

Conspectus

Harnessing visible light as the driving force for chemical transformations generally offers a more environmentally friendly alternative compared with classical synthetic methodology. The transition metal-based photocatalysts commonly employed in photoredox catalysis absorb efficiently in the visible spectrum, unlike most organic substrates, allowing for orthogonal excitation. The subsequent excited states are both more reducing and more oxidizing than the ground state catalyst and are competitive with some of the more powerful single-electron oxidants or reductants available to organic chemists yet are simply accessed via irradiation. The benefits of this strategy have proven particularly useful in radical chemistry, a field that traditionally employs rather toxic and hazardous reagents to generate the desired intermediates.
In this Account, we discuss our efforts to leverage visible light photoredox catalysis in radical-based bond-forming and bond-cleaving events for which few, if any, environmentally benign alternatives exist. Mechanistic investigations have driven our contributions in this field, for both facilitating desired transformations and offering new, unexpected opportunities. In fact, our total synthesis of (+)-gliocladin C was only possible upon elucidating the propensity for various trialkylamine additives to elicit a dual behavior as both a reductive quencher and a H-atom donor. Importantly, while natural product synthesis was central to our initial motivations to explore these photochemical processes, we have since demonstrated applicability within other subfields of chemistry, and our evaluation of flow technologies demonstrates the potential to translate these results from the bench to pilot scale.
Our forays into photoredox catalysis began with fundamental methodology, providing a tin-free reductive dehalogenation that exchanged the gamut of hazardous reagents previously employed for such a transformation for visible light-mediated, ambient temperature conditions. Evolving from this work, a new avenue toward atom transfer radical addition (ATRA) chemistry was developed, enabling dual functionalization of both double and triple bonds. Importantly, we have also expanded our portfolio to target clinically relevant scaffolds. Photoredox catalysis proved effective in generating high value fluorinated alkyl radicals through the use of abundantly available starting materials, providing access to libraries of trifluoromethylated (hetero)arenes as well as intriguing gem-difluoro benzyl motifs via a novel photochemical radical Smiles rearrangement. Finally, we discuss a photochemical strategy toward sustainable lignin processing through selective C–O bond cleavage methodology. The collection of these efforts is meant to highlight the potential for visible light-mediated radical chemistry to impact a variety of industrial sectors.
Introduction
Visible light photoredox catalysis has become a prominent sector of synthetic methodology in the last several years due to its mild nature, its high functional group compatibility, and the unique mechanistic approaches it enables.1 Common photocatalysts include Ru(II) or Ir(III) complexes, which undergo metal-to-ligand charge transfer (MLCT) upon irradiation with visible light. Intersystem crossing reveals a relatively long-lived excited state (e.g., for Ru(bpy)32+*, τ = 1100 ns1), allowing for productive outer-sphere electron transfers to take place.2 Both the quenching of the excited state photocatalyst and the subsequent return to the original oxidation state afford opportunities to utilize the metal in single electron transfer (SET) processes (Figure 1). Because this quenching can be performed in both oxidative and reductive manifolds (generating Ru(III) or Ru(I), respectively), this mode of catalysis offers significant flexibility. Additionally, altering the metal (Ru, Ir, Cu, Cr, etc.) or ligand leads to predictable changes in redox potentials, allowing one to tailor the catalyst to one’s needs.3 Importantly, these photochemical methods offer unusually mild entry to radical reaction manifolds, as they generally operate at ambient temperatures, employ bench-stable reagents, and typically display higher functional group tolerance than traditional methods. In contrast, classical approaches tend to require hazardous radical initiators (e.g., AIBN, BEt3), toxic reagents (e.g., Bu3SnH), and in many cases, high temperatures. Photoredox catalysis has also proven to be uniquely well-suited to operate in flow, because the more uniform light penetration relative to batch processes allows for efficient catalyst excitation.4 The enhanced scalability afforded by continuous flow processing has helped drive increasing interest from industry,5 given the promise of reduced waste streams and more efficient material throughput. The benefits of employing safer and more sustainable methods are amplified upon transitioning from discovery to process scale, further incentivizing the design and application of novel visible light-mediated methodologies toward both natural and non-natural scaffolds of interest to pharmaceutical and agrochemical domains.6
Figure 1.

Oxidative and reductive quenching cycles within photoredox catalysis (A) and structures of common transition metal photocatalysts (B).
Initial Methodology and Applications to Total Synthesis
The aforementioned advantages of photoredox catalysis give synthetic chemists the opportunity to design and access new and more challenging reactions in an environmentally benign fashion. However, at the outset of our research program, the bulk of today’s photoredox catalysts were exclusive to materials science and photovoltaics.1 Alongside other pioneering works from MacMillan and Yoon,7 our group was interested in applying photoredox catalysis toward novel bond disconnections in complex molecule synthesis, initially targeting radical C–C bond forming reactions.8 Indole- and pyrrole-based systems took our attention due to their abundance in natural products and biologically active compounds (e.g., actinophyllic acid (5), Figure 2A). The alkaloid natural product (+)-gliocladin C (7)9 was an early target, motivated by the potential for a strategic C3–C3′ radical coupling reaction between a pyrroloindoline and an indole.10,11 Specifically, it was anticipated that the combination of Ru(bpy)3Cl2 (1) and N,N-diisopropylethylamine (DIPEA) would lead to reductive quenching of the photoexcited catalyst to generate the Ru(I) species, which would subsequently reduce the C–Br bond of 3-bromopyrroloindoline 8 to form the desired tertiary radical and facilitate the designed intermolecular coupling. The predicted reduction occurred, but the desired coupling was not observed. Instead, the hydrodehalogenated pyrroloindoline 9 was obtained as single product (Figure 2B).12
Figure 2.
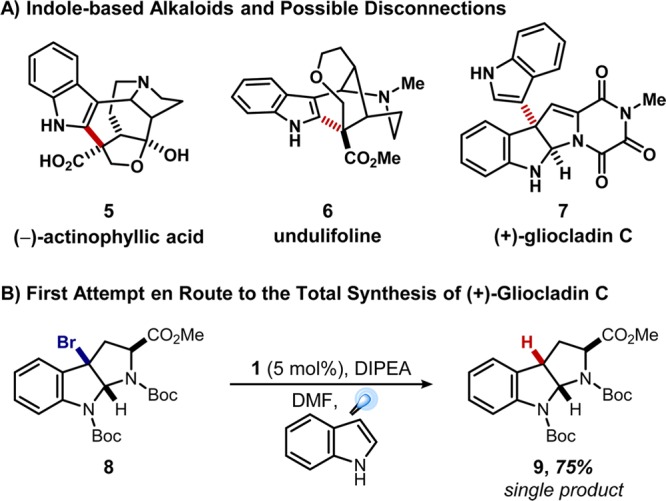
Initial attempt at the total synthesis of (+)-gliocladin C.
Subsequent investigation rationalized these results through the discovery that the amine was not only an effective quencher of the excited state photocatalyst but also a potent H-atom donor. We have subsequently leveraged this knowledge by productively employing α-amino radical cations and iminium intermediates in methodology,13 total synthesis,14 and the synthesis of pharmaceuticals.15,16
While the first attempt en route to (+)-gliocladin C gave no traces of the desired C–C bond coupling, the formation of this dehalogenated product 9 served as the foundation for further development of a general tin-free, visible light-mediated hydrodehalogenation protocol. Applying the Ru(bpy)32+ conditions to a range of different activated alkyl bromides and chlorides afforded the hydrodehalogenation products in excellent yields without the need for tin hydrides or hazardous radical initiators.12 As expected, aryl or alkenyl iodides were completely unreactive (Figure 3A, left), given their exceptionally negative reduction potentials (−2.24 V vs SCE for iodobenzene17). However, several Ir(III)-based photocatalysts offer significantly more reducing power than Ru(bpy)3+, potentially allowing one to dehalogenate less activated systems. Indeed, by employing the oxidative quenching cycle of fac-Ir(ppy)3 (4), we achieved deiodination of unactivated alkyl, vinyl, and aryl iodides, with good functional group tolerance (Figure 3A, right).18 Of note, reduction potentials are conventionally reported as peak potentials, yet these redox processes actually occur over a range of potentials (generally several hundred millivolts), enabling electron transfers that appear impossible based on literature values; thus while the excited state of 3 would seem to still be insufficiently reducing to affect aryl iodides, the reduction proceeds cleanly. Importantly, the application of flow technologies in both cases demonstrated superior performance relative to batch, allowing shorter reaction times with up to 20-fold decrease in the photocatalyst loading (Figure 3B).19
Figure 3.

Catalyst effects in reductive dehalogenation methods and the effectiveness of continuous flow processing.
As expected, while developing the reductive dehalogenation chemistry, a number of substrates with pendant olefins were found to readily undergo cyclization prior to H-atom abstraction (as seen in Figure 3). Because our goals for the dehalogenative chemistry ultimately focused on C–C bond-forming reactions, we were highly intrigued by these observations and sought to develop more generalized intramolecular cyclization conditions. A key discovery toward this end was that malonate radicals were much less likely to abstract hydrogen when triethylamine was used in place of DIPEA. For example, bromomalonate derivative 10 afforded 60% of the cyclization product 12 as the sole isolated product when irradiated in the presence of triethylamine as opposed to the 1.3:1 mixture of cyclization product 12 to hydrodehalogenated product 11 obtained when employing DIPEA (Scheme 1).20
Scheme 1. Divergent Reactivity of Trialkylamine Additives in the Photocatalytic Reduction of Alkylbromomalonates.

Armed with this knowledge, we explored the utility of this photochemical methodology within the context of classical cascade processes. Radical cascades are one of the most powerful tools for accessing complex structures in a single step,21 if the substrate is stable to the conditions for radical initiation. Gratifyingly, our mild, visible light-mediated methods for generating carbon-centered radicals proved highly effective in a number of radical cascade processes,20,22 generating fused tetracycle 14 from bromomalonate 13 and tricyclic compound 16 from alkenyne 15 in good yields as single diastereomers (Figure 4A). While exploring new avenues to vinylcyclopropanes, we discovered a particularly interesting cascade in which a [3,3]-sigmatropic rearrangement was induced upon achieving our designed cyclization. Ir(III) photocatalyst 3 was found to be optimal for reducing 1-amidobromocyclopropanes (e.g., 17); however, upon cyclization into the pendant alkyne, newly formed vinylcyclopropane (18) was properly disposed to undergo a subsequent divinylcyclopropane rearrangement, ultimately providing the seven-membered ring byproduct 19 after rearomatization in 69% yield (Figure 4B).23 Collectively, these results demonstrate that the already known utility of radical cascades to generate rapid gains in molecular complexity can be accessed with the mild conditions provided by photoredox catalysis.
Figure 4.
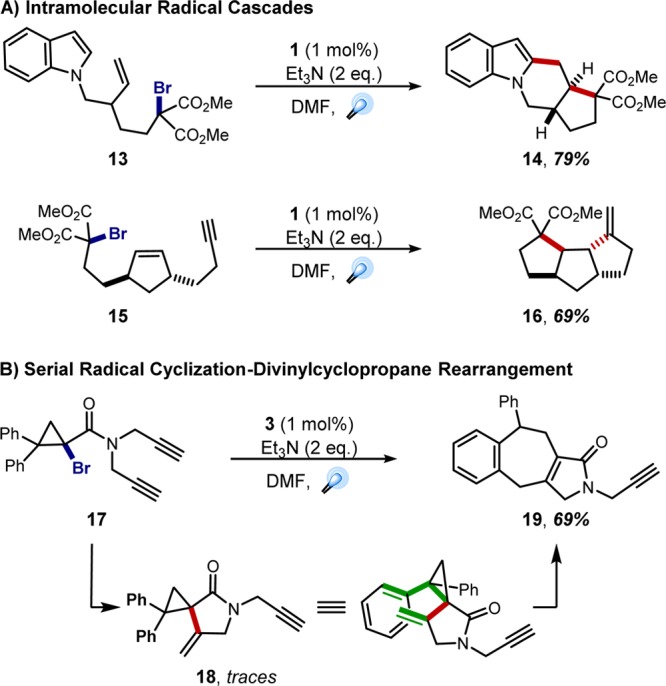
Applications of photoredox catalysis in intramolecular radical cascades.
Having developed a body of expertise utilizing the reductive dehalogenation strategy in intramolecular C–C bond-forming reactions, we turned our attention to intermolecular additions. The coupling of indoles with malonate radicals was initially our primary focus, given that malonate-like motifs are common C2-substituents in bioactive indole alkaloids, such as actinophyllic acid (5) or undulifoline (6) (Figure 2A). Initial efforts employed N,N-diphenyl-4-methoxyaniline as the reductive quencher (reducing the likelihood of H-atom abstraction pathways), which facilitated the coupling of malonate radicals to an extensive range of indole and pyrrole derivatives in good yields (Figure 5, left).24 The application of this methodology in flow proved to be extremely efficient, achieving comparable reaction yields with only 1 min of residence time. As many of the indole alkaloids that inspired this work contain quaternary carbon centers adjacent to C2, a complementary method employing the more challenging tertiary malonate radicals was developed.25 Avoiding the use of reductive quenching additives eliminated concerns over deleterious H-atom abstraction pathways. This was accomplished by directly reducing bromomalonate 20b via oxidative quenching of the strongly reducing fac-Ir(ppy)3 photocatalyst (4) (Figure 5, right), providing the targeted quaternary carbon centers in good to high yields.
Figure 5.

Intermolecular radical addition of secondary and tertiary radicals to electron-rich heterocycles.
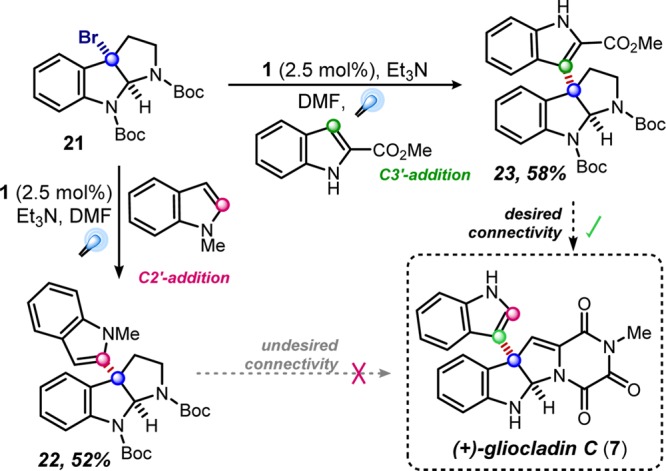
With a greater understanding of these reductive dehalogenation processes, we refined our approach toward the total synthesis of (+)-gliocladin C (7). Revisiting our strategic C3–C3′ coupling with triethylamine as the reductive quencher, we found that hydrodehalogenation of 3-bromopyrroloindoline 21 was avoided, but the coupling with N-methylindole exclusively formed the C3–C2′ adduct 22 (Scheme 2). This issue was overcome through the use of 2-methoxycarbonylindole, generating the desired C3′-addition product (23) in 58% yield.26
Scheme 2. Observed Regioselectivities in Intermolecular Pyrrole–Pyrroloindoline Couplings.
Transitioning these results toward the gliocladin scaffold, we prepared bromopyrroloindoline 25 from d-tryoptophan and employed the dehalogenative coupling conditions with 2-formylindole. The desired C3–C3′ coupling was observed, though the reaction stalled while employing triethylamine. Switching to an analogous amine with lower vapor pressure, n-tributylamine, ameliorated this issue, generating coupled product 26 in 82% yield on gram-scale. The subsequent deformylation, assemblage of the triketopiperazine moiety under microwave conditions, and global deprotection provided (+)-gliocladin C in 35% overall yield over 10 steps (Scheme 3).26
Scheme 3. Total Synthesis of (+)-Gliocladin C.

In parallel with the above work, a new avenue for intermolecular C–C bond-forming processes evolved from the observation of intriguing byproducts in our early intramolecular cyclization efforts. Upon exposure of cyclopentene 29 to the optimized malonate addition protocol (vide supra, Scheme 1), 85% of cyclized product was isolated, but this proved to be a 1:9 mixture of the anticipated reduced species 30a and bromide 30b (Figure 6).27 Discovering these Kharasch-type byproducts28 sparked our interest in atom transfer radical addition (ATRA) chemistry, since this offered the potential for a uniquely efficient and economical method for dual functionalization of double (and perhaps triple) bonds. Similar to the intermolecular malonate–indole coupling detailed above, these transformations are redox neutral, theoretically eliminating the need for additives and reducing the likelihood of deleterious off-target reactivity. However, this would again necessitate direct oxidative quenching of the excited state photocatalyst with the alkyl bromide.
Figure 6.
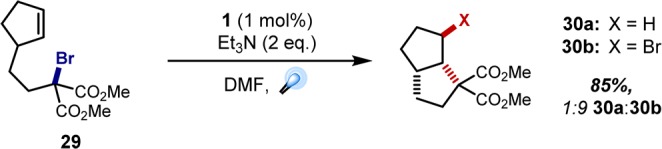
Preliminary observation of Kharasch-type products in photoredox catalysis.
Gratifyingly, preliminary optimization for the ATRA of diethyl bromomalonate across terminal olefins revealed that the heteroleptic Ir(III) photocatalyst [Ir{dF(CF3)ppy}2(dtbbpy)](PF6) (2) was effective when LiBr was added to activate the bromomalonate for reduction (Figure 7A).29 This method tolerated a variety of functional groups while generally providing the radical transfer products in ≥90% yield. Further optimization avoided the need for additives through the use of [Ru(bpy)3]Cl2 (1) in DMSO. The scope of the ATRA chemistry was expanded under these conditions, employing both new halides (e.g., CCl4, TsCl) and previously recalcitrant olefins (i.e., strained 1,2-disubstituted olefins, styrenes; Figure 7B).27 This strategy also proved effective for the iodoperfluoroalkylation of olefins and alkynes. This represented a new approach for fluorous tagging,30 which we demonstrated through post-transformational labeling and fluorous phase removal of problematic byproducts. For instance, Wittig olefination of aldehyde 32 with phosphonium salt 31 proceeded cleanly to styrene 34 (∼1:1 dr); the crude mixture was then exposed to the optimized fluorous tagging ATRA methodology, allowing for easy removal of the phosphine oxide byproduct via fluorous solid phase extraction (F-SPE). The irradiation step also facilitated the isomerization of the stilbene to predominantly the Z-olefin via triplet sensitization (35; Figure 7C).27,31
Figure 7.
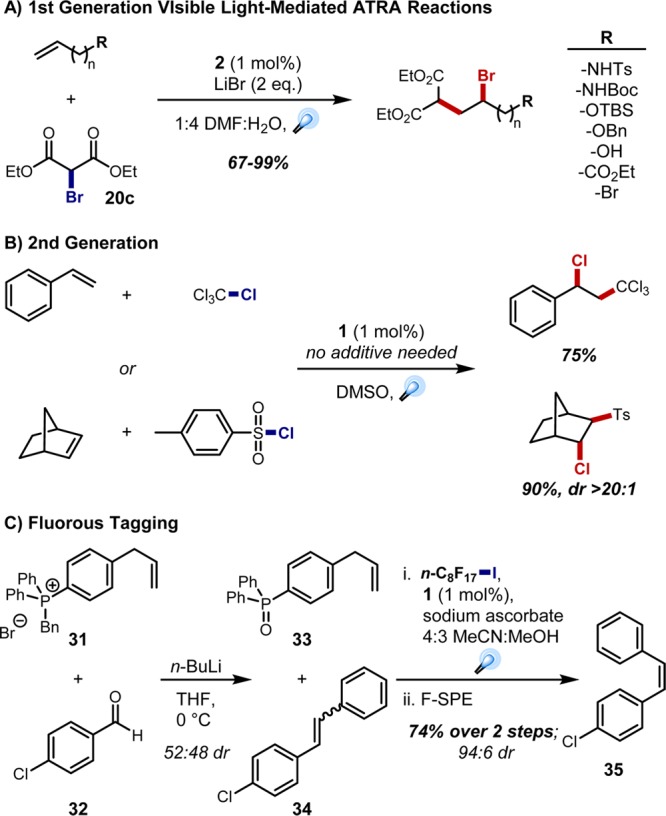
Optimized procedures for visible light-mediated ATRA and applications in fluorous tagging.
At this time, it was unclear whether a radical–polar crossover mechanism was affording the ATRA products or radical propagation pathways were driving the reaction forward; interestingly, experimentation would ultimately reveal evidence for both mechanisms. Supporting a polar mechanism, we observed a small amount of tetrahydrofuran byproduct 38 in ATRA additions to pentenol 36a. Notably, isolated bromide 37a could not be converted to tetrahydrofuran 38 upon exposure to the reaction conditions or after heating in toluene (Figure 8A), suggesting that a carbocation intermediate is necessary for its production. This cation could arise via radical addition to the olefin and oxidation of the resultant radical, returning the oxidized catalyst to its ground state. However, subsequent crossover studies would support a propagative pathway. Hexenol 36b and bromoacetate 20d alone provided no reaction under the optimized conditions, because this halide cannot be directly reduced by the excited state photocatalyst. In contrast, the bromoacetate-derived ATRA product 39 could be generated upon employing a mixture of bromomalonate 20c and bromoacetate 20d, indicating the involvement of radical propagation pathways (Figure 8B,C). Corroborating this complexity, Yoon and co-workers have recently demonstrated that a number of methods previously believed to proceed through fully catalyst-controlled mechanistic cycles are actually largely driven by radical propagation mechanisms.32 Importantly, the balance between closed catalytic cycles and open chain processes depends not just on the transformation as a whole but also on specific conditions, including scale.33 Demonstrating the viability of a given method on both discovery and preparative scales is thus critical to proving its potential to impact industrial processes.
Figure 8.
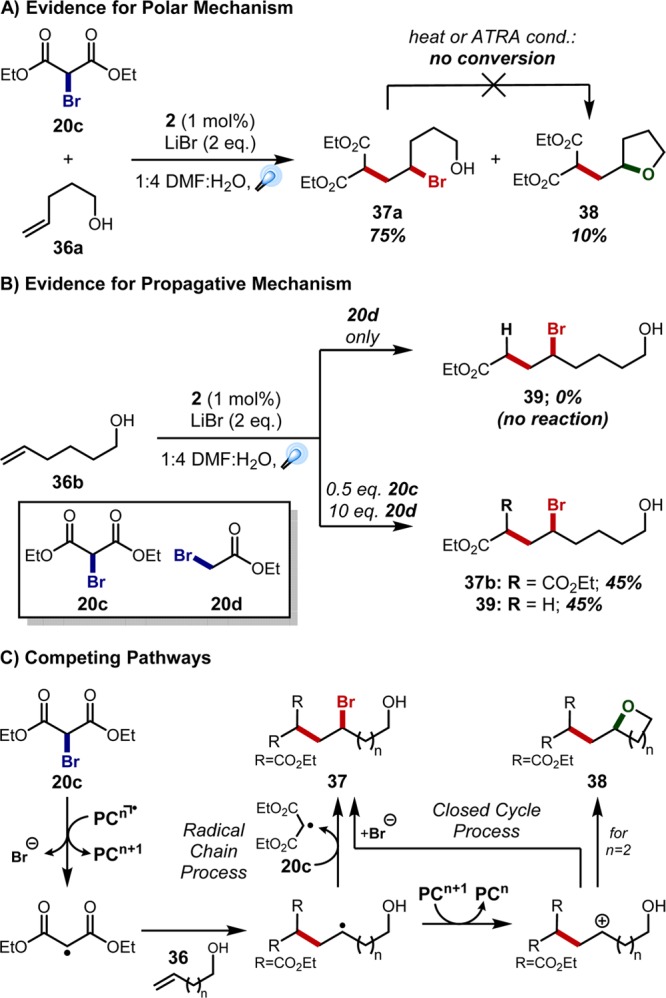
Competing mechanisms within photochemical ATRA methodology.
Targeting Pharmaceutically Relevant Scaffolds
As noted above, a significant driving force behind our initial photochemical efforts was to enable new bond disconnections toward biologically active natural products. This mentality has continuously resonated within the group, but concurrent with and complementary to amine oxidation efforts, which were beginning to realize this goal,14,16 we began pursuing avenues toward the diversification of non-natural clinically relevant scaffolds. Specifically, we sought to leverage the unique capabilities of photoredox catalysis to access a variety of fluoroalkyl radical species34 for the late-stage modification of therapeutic leads. Fluorinated functional groups (the trifluoromethyl group in particular) have become increasingly popular over the past few decades,35 because these motifs have little effect on size and shape of lead compounds yet can offer dramatic modifications to physicochemical properties. The following details our efforts toward trifluoromethylated and difluorobenzylated scaffolds and the promising potential to translate these and related methods to the process scale.
In regards to trifluoromethylation, the gamut of methodology for introducing the CF3 group highlights the demand for reliable access to such products.36 A number of methods have proven effective on the discovery scale, though these typically employ specialized reagents37 with weakened X–CF3 bonds to facilitate in situ generation of the operative trifluoromethylation intermediate. In hopes of providing an approach tailored to industrial scales, our design focused on using abundantly available CF3 sources and eliminating the need for prefunctionalized substrates.38 Fluoroform (a byproduct of Teflon production) offers one option,39 though environmental concerns (greenhouse gas; atmospheric lifetime of 254 years40) may supersede the cost benefits. Alternatively, trifluoroacetic acid and its anhydride (TFA, TFAA) are stable liquids and handled with relative ease. However, accessing the economic and operational practicality of these reagents in trifluoromethylation chemistry necessitates the activation of a highly stable C–C bond. Prior fragmentations of TFA required harsh thermolysis (140–210 °C with Cu salts41) or oxidation of trifluoroacetate at potentials incompatible with standard solvents and many substrates (for F3CO2Na: Eox > +2.4 V vs SCE in MeCN).42 As a result, TFA and TFAA have proven useful only in limited contexts, often requiring halogenated coupling partners41 and (super)stoichiometric metal promoters.41,43 We sought to alter this paradigm by providing a novel mode of C–C activation through the introduction of a “redox trigger”, a component that would generate a modified trifluoroacetate in situ such that the redox potential lies within the reach of photochemical methods. A pyridine N-oxide (PNO)–TFAA system was designed, because the acylated PNO offers a weak N–O bond suspected to be readily amenable to reduction (Figure 9A). Indeed, cyclic voltammetry measurements revealed a half-cell potential of −1.10 V vs SCE in MeCN for the acylated intermediate 39, with an onset potential of −0.86 V.44 These values lie well within the ±1.3 V window available with Ru(bpy)32+ catalytic cycles. Gratifyingly, this PNO–TFAA combination proved effective for the trifluoromethylation of a variety of (hetero)arenes (including those with Lewis basic functionality) as well as olefins when irradiated in the presence of photocatalyst 1 (Figure 9B).
Figure 9.
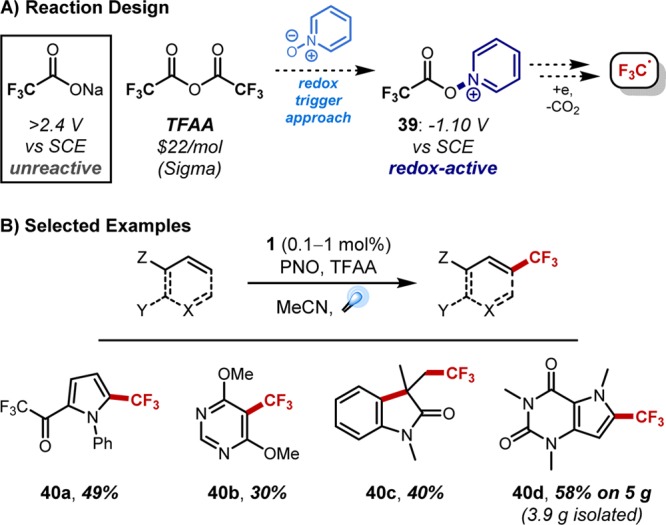
Accessing and implementing trifluoromethyl radicals derived from trifluoroacetic anhydride via visible light photoredox catalysis.
The putative mechanism proceeds through direct oxidative quenching of the excited state photocatalyst with the acylated PNO 39, liberating pyridine and the carboxyl radical 41, which rapidly decomposes to CO2 and the desired CF3 radical. Addition of this radical to the substrate of interest (i.e., clinically relevant arene or heteroarene substructures), oxidation of the resultant radical, and base-mediated rearomatization generates the trifluoromethylated substrate and closes the catalytic cycle (Figure 10).
Figure 10.
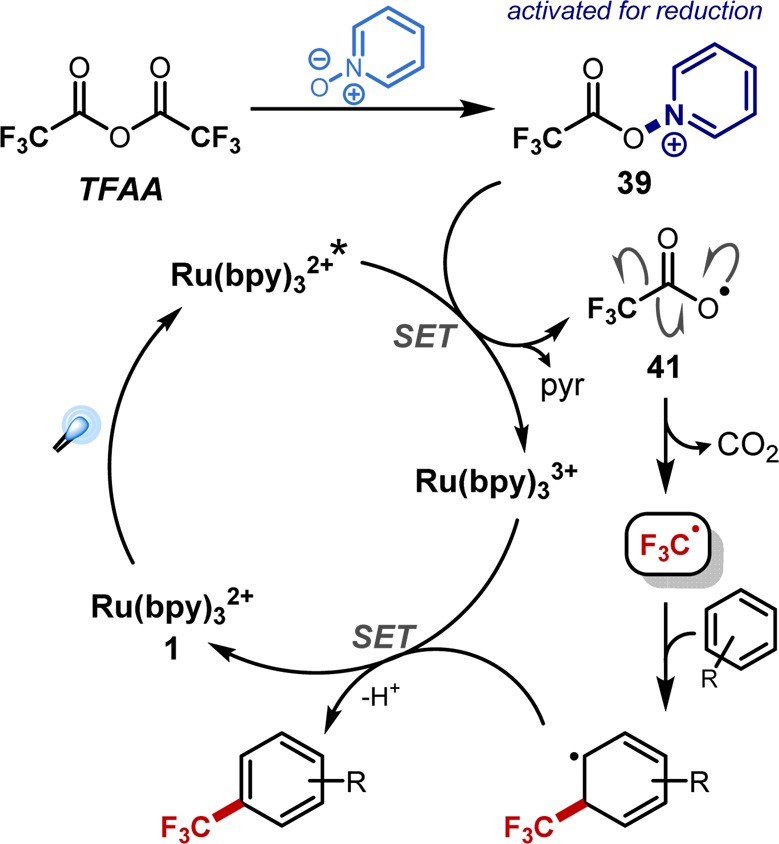
Putative mechanism of photochemical trifluoromethylation with TFAA–PNO system.
Importantly, this chemistry was readily translated to multigram scales for a number of substrates, including MIDA boronate 42, which was shown to be viable in subsequent cross-coupling chemistry (Figure 11A). In addition, trifluoromethylated 2-chloropyridine 47, a key intermediate in anti-infective programs at Boehringer Ingelheim, was prepared through this methodology (Figure 11B).45 Significantly, transitioning this new trifluoromethylation method to continuous flow processing improved the scalability.46N-Boc-pyrrole (48) was trifluoromethylated in 57% yield (5:1 mono/bis) when run on 18 g scale under batch conditions over 15 h (Figure 11C). The same reaction on 23 g scale, when run in flow with a 10 min residence time, afforded 71% of the product mixture. We are currently investigating continuous flow variants of this methodology for kilogram scale preparations of pharmaceutically relevant intermediates, work that will be reported in due course.
Figure 11.

Applications of visible light-mediated trifluoromethylation.
In addition to trifluoromethylation, we are also interested in developing reliable methods toward alternative and unique fluoroalkyl motifs to facilitate drug discovery programs. One specific challenge for which we have provided a solution is the gem-difluorobenzyl functional group. This work was inspired by recent efforts toward ORL-1 antagonists (ORL-1 = opioid receptor-like 1; target indications for depression and obesity) framed around spirocyclic piperidine 52. The reported route required just three steps to build the spirocyclic core 51 from alcohol 50, but four additional operations were needed to incorporate the gem-difluoro motif, relying on 2.6 equiv of Deoxo-Fluor (Figure 12).47 Circumventing this nonoptimal sequence could greatly increase material throughput for lead optimization and evaluation. Toward this end, we designed a photochemical radical Smiles rearrangement to generate difluorinated alcohol 53, a substrate to be used in lieu of previous intermediate 50.
Figure 12.

Redesigned route toward ORL-1 antagonist intermediate 52.
While radical Smiles rearrangements had previously been reported,48 these generally required harsh/hazardous conditions, usually employing AIBN in high loadings (up to 50 mol %49) as the radical initiator, presumably due to inefficient propagation processes. In contrast, our initially optimized method operated at ambient temperature with catalyst loadings as low as 0.01 mol %, requiring only tributylamine and formic acid as additives (Figure 13).50 The requisite 2-bromo-2,2-difluoroethanol is readily available from the corresponding ethyl ester 55 and couples efficiently with sulfonyl chlorides to provide the Smiles rearrangement-precursor. Notably, the wealth of commercially available sulfonyl chlorides suggests that this strategy can be used to diversify a vast range of (hetero)arene subscaffolds in a regioselective manner (note that thiophene substrates such as 53 are preferentially labeled at the 2- and 5-positions in the intermolecular fluoroalkylation strategies shown above). Indeed, a variety of heteroaryl substrates were amenable to this chemistry, many of which proceeded in good to high yields (Figure 13).
Figure 13.
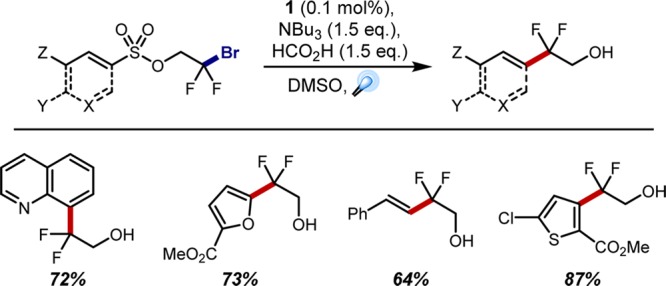
Selected examples of photochemical radical smiles rearrangement products.
Importantly, this methodology was applied toward the target substrate of interest. Initial scale-up revealed that Smiles precursor 56 was readily converted to thiophene 57 in 69% yield on 15 g scale (Scheme 4). The C2 methyl ester was saponified immediately after rearrangement as purification of the carboxylic acid proved to be operationally easier; decarboxylation then generated the targeted difluoroethanol intermediate 53 in just four steps from commercial materials. Recently, a more rigorous investigation of this method on preparative scale was undertaken, revealing a number of key insights on the mechanism.51 It was shown to be highly propagative, and this chain process was found to be highly sensitive to oxygen. Rigorous degassing allowed for catalyst loadings as low as 0.01 mol %; purely thermal conditions were also shown to be viable, though this was highly substrate-dependent and required elevated temperatures relative to the photochemical route (65–70 °C vs 25 °C, respectively). Importantly, this work demonstrated that the Smiles rearrangement of thiophene 56 could be carried out on 100 g scale, suggesting industrial applicability for this regiochemical incorporation of benzylic gem-difluoro motifs.
Scheme 4. Preparative Scale Synthesis of Difluorinated Intermediate 53.

Applications in Biofeedstock Processing
Alongside our interest in the synthesis of pharmaceutically relevant scaffolds (previous section), our group has pursued alternative means of impacting industrial processes through our efforts in conversion of biomass into value-added chemicals.52 In particular, we are highly interested in achieving the controlled depolymerization of lignin, one of the most abundant feedstocks for aromatic commodity compounds.
Lignin is a stable, branched biopolymer that is part of the cellular wall of plants and is primarily responsible for providing both rigidity and protection against environmental conditions. Its structure is primarily comprised of three different cinnamyl alcohols, coupling together to form a diverse array of motifs within the polymer chain (Figure 14). This stability and highly varied connectivity has precluded attempts to cleanly isolate high value compounds through degradative processing; more commonly, low yields of functional chemicals (∼20% of syngas) are obtained, while producing bulk quantities of intractable byproducts.53
Figure 14.

Structural representation of lignin.
A wealth of academic research has sought to ameliorate these biomass processing issues, be it through oxidative, reductive, or redox-neutral approaches.54,55 Many of these target the β-O-4 linkage, because this is the most abundant (45–65% of all linkages) and thus the most sensible starting point in lignin degradation efforts. Upon recognizing that the key β-O-4 bond is weakened following oxidation of the benzylic position (∼14 kcal/mol56), we reasoned that photoredox catalysis could provide a mild means of cleaving that critical bond. A two-step procedure was designed in which selective oxidation of the α-carbon would be accomplished with [4-AcNH-TEMPO]BF454b followed by photochemical reductive cleavage (Figure 15A).57
Figure 15.

Two-step protocol for the degradation of lignin model systems.
Through this strategy, we were able to efficiently degrade a range of lignin model systems, isolating the fragmentation products in excellent yields when employing photocatalyst 3 under reductive quenching conditions (Figure 15B).58 Significantly, flow technologies proved beneficial for this system as well, affording significant improvements in terms of reaction time and photocatalyst loading (0.03 mol % 3, 33 min residence time). The ability to reduce catalyst loading upon transitioning from batch to flow is a common advantage of flow processing, owing this improvement to the more efficient irradiation.
Conclusion
Our interest in photoredox catalysis originally centered on its potential to enable new and efficient strategies for the formation of organic free radicals and their application in complex molecule synthesis. While that interest remains, the summation of our work provided above highlights the potential for these photochemical strategies to address needs and challenges in fields beyond the realm of total synthesis. Both the fluoroalkylation methods and lignin degradation efforts were motivated by similar principles: a high demand from industry and a lack of sustainable, environmentally benign alternatives. Industry’s increasing investment in photochemical operations (especially those coupled with continuous flow processing4,5,59) suggests that visible light-mediated methods will soon provide impactful advances in the pharmaceutical, agrochemical, and commodity chemical sectors.
Acknowledgments
NSF (Grants CHE-1440118 and CHE-1565782), NIH-NIGMS (Grant R01-GM096129), Fundación Ramón Areces (I.B.), Eli Lilly, Novartis, Boston University, and the University of Michigan are gratefully acknowledged for financial support of this research.
Biographies
Daryl Staveness received a B.S. in Chemistry from the University of Wisconsin—Madison in 2009 and a Ph.D. from Stanford University in 2015 under the supervision of Prof. Paul Wender. He is currently pursuing postdoctoral studies in the lab of Prof. Corey Stephenson.
Irene Bosque received a B.S. in Chemistry in 2010 and an M.Sc. in 2012 from the University of Alicante after an internship at Karolinska Institute, Stockholm. She received a Ph.D. in 2014 from the University of Alicante under the supervision of Prof. José Carlos González-Gómez. She is currently a postdoctoral fellow with Prof. Corey Stephenson.
Corey Stephenson earned his Ph.D. from the University of Pittsburgh under Prof. Peter Wipf before pursuing postdoctoral studies at ETH Zurich with Prof. Erick Carreira. He began his independent career in 2007 at Boston University and moved to the University of Michigan in 2013. His research interests are broadly focused on catalysis, complex molecule synthesis, and biomass degradation.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Photoredox Catalysis in Organic Chemistry”.
References
- For recent reviews on the field of photoredox catalysis, see:; a Prier C.; Rankic D.; MacMillan D. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Douglas J.; Nguyen J.; Cole K.; Stephenson C. Enabling Novel Photoredox Reactivity via Photocatalyst Selection. Aldrichimica Acta 2014, 47, 15–25. [Google Scholar]
- Juris A.; Balzani V.; Belser P.; von Zelewsky A. Characterization of the Excited State Properties of Some New Photosensitizers of the Ruthenium (Polypyridine) Family. Helv. Chim. Acta 1981, 64, 2175–2182. 10.1002/hlca.19810640723. [DOI] [Google Scholar]
- Tucker J.; Stephenson C. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- Cambié D.; Bottecchia C.; Straathof N.; Hessel V.; Noël T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- Porta R.; Benaglia M.; Puglisi A. Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Process Res. Dev. 2016, 20, 2–25. 10.1021/acs.oprd.5b00325. [DOI] [Google Scholar]
- Kärkäs M.; Porco J. Jr.; Stephenson C. Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 10.1021/acs.chemrev.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nicewicz D.; MacMillan D. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ischay M.; Anzovino M.; Du J.; Yoon T. Efficient Visible Light Photocatalysis of [2 + 2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- Hart D. Free-Radical Carbon–Carbon Bond Formation in Organic Synthesis. Science 1984, 223, 883–887. 10.1126/science.223.4639.883. [DOI] [PubMed] [Google Scholar]
- Usami Y.; Yamaguchi J.; Numata A. Gliocladins A - C and Glioperazine; Cytotoxic Dioxo- or Trioxopiperazine Metabolites from a Gliocladium Sp. Separated from a Sea Hare. Heterocycles 2004, 63, 1123–1129. 10.3987/COM-04-10037. [DOI] [Google Scholar]
- Lathrop S.; Kim J.; Movassaghi M. Radical-mediated Dimerization and Oxidation Reactions for the Synthesis of Complex Alkaloids. Chimia 2012, 66, 389–393. 10.2533/chimia.2012.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzumi S.; Mochizuki S.; Tanaka T. Photocatalytic reduction of phenacyl halides by 9,10-dihydro-10-methylacridine: control between the reductive and oxidative quenching pathways of tris(bipyridine)ruthenium complex utilizing an acid catalysis. J. Phys. Chem. 1990, 94, 722–726. 10.1021/j100365a039. [DOI] [Google Scholar]
- Narayanam J.; Tucker J.; Stephenson C. Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- Condie A.; González-Gómez J.; Stephenson C. Visible-Light Photoredox Catalysis: Aza-Henry Reactions via C-H Functionalization. J. Am. Chem. Soc. 2010, 132, 1464–1465. 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]
- Beatty J.; Stephenson C. Synthesis of (−)-Pseudotabersonine, (−)-Pseudovincadifformine, and (+)-Coronaridine Enabled by Photoredox Catalysis in Flow.. J. Am. Chem. Soc. 2014, 136, 10270–10273. 10.1021/ja506170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J.; Cole K.; Stephenson C. Photoredox Catalysis in a Complex Pharmaceutical Setting: Toward the Preparation of JAK2 Inhibitor LY2784544. J. Org. Chem. 2014, 79, 11631–11643. 10.1021/jo502288q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- These efforts were recently reviewed in a separate Account:Beatty J.; Stephenson C. Amine functionalization via oxidative photoredox catalysis: Methodology development and complex molecule synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause L.; Robert M.; Savéant J.-M. Can Single-Electron Transfer Break an Aromatic Carbon–Heteroatom Bond in One Step? A Novel Example of Transition between Stepwise and Concerted Mechanisms in the Reduction of Aromatic Iodides. J. Am. Chem. Soc. 1999, 121, 7158–7159. 10.1021/ja991365q. [DOI] [Google Scholar]
- Nguyen J.; D’Amato E.; Narayanam J.; Stephenson C. Engaging Unactivated Alkyl, Alkenyl and Aryl Iodides in Visible-Light-Mediated Free Radical Reactions. Nat. Chem. 2012, 4, 854–859. 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]
- Nguyen J.; Reiß B.; Dai C.; Stephenson C. Batch to flow deoxygenation using visible light photoredox catalysis. Chem. Commun. 2013, 49, 4352–4354. 10.1039/C2CC37206A. [DOI] [PubMed] [Google Scholar]
- Tucker J.; Narayanam J.; Krabbe S. W.; Stephenson C. Electron Transfer Photoredox Catalysis: Intramolecular Radical Addition to Indoles and Pyrroles. Org. Lett. 2010, 12, 368–371. 10.1021/ol902703k. [DOI] [PubMed] [Google Scholar]
- Sebren L.; Devery J.; Stephenson C. Catalytic Radical Domino Reactions in Organic Synthesis. ACS Catal. 2014, 4, 703–716. 10.1021/cs400995r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker J.; Nguyen J.; Narayanam J.; Krabbe S.; Stephenson C. Tin-free radical cyclization reactions initiated by visible light photoredox catalysis. Chem. Commun. 2010, 46, 4985–4987. 10.1039/c0cc00981d. [DOI] [PubMed] [Google Scholar]
- Tucker J.; Stephenson C. Tandem Visible Light-Mediated Radical Cyclization-Divinylcyclopropane Rearrangement to Tricyclic Pyrrolidinones. Org. Lett. 2011, 13, 5468–5471. 10.1021/ol202178t. [DOI] [PubMed] [Google Scholar]
- Furst L.; Matsuura B.; Narayanam J.; Tucker J.; Stephenson C. Visible Light-Mediated Intermolecular C-H Functionalization of Electron-Rich Heterocycles with Malonates. Org. Lett. 2010, 12, 3104–3107. 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]
- Swift E.; Williams T.; Stephenson C. Intermolecular Photocatalytic C-H Functionalization of Electron-Rich Heterocycles with Tertiary Alkyl Halides. Synlett 2016, 27, 754–758. 10.1055/s-0035-1561320. [DOI] [Google Scholar]
- Furst L.; Narayanam J.; Stephenson C. Total Synthesis of (+)-Gliocladin C Enabled by Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed. 2011, 50, 9655–9659. 10.1002/anie.201103145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallentin C.; Nguyen J.; Finkbeiner P.; Stephenson C. Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]
- Kharasch M.; Skell P.; Fisher P. Reactions of Atoms and Free Radicals in Solution. XII. The Addition of Bromo Esters to Olefins. J. Am. Chem. Soc. 1948, 70, 1055–1059. 10.1021/ja01183a053. [DOI] [Google Scholar]
- Nguyen J.; Tucker J.; Konieczynska M.; Stephenson C. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran D. Fluorous Reverse Phase Silica Gel. A New Tool for Preparative Separations in Synthetic Organic and Organofluorine Chemistry. Synlett 2001, 2001, 1488–1496. 10.1055/s-2001-16800. [DOI] [Google Scholar]
- Wrighton M.; Markham J. Quenching of the Luminescent State of Tris(2,2′-bipyridine)ruthenium(II) by Electronic Energy Transfer. J. Phys. Chem. 1973, 77, 3042–3044. 10.1021/j100644a002. [DOI] [Google Scholar]
- Cismesia M.; Yoon T. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. 10.1039/C5SC02185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Further discussion can be found in a perspective article highlighting ref (32), see:Kärkäs M.; Matsuura B.; Stephenson C. Enchained by visible light-mediated photoredox catalysis. Science 2015, 349, 1285–1286. 10.1126/science.aad0193. [DOI] [PubMed] [Google Scholar]
- Koike T.; Akita M. Trifluoromethylation by Visible-Light-Driven Photoredox Catalysis. Top. Catal. 2014, 57, 967–974. 10.1007/s11244-014-0259-7. [DOI] [Google Scholar]
- Gillis E.; Eastman K.; Hill M.; Donnelly D.; Meanwell N. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Alonso C.; Martínez de Marigorta E.; Rubiales G.; Palacios F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. 10.1021/cr500368h. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Matoušek V.; Pietrasiak E.; Schwenk R.; Togni A. One-pot synthesis of hypervalent iodine reagents for electrophilic trifluoromethylation. J. Org. Chem. 2013, 78, 6763–6768. 10.1021/jo400774u. [DOI] [PubMed] [Google Scholar]; b Umemoto T.; Ishihara S. Effective methods for preparing S-(trifluoromethyl)dibenzothiphenium salts. J. Fluorine Chem. 1998, 92, 181–187. 10.1016/S0022-1139(98)00276-0. [DOI] [Google Scholar]; c Ji Y.; Brueckl T.; Baxter R.; Fujiwara Y.; Seiple I.; Su S.; Blackmond D.; Baran P. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411–14415. 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Morimoto H.; Tsubogo T.; Litvinas N.; Hartwig J. A Broadly Applicable Copper Reagent for Trifluoromethylations and Perfluoroalkylations of Aryl Iodides and Bromides. Angew. Chem., Int. Ed. 2011, 50, 3793–3798. 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For notable methods that avoid the use of pre-functionalized substrates, see ref (34)c andNagib D.; MacMillan D. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lishchynskyi A.; Novikov M.; Martin E.; Escudero-Adan E.; Novak P.; Grushin V. Trifluoromethylation of Aryl and Heteroaryl Halides with Fluoroform-Derived CuCF3: Scope, Limitations, and Mechanistic Features. J. Org. Chem. 2013, 78, 11126–1146. 10.1021/jo401423h. [DOI] [PubMed] [Google Scholar]
- McCulloch A.; Lindley A. Global emmissions of HFC-23 estimated to year 2015. Atmos. Environ. 2007, 41, 1560–1566. 10.1016/j.atmosenv.2006.02.021. [DOI] [Google Scholar]
- a Matsui K.; Tobita E.; Ando M.; Kondo K. A Convenient Trifluoromethylation of Aromatic Halides with Sodium Trifluoroacetate. Chem. Lett. 1981, 1719–1720. 10.1246/cl.1981.1719. [DOI] [Google Scholar]; b Chen M.; Buchwald S. L. Rapid and Efficient Trifluoromethylation of Aromatic and Heteroaromatic Compounds Using Potassium Trifluoroacetate Enabled by a Flow System. Angew. Chem., Int. Ed. 2013, 52, 11628–11631. 10.1002/anie.201306094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depecker C.; Marzouk H.; Trevin S.; Devynck J. Trifluoromethylation of aromatic compounds via Kolbe electrolysis in pure organic solvent. Study on laboratory and pilot scale. New J. Chem. 1999, 23, 739–742. 10.1039/a901305i. [DOI] [Google Scholar]
- Shi G.; Shao C.; Pan S.; Yu J.; Zhang Y. Silver-Catalyzed C–H Trifluoromethylation of Arenes Using Trifluoroacetic Acid as the Trifluoromethylating Reagent. Org. Lett. 2015, 17, 38–41. 10.1021/ol503189j. [DOI] [PubMed] [Google Scholar]
- Beatty J.; Douglas J.; Cole K.; Stephenson C. A scalable and operationally simple radical trifluoromethylation. Nat. Commun. 2015, 6, 7919–7924. 10.1038/ncomms8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J.; Frutos R.; Patel N.; Qu B.; Sun X.; Tampone T.; Gao J.; Sarvestani M.; Eriksson M.; Haddad N.; Shen S.; Song J.; Senanayake C. Development of a Safe and Economical Synthesis of Methyl 6-Chloro-5-(trifluoromethyl)nicotinate: Trifluoromethylation on Kilogram Scale. Org. Process Res. Dev. 2013, 17, 940–945. 10.1021/op400061w. [DOI] [Google Scholar]
- Rehm T. Photochemical Fluorination Reactions-A Promising Research Field for Continuous-Flow Synthesis. Chem. Eng. Technol. 2016, 39, 66–80. 10.1002/ceat.201500195. [DOI] [Google Scholar]
- DeBaillie A.; Jones C.; Magnus N.; Mateos C.; Torrado A.; Wepsiec J.; Tokala R.; Raje P. Synthesis of an ORL-1 Receptor Antagonist via a Radical Bromination and Deoxyfluorination to Afford a gem-Difluorospirocycle. Org. Process Res. Dev. 2015, 19, 1568–1575. 10.1021/op5000094. [DOI] [Google Scholar]
- Chen Z.; Zhang X.; Tu Y. Radical aryl migration reactions and synthetic applications. Chem. Soc. Rev. 2015, 44, 5220–5245. 10.1039/C4CS00467A. [DOI] [PubMed] [Google Scholar]
- Tada M.; Shijima H.; Nakamura M. Smiles-type Free Radical Rearrangement of Aromatic Sulfonates and Sulfonamides: Syntheses of Arylethanols and Arylethylamines. Org. Biomol. Chem. 2003, 1, 2499–2505. 10.1039/b303728b. [DOI] [PubMed] [Google Scholar]
- Douglas J.; Albright H.; Sevrin M.; Cole K.; Stephenson C. A Visible Light-Mediated Radical Smiles Rearrangement and its Application to the Synthesis of a Difluoro-Substituted Spirocyclic ORL-1 Antagonist. Angew. Chem., Int. Ed. 2015, 54, 14898–14902. 10.1002/anie.201507369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J.; Sevrin M.; Cole K.; Stephenson C. Preparative Scale Demonstration and Mechanistic Investigation of a Visible Light-Mediated Radical Smiles Rearrangement. Org. Process Res. Dev. 2016, 20, 1148–1155. 10.1021/acs.oprd.6b00126. [DOI] [Google Scholar]
- Vennestrøm P.; Osmundsen C.; Christensen C.; Taarning E. Beyond Petrochemicals: The Renewable Chemicals Industry. Angew. Chem., Int. Ed. 2011, 50, 10502–10509. 10.1002/anie.201102117. [DOI] [PubMed] [Google Scholar]
- Zakzeski J.; Bruijnincx P.; Jongerius A.; Weckhuysen B. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Lancefield C.; Ojo O.; Tran F.; Westwood N. Isolation of Functionalized Phenolic Monomers through Selective Oxidation and C–O Bond Cleavage of the β-O-4 Linkages in Lignin. Angew. Chem., Int. Ed. 2015, 54, 258–262. 10.1002/anie.201409408. [DOI] [PubMed] [Google Scholar]; b Rahimi A.; Ulbrich A.; Coon J.; Stahl S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252. 10.1038/nature13867. [DOI] [PubMed] [Google Scholar]; c vom Stein T.; den Hartog T.; Buendia J.; Stoychev S.; Mottweiler J.; Bolm C.; Klankermayer J.; Leitner W. Ruthenium-Catalyzed C–C Bond Cleavage in Lignin Model Substrates. Angew. Chem., Int. Ed. 2015, 54, 5859–5863. 10.1002/anie.201410620. [DOI] [PubMed] [Google Scholar]
- Kärkäs M.; Matsuura B.; Monos T.; Magallanes G.; Stephenson C. Transition-metal catalyzed valorization of lignin: the key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. 10.1039/C5OB02212F. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chmely S.; Nimlos M.; Bomble Y.; Foust T.; Paton R.; Beckham G. Computational Study of Bond Dissociation Enthalpies for a Large Range of Native and Modified Lignins. J. Phys. Chem. Lett. 2011, 2, 2846–2852. 10.1021/jz201182w. [DOI] [Google Scholar]
- Nguyen J.; Matsuura B.; Stephenson C. A Photochemical Strategy for Lignin Degradation at Room Temperature. J. Am. Chem. Soc. 2014, 136, 1218–1221. 10.1021/ja4113462. [DOI] [PubMed] [Google Scholar]
- Monos T.; Magallanes G.; Sebren L.; Stephenson C. Visible Light Mediated Reductions of Ethers, Amines and Sulfides. J. Photochem. Photobiol., A 2016, 328, 240–248. 10.1016/j.jphotochem.2016.05.014. [DOI] [Google Scholar]
- Selected implementations of flow-based photochemical methods toward clinically relevant compounds:; a Levesque F.; Seeberger P. Continuous-Flow Synthesis of the Anti-Malaria Drug Artemisinin. Angew. Chem., Int. Ed. 2012, 51, 1706–1709. 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]; b Yayla H.; Peng F.; Mangion I.; McLaughlin M.; Campeau L.; Davies I.; DiRocco D.; Knowles R. Discovery and mechanistic study of a photocatalytic indoline dehydrogenation for the synthesis of elbasvir. Chem. Sci. 2016, 7, 2066–2073. 10.1039/C5SC03350K. [DOI] [PMC free article] [PubMed] [Google Scholar]