MC, et al. A defined zebrafish line for high-throughput genetics and genomics: NHGRI-1. Genetics. 2014;198:167–170. doi: 10.1534/genetics.114.166769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes & Development. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 90.Kawakami K, et al. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proceedings of the National Academy of Sciences. 2000;97(21):11403–11408. doi: 10.1073/pnas.97.21.11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keng VW, et al. Efficient Transposition of Tol2 in the Mouse Germline. Genetics. 2009;183(4):1565–1573. doi: 10.1534/genetics.109.100768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kwan KM, et al. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Developmental Dynamics. 2007;236(11):3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]