

Abstract
We report the development of a new class of guanidine-containing peptides as multifunctional ligands for transition-metal catalysis and its application in the remote desymmetrization of diarylmethanes via copper-catalyzed Ullman cross-coupling. Through design of these peptides, high levels of enantioinduction and good isolated yields were achieved in the long-range asymmetric cross-coupling (up to 93:7 er and 76% yield) between aryl bromides and malonates. Our mechanistic studies suggest that distal stereocontrol is achieved through a Cs-bridged interaction between the Lewis-basic C-terminal carboxylate of the peptides with the distal arene of the substrate.

Introduction
Catalytic reactions that selectively create one or more new chiral elements depend on close communication between the catalyst and the substrate. In general, chirality is established by the chiral catalyst as part of the bond forming process itself, or commonly when the generated stereogenic center is proximal to the newly constructed bond. Chirality can also be created over longer ranges through bond-formation at enantiotopic sites which are several bonds removed from the stereogenic center. However, it is challenging to differentiate these distant enantiotopic sites, and enzymes or catalysts of comparable dimensions are often required.1 We reported one such case, employing short peptide catalyst 2 to break symmetry in bis(phenol) 1a viaO-acylation, wherein the newly formed stereogenic center is five bonds away from the site of bond formation, and the enantiotopic sites are separated by nearly a full nanometer (eq 1).2 To date, only a few examples using small molecule catalysts3 for remote stereoinduction have been reported, and employment of transition-metal catalysis for a related long-range asymmetric induction has remained elusive.
To accomplish such challenging processes, we endeavored to develop new chiral ligands for transition-metals that would allow distal stereocontrol in the desymmetrization of compound 3 (eq 2). We questioned whether such asymmetric induction could be achieved using simple peptides that closely interacted with the substrate, and that housed a defined metal-binding moiety that
 |
(eq 1) |
 |
(eq 2) |
 |
(eq 3) |
would confer high activity to go along with any observed enantioselectivity (eq 2). As boundary conditions for our study, we wished to use abundant and inexpensive first-row transition metal complexes as catalysts. We chose to focus on the Cu-catalyzed Ullman cross-coupling reaction between malonate derivatives and aryl halides (the Hurtley reaction; eq 3).4 Ma and co-workers have established the enantioselective variant of the process employing 2-methylacetoacetates and simple amino acid ligands such as hydroxyproline.5 Herein, we report a new class of peptide-based ligands that has culminated in efficient symmetry-breaking cross-coupling reactions for the preparation of enantioenriched, drug-like diarylmethane derivatives 4.6 As a related system to eq 1, these processes fall into the category of reactions with distal pro-stereogenic elements, and our experiments show that the peptide-based catalysts are well-suited for these metal-catalyzed processes.
The ability to modulate ligands is essential to optimizing asymmetric induction.7 We imagined that amino acids and peptide-based metal complexes could be a suitable platform for achieving long-range asymmetric induction (eq 2, a–c).8 We proposed that the dimensions of the catalyst could be engineered to adopt an optimal length and geometry (eq 2, a) such that differentiation of the distal enantiotopic sites would be possible. In order to fine-tune the Cu center, we speculated that bulky and electron-rich guanidines (eq 2, b) could facilitate the Ullman cross-coupling reaction under mild conditions. Despite many well-known nitrogen-based ligands for asymmetric transition-metal catalysts,9 there are few examples of fully substituted guanidines as chiral ligands (eq 2, c).10 Notably, pentasubstituted guanidines, unlike primary or secondary amines, should not themselves be competitive substrates for Ullman-type reactions.11 Additionally, we were attracted to guanidine derivatives due to their ease of synthesis and bench-stability.
Results and Discussion
Catalyst optimization
Our studies began with an evaluation of the desymmetrization reaction shown in Scheme 1. Treatment of the symmetrical compound 3a with diethyl malonate (1.1 equiv) in the presence of CuI (20 mol%) and various ligands provided an initial set of encouraging results.12 The use of proline (L1) (Ma-type conditions) provided reasonable levels of conversion, and a modest level of enantioselectivity for the desired product 4a (41:59 er; Scheme 1).13 N,N-Dimethylphenylalanine (L2) also provided good conversion, but a nearly racemic product. In comparison, the application of the pentasubstituted guanidine L3 led to a striking result, wherein the product 4a could be isolated with an appreciable er of 89:11. Unfortunately, when the catalyst loading was reduced to 5 mol%, the level of selectivity declined to 58:42 er. Further studies revealed that asparagine derivative L4 delivered er of 85:15 with 20 mol% of the catalytic system, which diminished to only 72:28 er when 5 mol% was used. We deemed the results to be sufficiently encouraging and robust to stimulate further studies, on the basis of our ligand design, through decoration of the side chain of L4 with peptide residues.
Scheme 1.

As with many studies of peptide-based organocatalysts, we imagined that incorporation of the metal-ligating residue into a peptide capable of forming a secondary structure (e.g., a β-turn) could be beneficial in terms of enhancing catalyst performance.16 Furthermore, we hypothesized that a C-terminal amide or a carboxylate could interact with the substrate, perhaps through hydrogen bonding (c.f., eq 2).17
Table 1 lists the results of experiments in which the complexity of the ligand was gradually increased. Catalysts with secondary amide functionality at the C-terminal position of the ligand (entries 1–4), exhibited only modest changes in selectivity, with ers for the mono-functionalized product 4a ranging from 69:31 to 76:24. However, a significant enhancement in enantioselectivity was observed with elongated catalysts in which the C-terminal position was simply exchanged for a Li carboxylate (entries 5–8). Notably, the catalyst derived from ligand L10 provided the highest observed er (entry 6, 93:7), while also producing the highest level of conversion to the product 4a (66%). In the presence of a large excess Cs ions, the Li carboxylates seem likely to undergo salt-exchange in situ to form Cs carboxylates.
Table 1.

| entry | peptide | 3ac % remaining | 4ac % conv. | 4a er | 7a % conv. |
|---|---|---|---|---|---|
| 1 | L5 | 41 | 47 | 75:25 | 12 |
| 2 | L6 | 34 | 48 | 76:24 | 18 |
| 3 | L7 | 26 | 43 | 69:31 | 31 |
| 4 | L8 | 23 | 47 | 76:24 | 30 |
|
| |||||
| 5 | L9 | 35 | 49 | 77:23 | 15 |
| 6 | L10 | 19 | 66 | 93:7 | 15 |
| 7 | L11 | 23 | 61 | 90:10 | 16 |
| 8 | L12 | 26 | 57 | 86:14 | 17 |
Reaction conditions: substrate (0.4 mmol), diethyl malonate (1.1 equiv), CuI (5 mol%), peptide (10 mol%), Cs2CO3 (3.2 equiv), and DMF (1.6 mL).
Conversions were determined by 1H NMR and enantiomeric ratios were determined by chiral high performance liquid chromatography analysis, see supporting information for details.
Contains 5–10% of inseparable impurity 8. See eq 4 and supporting information for details.
During the course of these initial experiments, we discovered that reaction efficiencies were compromised by the formation of a persistent byproduct. While the mono-functionalized bromide 4a could be isolated as the major product, a trace amount of the corresponding iodide 8 (eq 4) was also observed, which presumably originated from cross-coupling of the pre-catalyst iodide ligand and substrate 3a.18 Although 4a and 8 could be analyzed as a mixture in a chiral HPLC analysis, isolated yields were eroded. Based on the identification of this iodide byproduct, we employed Cu(MeCN)4BF4 as the Cu source, as delineated in Table 2.19 This straightforward solution completely eliminated the byproduct, and allowed for less complicated reaction optimization and purification.
 |
(eq 4) |
Table 2.

| entry | peptide | 3a % remaining | 4a % conv. | 4a er | 7a % conv. |
|---|---|---|---|---|---|
| 1 | L10 | 13 | 75 | 91:9 | 12 |
| 2 | L13 | 13 | 65 | 86:14 | 22 |
| 3 | L6 | 12 | 66 | 89:11 | 22 |
Reaction conditions: substrate (0.4 mmol), diethyl malonate (1.1 equiv), Cu(MeCN)4BF4 (5 mol%), peptide (10 mol%), Cs2CO3 (3.2 equiv), DMF/Tol (0.5/1.1 mL).
Conversions were determined by 1H NMR and enantiomeric ratios were determined by chiral high performance liquid chromatography analysis, see supporting information for details.
As shown in Table 2 (entry 1), the catalyst derived from Cu(MeCN)4BF4 and L10 allowed for efficient conversion to the desymmetrized product 4a, which exhibited a 91:9 er at 75% conversion. As part of the optimization, we also determined that a mixture of DMF/toluene was a suitable solvent system for the reaction. Notably, a higher ratio of 4a to 7a was obtained under the newly optimized conditions. After an extensive base screen, we discovered that Cs2CO3 was essential for both reactivity and selectivity.20 Moreover, alterations to the catalyst C-terminus were evaluated. Exchange of the Li carboxylate (L10, entry 1) for a methyl ester (L13, entry 2), or N-methyl amide (L6, entry 3) resulted in noticeable decreases in both enantioselectivity (86:14, 89:11, respectively) and the 4a to 7a ratio. In accordance with our initial hypothesis, the extensive studies of the peptide sequence21 (which in turn affects the dimensions of the peptide through its secondary structure) indicates that the peptides serve multiple functions (c.f., eq 2), including a potential interaction between the C-terminal carboxylate moiety of the peptide and the substrate. Mechanistic studies regarding these aspects are discussed in more detail in a following section.
Substrate scope
With an effective catalyst in hand, we turned our attention to the scope of the reaction. As shown in Table 3, simple variations to the malonate structure do not greatly impact the reaction outcomes. Diethyl (entry 1), dimethyl (entry 2) di-iso-propyl (entry 3) and dibenzyl (entry 4) malonates all deliver the corresponding desymmetrized products 4a–4e with comparable levels of conversion, isolated yield and er. However, the introduction of an α-methyl group (entry 5) leads to a much slower reaction, and only 12% isolated yield, although the er is 93:7 in this case.22 Gratifyingly after a single crystallization, the functionalized diarylmethanes (4a and 4c) can be obtained in essentially enantiopure forms with good recovery of material (62% and 71%, respectively). Moreover, these enantioenriched drug-like scaffolds contain synthetically versatile functional groups (unreacted aryl bromide, malonate, anilines) that can be easily modified to generate pharmaceutically-related motifs such as N-heterocycles23 and aryl halides.24
Table 3.

| entry | Malonate | 3a % remaining | 4 % conv.c | 4 er | 7 % conv.c |
|---|---|---|---|---|---|
| 1 |
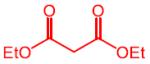
|
11 | 4a, 76 (74) | 92:8 | 7a, 13 |
| After a single recrystallization | (62)d | >99:1 | |||
|
| |||||
| 2 |
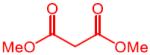
|
12 | 4b, 72 (70) | 91:9 | 7b, 16 |
|
| |||||
| 3 |
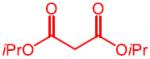
|
23 | 4c, 71 (68) | 92:8 | 7c, 5 |
| After a single recrystallization | (71)d | >99:1 | |||
|
| |||||
| 4 |

|
15 | 4d, 72 (70) | 10:90e | 7d, 13 |
|
| |||||
| 5 |

|
90 | 4e, 10 (3) | 7:93e | - |
Reaction conditions: substrate (0.4 mmol), malonate (1.1 equiv), Cu(MeCN)4BF4 (5 mol%), peptide (10 mol%), Cs2CO3 (3.2 equiv), DMF/Tol (0.5/1.1 mL).
Conversions were determined by 1H NMR and enantiomeric ratios were determined by chiral high performance liquid chromatography analysis, see supporting information for details.
The numbers in parentheses are isolated yields.
The numbers in parentheses are % recovery yields.
Cause of the reversed order of elution of enantiomers undetermined.
The scope of the diarylmethane starting material was also evaluated, and provides fascinating results (Table 4). As with our studies of desymmetrization through organocatalytic O-acylation, the corresponding cross-coupling reactions respond dramatically to alterations to the substituent located along the mirror plane of the substrate.25 For example, whereas the tert-butyl substituent gives the best result (entry 1, 92:8 er), decreasing steric size leads to substrates that exhibit lower selectivity. Cyclohexyl (entry 2, 78:22 er) and methyl (entry 3, 52:48 er) typify this trend. Aryl-substituted compounds also exhibit lower selectivity (entries 4–6). In our previous studies of this substrate class, we proposed that the molecule could exist in different “propeller like” conformations that were influenced by the central substituent. The presently reported data appear to follow this trend despite the markedly different reaction mechanisms at play. The exact nature of these substituent effects remains unknown. Yet, in the mechanistic studies of the process shown in eq 1, the Sigman group suggests that correlations between central substituents and selectivity may be found in this class of substrate, and most closely correspond to conventional steric parameterization26
Table 4.

| entry | R″ | 3 % remainingc | 4 % conv.c | 4 er | 7 % conv.c |
|---|---|---|---|---|---|
| 1 |

|
3a, 11 | 4a, 76 (74) | 92:8 | 7a, 13 |
| 2d |

|
3f, (52) | 4f, (30) | 78:22 | 7f, (16) |
| 3d |

|
3g, (32) | 4g, (50) | 52:48 | 7g (18) |
| 4 |
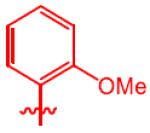
|
3h, 22 | 4h, 54 (45) | 57:43 | 7h, 24 |
| 5 |

|
3i, 33 | 4i, 54 (39) | 37:63e | 7i, 13 |
| 6 |

|
3j, 41 | 4j, 47 (47) | 65:35 | 7j, 12 |
Reaction conditions: substrate (0.4 mmol), diethyl malonate (1.1 equiv), Cu(MeCN)4BF4 (5 mol%), peptide (10 mol%), Cs2CO3 (3.2 equiv), DMF/Tol (0.5/1.1 mL).
Conversions were determined by 1H NMR and enantiomeric ratios were determined by chiral high performance liquid chromatography analysis, see supporting information for details.
The numbers in parentheses are isolated yields.
Product distributions are determined with isolated yields.
Cause of the reversed order of elution of enantiomers undetermined.
Mechanism-driven experiments
With the goal of understanding how elements of catalyst and substrate might interact to afford the observed selectivity, we first examined a number of substituents at the anilino nitrogen (eq 5). Notably, both the trifluoroacetyl group and its amide proton proved to be critical for reactivity. Neither acetamide 9 nor N-methyl amide 10 participates in the reaction, and no cross-coupled product is observed under the optimized conditions (eq 5). It seems possible that the presence of the amide proton is necessary for deprotonation or hydrogen bonding and that there are stringent electronic requirements for the proton, likely related to pKa or hydrogen bonding capacity. With these results in mind, we subjected the Cs-salt of the parent substrate (11) to the Cu-catalyzed cross-coupling conditions (eq 6),27 which gave essentially identical results as shown in Table 3, entry 2.
Based on these results, we presumed that ionic-type interactions are operative between the amides of the parent substrate (deprotonated in situ) and both termini of Cu/peptide complex (Figure 1a). Furthermore, taking into account the importance of C-terminal carboxylate on the peptide L10 (Table 2), we specu-
 |
(eq 5) |
 |
(eq 6) |
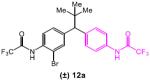
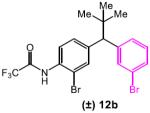
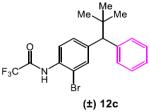
lated that the long-range asymmetric induction could be achieved through a Cs-ion bridged interaction between the C-terminal carboxylate and a distal deprotonated amide (Figure 1a, highlighted in red). We thus designed mechanism-driven kinetic resolution experiments to investigate these hypotheses (Figure 1b). If both termini were participating in the formation of favorable diastereomeric transition state (Figure 1a and 1b, Scenario 1), then one could expect high krel values to be observed in kinetic resolutions when racemic substrates possessing both terminal amides were tested. However, in Scenario 2 (Figure 1b), this requirement for multifunctionality would lead to a low krel value. Unsymmetrical substrates 12a–c (Table 5) were prepared to evaluate these limiting cases.
Figure 1.

a) Proposed model for the interaction between C-terminal carboxylate of the peptide L10 and distal amide of diarylmethane. b) Prediction for mechanistically-driven kinetic resolution studies
Table 5.

| racemic substrate | entry | solvent | k rel |
|---|---|---|---|
|
1 | DMF/Tol | 5.3 |
| 2 | DMF | 3.5 | |
|
3 | DMF/Tol | 11.3 |
| 4 | DMF | 6.9 | |
|
5 | DMF/Tol | 10.3 |
| 6 | DMF | 4.8 |
Reaction conditions: racemic substrate (0.3 mmol), diethyl malonate (1.1 equiv), Cu(MeCN)4BF4 (5 mol%), peptide (10 mol%), Cs2CO3 (3.2 equiv), DMF/Tol (0.38/0.83 mL) or DMF (1.2 mL).
Conversions were determined by 1H NMR and enantiomeric ratios were determined by chiral high performance liquid chromatography analysis. See supporting information for details. Results are reported as an average of two runs.
The unexpected results shown in Table 5 caused us to reevaluate our initial hypothesis. Under the optimized conditions (entries 1, 3, 5), the unsymmetrical substrates exhibited a range of selectivity, though contrary to our prediction (Figure 1b). While we expected to obtain the highest krel with substrate 12a (bearing both CF3COHN groups), this compound gives the lowest selectivity in the series (krel = 5.3; entry 1). Instead, compounds 12b and 12c (lacking the distal CF3COHN group) exhibited notably higher krel values of 11.3 (entry 3) and 10.3 (entry 5), respectively. These trends were preserved, though the magnitudes were reduced, when DMF alone was employed as the solvent (entries 2, 4, 6). The results stimulated a new hypothesis, that a remote arene-catalyst interaction might be responsible for the asymmetric induction. Indeed, when the distal arene was replaced with a simple methyl or cyclohexyl group (as in substrate 12d and 12e, respectively), selectivity in the kinetic resolution was severely reduced (Scheme 2, krel = 1.6 and 1.2 based on 13d and 13e, respectively).
Scheme 2.
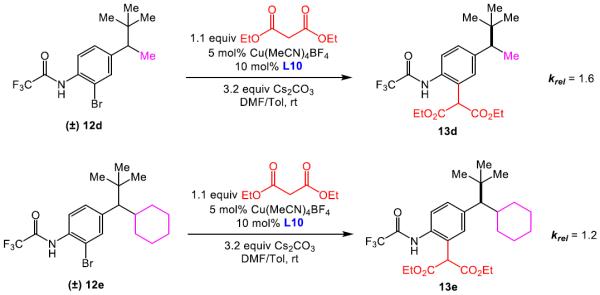
Kinetic resolution of racemic substrates that lacks a distal arene (12d and 12e)
These findings, as well as the observations discovered during our catalyst optimization (Tables 1 and 2), point to a possible non-covalent interaction between the C-terminus of peptide L10 and the distal arene, in which they are bridged by a Cs ion (Figure 2). These ions are known to form various cation-π complexes with arenes.28 For example, this type of molecular recognition has been used for selective extraction of Cs ions from nuclear waste.29 Based on the known binding energies for cation-π interactions, the Cs-bridge between the peptide and the distal arene may be delicate.30 However, utilization of weak coordination between substrates and catalysts have been demonstrated to be fruitful in asymmetric functionalization.31 Notably, substrate 12a is also capable of this interaction, although the trifluoroacetamide may introduce other deleterious effects. One possibility could be that the trifluoroacetamide displaces the Cs ion from its ideal position on the face of the arene, although this idea remains speculative. Additionally, due to likely changes in both the geometry and pKa of the trifluoroacetamide of substrate 12a, in comparison to compound 3a, we speculate that the distal trifluoroacetamide is less prone to deprotonation, which could modulate the interaction (e.g., via H-bonding) with the peptide. As for the local arene (shown in black, Figure 2), based on Yu and Musaev's mechanistic details on Pd-catalyzed C(sp3)–H bond arylation, we imagine that the Cs ion may remain bound between the carbonyl oxygen of the amide and the arene which allows for N-coordination of the Cu center to facilitate oxidative addition into the Ar–Br bond.32 It seems possible that in the parent compound 3a, the distal trifluoroacetamide is capable of binding a second Cs ion in a similar way (Figure 3a). It is also conceivable that even a third Cs ion could be involved, although our experiments do not resolve these possibilities.
Figure 2.
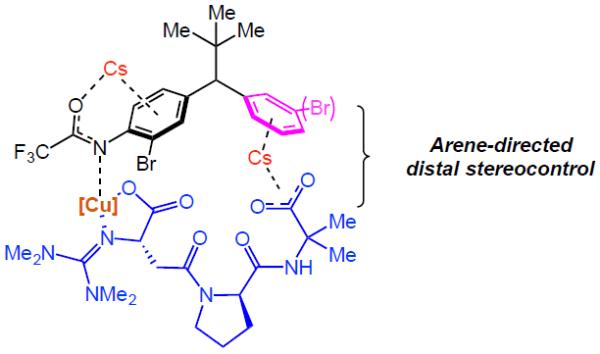
Arene-directed distal stereoinduction
Figure 3.

Current proposed models for interactions between different C-termini of the peptide ligand and the diarylmethane
In order to further understand the interactions between peptide ligand and substrate, kinetic resolution studies were performed using the same substrates as in Table 5, but with amide capped peptide L6. One apparent difference between peptides L6 and L10 is the C-terminal Lewis basicity, with the anionic carboxylate being relatively basic compared to the neutral amide. An ionic interaction between the catalyst and Cs ion would be relatively strong in the case of the carboxylate (Figure 3a), while the weaker amide interaction might be out-competed by Lewis basic solvent molecules such as DMF (Figure 3b).33 If the Cs ion bridge interaction proposed in Figure 2 is relevant for substrate-catalyst recognition, then this interaction could be attenuated by altering solvent conditions, as well.
Analysis of C-terminal amide-containing peptide L6 proved consistent with these notions. As shown in Table 6, (entries 1, 3, 5), experiments with ligand L6 revealed incrementally lower krel values in each of the cases examined. These effects were exacerbated under conditions in pure DMF, in which competition for non-covalent interactions would be expected to be greatest (entries 2, 4, 6). These observations support the assertion that the same type of interaction may be at play with both peptides L10 and L6, but to a lesser extent in the amide case. The combined results of Tables 5 and 6 are in agreement with the model shown in Figure 3b, where the C-terminus no longer interacts with the Cs ion due to competing solvent molecule interactions.
Table 6.
Mechanistic studies via kinetic resolution of unsymmetrical diarylmethanes with C-terminal amide peptidea,b
| racemic substrate | entry | solvent | k rel |
|---|---|---|---|

|
1 | DMF/Tol | 3.3 |
| 2 | DMF | 1.6 | |

|
3 | DMF/Tol | 8.4 |
| 4 | DMF | 1.8 | |

|
5 | DMF/Tol | 7.5 |
| 6 | DMF | 1.8 |
Reaction conditions: racemic substrate (0.3 mmol), diethyl malonate (1.1 equiv), Cu(MeCN)4BF4 (5 mol%), peptide (10 mol%), Cs2CO3 (3.2 equiv), DMF/Tol (0.38/0.83 mL) or DMF (1.2 mL).
Conversions were determined by 1H NMR and enantiomeric ratios were determined by chiral high performance liquid chromatography analysis. See supporting information for details. Results are reported as an average of two runs.
Conclusion
In summary, we have developed a new peptide-based ligand class for the enantioselective desymmetrization of diarylmethane-based aryl bromides. Our design criteria of achieving remote asymmetric induction in the Cu-catalyzed desymmetrizations were satisfied through the development of a guanidinylated peptide, which upon optimization of its dimensions and sequence, allowed for the observation of substantial levels of selectivity. The basis of the asymmetric induction proved to be a complex interplay of effects, consistent with the critical role of each of the components required to achieve the best results. Remote interactions involving the catalyst and the arene that does not undergo cross-coupling were found to be essential, and seem embedded in an Cs-arene association. Evaluating the generality of the guanidinylated peptides as ligands for various other asymmetric reactions, including cross-couplings in complex molecular settings,34 is one next step for this work, and we plan to report on these studies in the near future.
Supplementary Material
ACKNOWLEDGMENT
We thank the Boehringer-Ingelheim-Pfizer-Glaxo-Smith-Kline-Abbvie Non-Precious Metal Catalysis Consortium for general discussions in this area. We are grateful to Pfizer and Boehringer-Ingelheim for partial support of this research. Additional support from the National Institute of General Medical Science of the National Institutes of Health (GM-068649) is gratefully acknowledged.
Footnotes
Supporting Information Additional figures, experimental details, characterization data for all catalysts, substrates, and products, and crystallographic information. X-ray data of 3a, SL1, SL2, SL11, SL20. The Supporting Information is available free of charge on the ACS Publication website.
The authors declare no competing financial interests. Crystallographic data are deposited with the Cambridge Crystallographic Data Centre under the accession number CCDC 1471490 (3a), CCDC 1471491 (SL1), CCDC 1471492 (SL20), CCDC 1471493 (SL11), and CCDC 1471494 (SL2).
REFERENCES
- 1.(a) Yang J, Breslow R. Angew. Chem. Int. Ed. 2000;39:2692–2694. doi: 10.1002/1521-3773(20000804)39:15<2692::aid-anie2692>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]; (b) Garcia-Urdiales E, Alfonso I, Gotor V. Chem. Rev. 2005;105:313–354. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]; (c) Garcia-Urdiales E, Alfonso I, Gotor V. Chem. Rev. 2011;111:110–180. doi: 10.1021/cr100330u. [DOI] [PubMed] [Google Scholar]; (d) Hughes DL, Bergan JJ, Amato JS, Bhupathy M, Leazer JL, McNamara JM, Sidler DR, Reider PJ, Grabowski EJJ. J. Org. Chem. 1990;55:6252–6259. [Google Scholar]
- 2.(a) Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2006;128:16454–16455. doi: 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]; (b) Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2008;130:16358–16365. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Iorio N, Righi P, Mazzanti A, Mancinelli M, Ciogli A, Bencivenni G. J. Am. Chem. Soc. 2014;136:10250–10253. doi: 10.1021/ja505610k. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ullman F. Chem. Ber. 1901;34:2174. [Google Scholar]; (b) Hurtley WRH. J. Chem. Soc. 1929:1870. [Google Scholar]; (c) Monnier F, Taillefer M. Angew. Chem. Int. Ed. 2009;48:6954–6971. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- 5.Xie X, Chen Y, Ma D. J. Am. Chem. Soc. 2006;128:16050–16051. doi: 10.1021/ja066991j. [DOI] [PubMed] [Google Scholar]
- 6.(a) Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chem. Soc. Rev. 2006;35:454–470. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]; (b) Chu L, Wang X-C, Moore CE, Rheingold AL, Yu J-Q. J. Am. Chem. Soc. 2013;135:16344–16347. doi: 10.1021/ja408864c. [DOI] [PubMed] [Google Scholar]; (c) Chu W-D, Zhang L-F, Bao X, Zhao X-H, Zeng C, Du J-Y, Zhang G-B, Wang F-X, Ma X-Y, Fan C-A. Angew. Chem. Int. Ed. 2013;52:9229–9233. doi: 10.1002/anie.201303928. [DOI] [PubMed] [Google Scholar]; (d) Cheng X-F, Li Y, Su Y-M, Yin F, Wang J-Y, Sheng J, Vora HU, Wang X-S, Yu J-Q. J. Am. Chem. Soc. 2013;135:1236–1239. doi: 10.1021/ja311259x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Laforteza BN, Chan KSL, Yu J-Q. Angew. Chem. Int. Ed. 2015;54:11143–11146. doi: 10.1002/anie.201505204. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lee T, Wilson TW, Berg R, Ryberg P, Hartwig JF. J. Am. Chem. Soc. 2015;137:6742–6745. doi: 10.1021/jacs.5b03091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Guduguntla S, Hornillos V, Tessier R, Fananas-Mastral M, Feringa BL. Org. Lett. 2016;18:252–255. doi: 10.1021/acs.orglett.5b03396. [DOI] [PubMed] [Google Scholar]
- 7.Engle KM, Yu J-Q. J. Org. Chem. 2013;78:8927–8955. doi: 10.1021/jo400159y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews of peptide-metal complexes in asymmetric catalysis see: Deuss PJ, den Heeten R, Laan W, Kamer PCJ. Chem. Eur. J. 2011;17:4680–4698. doi: 10.1002/chem.201003646.. Ball ZT. Acc. Chem. Res. 2013;46:560–570. doi: 10.1021/ar300261h.. For selected examples of peptide metal complexes in asymmetric catalysis see: Brown KM, Degrado SJ, Hoveyda AH. Angew. Chem. In. Ed. 2005;44:5306–5310. doi: 10.1002/anie.200501251.. Friel DK, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2008;130:9942–9951. doi: 10.1021/ja802935w.
- 9.Caputo CA, Jones ND. Dalton Trans. 2007:4627–4640. doi: 10.1039/b709283k. [DOI] [PubMed] [Google Scholar]
- 10.For recent applications of less substituted guanidine ligands in asymmetric catalysis see: Zhu Y, Liu X, Dong S, Zhou Y, Li W, Lin L, Feng X. Angew. Chem. Int. Ed. 2014;53:1636–1640. doi: 10.1002/anie.201308501.; Tang Y, Chen Q, Liu X, Wang G, Lin L, Feng X. Angew. Chem. Int. Ed. 2015;54:9512–9516. doi: 10.1002/anie.201501918.; Chen Q, Tang Y, Huang T, Liu X, Lin L, Feng X. Angew. Chem. Int. Ed. 2016;55:5286–5289. doi: 10.1002/anie.201600711.; For reviews for the coordination chemistry of achiral guanidines see: Bailey PJ, Pace S. Coord. Chem. Rev. 2001;214:91–141.; Jones C. Coord. Chem. Rev. 2010;254:1273–1289.
- 11.Ma DW, Zhang YD, Yao JC, Wu SH, Tao FG. J. Am. Chem. Soc. 1998;120:12459–12467. [Google Scholar]
- 12.Our initial investigations showed that N,O- bidendate coordination was crucial for reactivity. Common N,N-bidendate ligands such as bipydrine and 1,2-diamines showed trace reactivity.
- 13.For the purpose of discussion conversions were omitted. For full details concerning conversion and product distribution see supporting information.
- 14.See supporting information for full details concerning the reaction conditions.
- 15.Absolute chemistry is currently unknown. Several attempts to determine absolute chemistry by X-ray crystallography were not successful.
- 16.(a) Miller SJ. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]; (b) Davie EAC, Mennen SM, Xu Y, Miller SJ. Chem. Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- 17.Knowles RR, Jacobsen EN. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20678–20685. doi: 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klapars A, Buchwald SL. J. Am. Chem. Soc. 2002;124:14844–14845. doi: 10.1021/ja028865v. [DOI] [PubMed] [Google Scholar]
- 19.The ratio of copper to peptide ligand was optimized during this process.
- 20.See supporting information for a base screening data.
- 21.See supporting information for full details concerning peptide ligand optimization.
- 22.At elevated temperatures the additional byproducts were observed with no increase in yield for the desired product 4e
- 23.(a) Maciver EE, Knipe PC, Cridland AP, Thompson AL, Smith MD. Chem. Sci. 2012;3:537–540. [Google Scholar]; (b) Maciver EE, Thompson S, Smith MD. Angew. Chem. Int. Ed. 2009;48:9979–9982. doi: 10.1002/anie.200905169. [DOI] [PubMed] [Google Scholar]; (c) Forbes IT. Tetrahedron Lett. 2001;42:6943–6945. [Google Scholar]
- 24.Johnston CP, Kothari A, Sergeieva T, Okovytyy SI, Jackson KE, Paton RS, Smith MD. Nature Chem. 2015;7:171–177. doi: 10.1038/nchem.2150. [DOI] [PubMed] [Google Scholar]
- 25.Gustafson JL, Sigman MS, Miller SJ. Org. Lett. 2010;12:2794–2797. doi: 10.1021/ol100927m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Harper KC, Bess EN, Sigman MS. Nat. Chem. 2012;4:366–374. doi: 10.1038/nchem.1297. [DOI] [PubMed] [Google Scholar]; (b) Milo A, Bess EN, Sigman MS. Nature. 2014;507:210–214. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- 27.The amount of Cs2CO3 was adjusted to account for the number of pre-deprotonation in substrate 11. Notably, we observed that at least 3.2 equiv of Cs2CO3 was required to match all acidic protons in the reaction mixture (2 equiv from 3a, 1.1 equiv from diethyl malonate, 0.1 equiv from L10). Subjecting substrate 3a under the optimized condition with <3.2 equiv of Cs2CO3 gave trace conversion which supports the hypothesis that formation of compound 11 is required prior the cross-coupling.
- 28.(a) Hoffmann D, Bauer W, Schleyer PV, Pieper U, Stalke D. Organometallics. 1993;12:1193–1200. [Google Scholar]; (b) Werner B, Krauter T, Neumuller B. Organometallics. 1996;15:3746–3751. [Google Scholar]; (c) Li R, Winter REK, Kramer J, Gokel GW. Supramol. Chem. 2010;22:73–80. [Google Scholar]; (d) Nath B, Baruah JB. Cryst. Growth Des. 2013;13:5146–5155. [Google Scholar]; (e) Grubel K, Brennessel WW, Mercado BQ, Holland PL. J. Am. Chem. Soc. 2014;136:16807–16816. doi: 10.1021/ja507442b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Horner DE, Crouse DJ, Brown KB, Weaver B. Nucl. Sci. Eng. 1963;17:234–246. [Google Scholar]; (b) Bryan JC, Delmau LH, Hay BP, Nicholas JB, Rogers LM, Rogers RD, Moyer BA. Struct. Chem. 1999;10:187–203. [Google Scholar]
- 30.Dougherty DA. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 31.Engle KM, Mei T-S, Wasa M, Yu J-Q. Acc. Chem. Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figg TM, Wasa M, Yu J-Q, Musaev DG. J. Am. Chem. Soc. 2013;135:14206–14214. doi: 10.1021/ja4053416. [DOI] [PubMed] [Google Scholar]
- 33.Jockusch RA, Lemoff AS, Williams ER. J. Am. Chem. Soc. 2001;123:12255–12265. doi: 10.1021/ja0106873. [DOI] [PubMed] [Google Scholar]
- 34.(a) Pathak TP, Miller SJ. J. Am. Chem. Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pathak TP, Miller SJ. J. Am. Chem. Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.