

Abstract
A multitude of micro- and nanoparticles have been developed to improve the delivery of systemically administered pharmaceuticals, which are subject to a number of biological barriers that limit their optimal biodistribution. Bioinspired drug-delivery carriers formulated by bottom-up or top-down strategies have emerged as an alternative approach to evade the mononuclear phagocytic system and facilitate the transport across the endothelial vessel wall. Here, we describe a method that leverages the advantages of bottom-up and top-down strategies to incorporate proteins derived from the leukocyte plasma membrane into lipid nanoparticles. The resulting proteolipid vesicles - which we refer to as leukosomes - retained the versatility and physicochemical properties typical of liposomal formulations, preferentially targeted inflamed vasculature, enabled the selective and effective delivery of dexamethasone to inflamed tissues, and reduced phlogosis in a localized model of inflammation.
A primary directive of nanotechnology is to develop drug delivery platforms that effectively reduce systemic toxicity of currently used drugs while retaining their pharmacological activity. To this purpose, organic (lipids1, polymers2) and inorganic (silicon3, silica4, gold5 and iron oxide6) materials have been manipulated at the micro- and nano-scale to synthesize drug carriers. In the development of such materials, bioinspired approaches have emerged as a way to counteract the extraordinary ability of our body to recognize, label, sequester, and clear foreign objects7. By combining properties of conventional synthetic nanoparticles with natural8–10 or biomimetic11, 12 materials, new solutions in the field of drug delivery were offered13. The development of bottom-up approaches allowed for the synthesis of bio-inspired delivery systems through surface functionalization using ligands and molecules able to bind the receptors of specific target cells. A pioneer in this field are polymersomes14, which reconstitute two important leukocyte-derived adhesion molecules (analogs of PSGL-1 and LFA-1 receptors) on their surface. Due to these two receptors, polymersomes selectively targeted the inflamed endothelium both in vitro and in vivo, suggesting their synergistic effect in replicating leukocyte targeting properties14. Similarly, platelet-like nanoparticles displayed surface-binding, site-selective adhesion and aggregation properties effectively mimicking platelets and their hemostatic functions15. Although bottom-up approaches provide superior physicochemical control over the final formulation, current chemical conjugation methods remain inadequate to reproduce the complexity of the cellular membrane on the surface of nanocarriers7. The simultaneous addition of multiple moieties on the surface of nanocarriers requires complex synthetic routes and purification protocols that complicate the synthetic process.
Top-down procedures were proposed to bridge the gap between synthetic nanoparticles and biological materials. In this scenario, the synthesis of cell ghosts derived from red blood cells8, platelets9, and stem cells16 facilitated the recapitulation of the biological complexity of the original cells on the carrier’s surface. Biomimetic coatings offer a one-step solution to bestow particles with multiple bioactive functions including evasion of the mononuclear phagocytic system (MPS) and negotiation across various biological barriers10, 17, 18. However, top-down approaches have their own limitations, such as issues in the control of physical parameters (i.e. size and homogeneity of the final formulation), poor control of the encapsulation, and retention of chemically different molecules (i.e. loading of hydrophilic, amphiphilic, and lipophilic small drugs), as well as difficulties in establishing a standardized protocol for their preparation and storage19, 20. Herein, we debut a biomimetic vesicle, the leukosome, constituted by proteins derived from the leukocytes’ plasmalemma integrated into a synthetic phospholipid bilayer, through a combined bottom-up and top-down approach.
Leukocyte membrane proteins’ incorporation in lipid vesicles
Membrane proteins contained in the leukosome’s surface were extracted from both primary and immortalized immune cells (Supplementary Fig. 1a,b). Once isolated, they were stable for one month when lyophilized and preserved at −20°C or −80°C (Supplementary Fig. 1c). We assembled a mixture of cholesterol and synthetic choline-based phospholipids (see Methods) that mimicked the physiologic composition of the plasmalemma21 and enriched it with purified proteins using the thin layer evaporation (TLE) method (Fig. 1a–c). Unilamellar vesicles were obtained by extrusion through cellulose acetate membranes (200 nm pore size), while unincorporated material was eliminated through dialysis. To hydrate the lipid film, three different weight-to-weight ratios of membrane proteins-to-synthetic phospholipids were evaluated (1:100, 1:300 and 1:600). Differential scanning calorimetry (DSC) was used to investigate the bilayer transition temperature (Tm), which provided insight into the thermodynamic changes of the leukosome bilayer following the incorporation of membrane proteins22. Compared to control liposomes (Tm = 36.57 °C), the 1:300 ratio (Tm = 40.76 °C) resulted in the highest incorporation of leukocyte membrane proteins, followed by the 1:600 (Tm = 39.96°C) and 1:100 ratios (Tm = 36.85°C) (Fig. 1d). The data suggested that the degree of protein integration within the lipid bilayer correlated to the increase of Tm, possibly due to a packing effect of the leukosome bilayer. However, at the higher ratio (1:100), we measured a Tm similar to control liposomes, suggesting the existence of a threshold above which leukosome bilayers could not be further enriched with proteins. As inferred by the Tm of 55°C in the thermogram (Fig. 1d), this phenomenon was likely due to the formation of protein aggregates as a result of the heating and vortexing steps during the TLE procedure. This was further confirmed by the extrusion assay23. The assay was based on the extrusion of lipid formulations through 50 nm pore membranes with a slight decrease in the vesicles’ diameter indicating a higher deformability of the bilayer24 (Fig. 1e). Here, the decrease in the leukosomes’ diameter correlated with the increase of protein content into the lipid bilayer (from 1:100 and 1:600 up to 1:300 protein-to-lipid ratio, Fig. 1f). Taken together, these results indicated that the 1:300 ratio provided the greatest compromise between stability, protein content, and membrane fluidity and was chosen for the assembly of leukosomes in all subsequent studies.
Figure 1. Leukosome synthesis and formulation.

a, Extraction of proteolipid material from murine J774 macrophages. b, Protein enrichment of the phospholipid film. c, Vesicular formulation of Leukosomes. d, DSC analysis* of leukosomes and liposomes revealed a change in bilayer transition temperature (Tm) after membrane proteins incorporation. e, Deformability index evaluation* demonstrated leukosome bilayer’s packing as a function of the protein-to-lipid ratio from 1:600 to 1:300. No change in vesicle deformability was noted at 1:100 protein-to-lipid ratio. f, Schematic of membrane proteins’ incorporation and of vesicle deformability dynamics. D1: vesicle diameter before extrusion; D2,3,4: vesicle diameter after extrusion (D1>D2>D3>D4). *All values are the average of at least 7 different measurements ± s.d. **p<0.01.
Physicochemical characterization of leukosomes
After extrusion and dialysis, dynamic light scattering (DLS), zeta potential, and cryo-TEM analyses were used to evaluate the size, homogeneity, surface charge, shape, and structure of leukosomes (Fig. 2). The biomimetic formulation of the proteolipid material produced leukosomes with homogeneous size (~120 nm, with >90% unilamellar vesicles), as demonstrated by cryo-TEM and DLS polydispersity index (PDI, Fig. 2a,b). Compared to liposomes, the decreased negativity of leukosomes (−19.4 mV vs. −13.8 mV, respectively) was attributed to the shielding effect of the membrane proteins toward the negative charge of the phosphate groups. High magnification cryo-TEM revealed a 1.3 fold-increase in bilayer thickness compared to control liposomes (Fig. 2c–e, Supplementary Fig. 2). Topographical analysis by atomic force microscopy (AFM) confirmed the increased surface roughness of leukosomes suggesting the presence of hinged structures in their bilayer25 (Fig. 2f–h). Viscoelastic properties of both liposomes and leukosomes were also investigated by calculating the increased Young’s modulus (Supplementary Fig. 3), as a measure of the higher stiffness of the material26. The elastic modulus for leukosomes demonstrated a slight increase (p<0.05) in stiffness when compared to liposomes (476 kPa vs. 423 kPa). Next, we identified the vibrational modes and chemical signatures of leukocyte membranes (black line), leukosomes (red) and liposomes (green) through Fourier transform infrared (FTIR) spectroscopy (Fig. 2i). Three protein absorption bands were present: the amide I band (1700–1600 cm−1) due to the C=O stretching vibrations, amide II (1580–1510 cm−1) associated with the N–H bending with a contribution of the C–N stretching vibrations, and amide III (1400–1200 cm−1) due to the N–H bending and stretching vibrations from C–Cα and C–N. In addition, the 1200–900 cm−1 region showed the absorption of protein-associated sugar chains indicating the incorporation of glycosylated proteins in the membranes27. To confirm the glycosylation of the proteolipid material we stained leukosomes with wheat germ agglutinin (WGA), a lectin that selectively binds N-acetyl-D-glucosamine and glycosylated sialic acid residues. Spectrofluorometric analysis verified the presence of glycosylated proteins on the surface of the leukosome showing the integration, correct orientation, and stabilization of membrane proteins in their post-transcriptionally modified state7 (Fig. 2j).
Figure 2. Characterization of leukosomes physicochemical features.

DLS and cryo-TEM analysis of a, leukosomes and b, liposomes showed size, zeta potential, polydispersity index values, and size homogeneity of the two formulations (Scale bar 100 nm). High magnification cryo-TEM micrographs of liposomes (c) and leukosomes (d) reveal a spherical shape for both vesicles, and a thicker bilayer for leukosomes (Scale bar 50 nm). e, Quantification of lipid bilayer showed a 1.3-fold increase of membrane thickness for leukosomes respect to liposomes. Atomic force microscopy images of f, liposomes and g, leukosomes reveal the presence of hinged structures on leukosome surface. h, Quantification of single particles’ surface roughness (Ra) showed a 4-fold increase in leukosomes. Error bars in e,h represent the s.d. of 60 random particles in 3 replicates. i, ATR/FTIR spectrum of J774 membrane (black), liposomes (green), and leukosomes (red), confirmed the presence of protein components of the J774 membrane in the leukosomes’ bilayer - broad band around ≈1620 cm−1 (highlighted by the dotted line). j, Wheat Germ Agglutinin assay showed the presence of glycosylated proteins on the leukosome surface. Liposomes and membranes were used as negative and positive control, respectively. *p<0.05; **p<0.01; ***p<0.001. Error bars represent the s.d. of 3 replicates.
Characterization of leukosome protein composition
The proteomic profiling of the leukosome resulted in the identification of 342 distinct proteins (Supplementary Table 1). Two thirds were small proteins (10–50 KDa, Supplementary Fig. 4a) and more than half were identified with high confidence scores (300–2000) with a sequence coverage ranging from 10% to 30% (Supplementary Fig. 4b). This allowed the classification of leukosome proteins as: integral or lipid-anchored (38%), cytoskeletal and/or junctional (30%), peripheral (21%), and vesicular or secreted proteins (11%) (Fig. 3a,b). The presence of proteins from other cellular compartments (primarily ribosomes and mitochondria) was attributed to the dynamic trafficking of proteins between internal organelles and the cell surface28,29 and was in line with previous reports30–33. A functional classification identified proteins involved in transport (48%), signaling (16%), immunity (12%), cell adhesion (9%), lipid metabolism (5%), and structure (4%) (Fig. 3c,d), the majority of which were associated to the leukocyte plasma membrane (Fig. 3e). The presence of critical leukocyte surface proteins, such as those involved in leukocyte adhesion to inflamed endothelium (e.g., LFA-1, Mac-1) and in self-tolerance (e.g., leukocyte common antigen (CD45)), was assessed on the surface of donor cells through fluorescence microscopy and flow cytometry (Supplementary Fig. 5a,b). PSGL-1, involved in leukocyte firm adhesion on substrates expressing P-selectin (i.e. platelets and endothelial cells34), and the marker-of-self CD4735 were also identified on the leukosome’s surface. Immunolabeling with antibodies directed against the extracellular domain of these proteins confirmed their presence and correct orientation within the leukosome’s bilayer (Fig. 3f). In addition, standards developed with fluorescently labeled antibodies for proteins of interest were prepared and incubated separately with liposomes and leukosomes. Theoretical calculations based on established criteria36 revealed a surface density of approximately 206, 149, 85, 144, 109, and 187 copies per μm2 of LFA-1, Mac-1, PSGL-1, CD18, CD45, and CD47, respectively (Supplementary Table 2). Taken together, these results confirmed the successful transfer of leukocyte membrane-based markers onto the surface of leukosomes in an amount sufficient to exert their activity14, 36.
Figure 3. Analysis of the leukocyte membrane proteins transferred to the leukosome’s lipid bilayer.
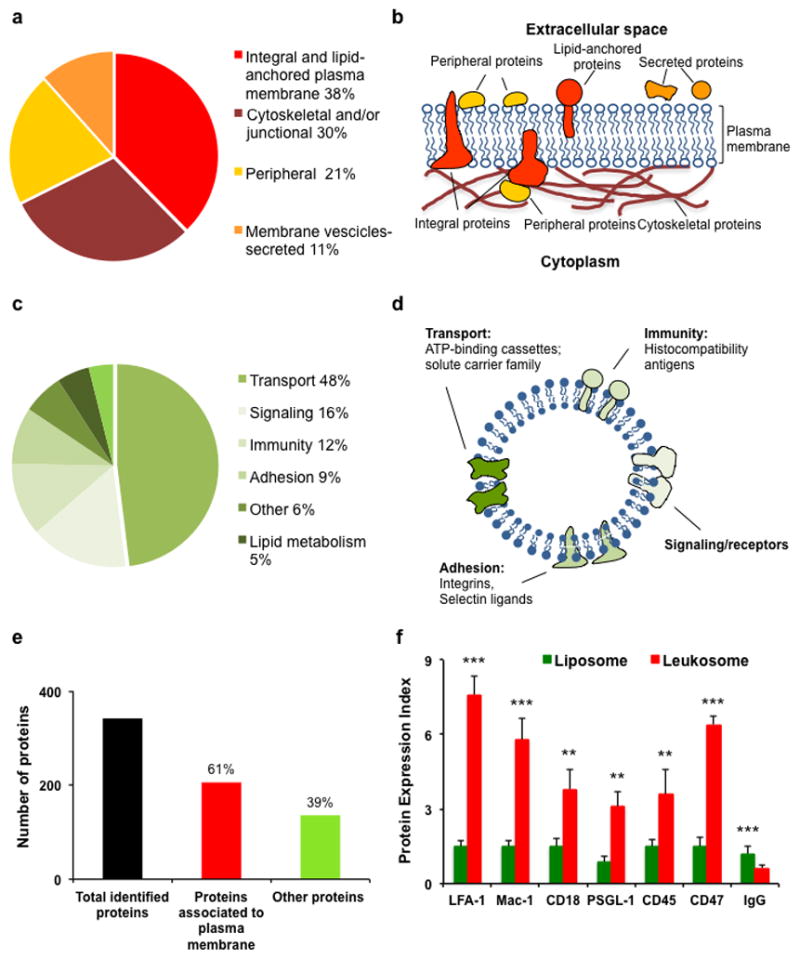
a, Pie chart of the proteins identified in the Leukosome classified according to UniProt/GO information and manually searching in literature; classification of sub-classes of plasma membrane-associated proteins. b, Schematic representation of the types of membrane and membrane-associated proteins identified. Integral proteins penetrate the membrane, while the peripheral ones are attached to one side. Cytoskeletal proteins are connected to plasma membranes thanks to the action of structural proteins that serve as anchors. Lipid anchored proteins are covalently bonded through a fatty acid to the plasma membrane. Secreted proteins are cycled between the outside and the inside of the cell through vesicles-mediated secretory pathways. c, Functional characterization of the integral and lipid-anchored plasma membrane proteins identified in the Leukosome; d, Schematic representation of leukosome bilayer enriched with molecules involved in transport, signaling, immunity, and adhesion; e, Number of total and plasma membrane-associated proteins identified in the leukosome formulation. The samples were analyzed in triplicate. f, Flow cytometry analysis validates LFA-1, Mac-1, CD18, PSGL-1, CD45, and CD47 presence and their correct orientation on the surface of leukosomes’ surface. The incubation of fluorescently labeled IgG with both liposomes and leukosomes revealed the absence of any unspecific binding of the antibody with vesicles’ surface, thus indicating the high selectivity of the assay. **P<0.01; ***p<0.001. Error bars represent the s.d. of 3 replicates.
Pharmaceutical features of leukosomes
From a pharmaceutical standpoint, leukosomes displayed similar stability properties as liposomes following storage at 4°C (Supplementary Fig. 6). DLS analysis revealed that empty leukosomes were stable for two weeks with no significant change in vesicle size (Supplementary Fig. 6a). After three weeks, a slight increase (<20%) in leukosome diameter was observed but yielded no significant increase in the PDI (Supplementary Fig. 6b). Leukosomes also retained loading and release properties similar to liposomes, as well as high versatility for the encapsulation of compounds with variable solubility37 (Fig. 4a). Dexamethasone (DXM), caffeine, and paclitaxel were chosen as model compounds to represent small molecules with hydrophilic, amphiphilic, and hydrophobic characteristics, respectively (Fig. 4b–d). Loading of the drugs into leukosomes did not affect their vesicular structure and shape (Supplementary Fig. 7a) and did not significantly affect vesicle size. DXM and caffeine encapsulation produced a minimal impact on surface charge with paclitaxel exhibited the most pronounced effect (Supplementary Fig. 7b). Specifically, paclitaxel increased the leukosomes’ surface charge to 15 mV. As previously shown for other hydrophobic drugs38, paclitaxel intercalates among the hydrophobic tails of the lipid bilayer which likely induced a structural rearrangement of the membrane, possibly through the exposure of the choline groups to the outer bilayer surface39. Additionally, a first-order kinetic release profile was observed for DXM and caffeine (Fig. 4b,c), while paclitaxel displayed zero-order kinetics (Fig. 4d). This slight delay in the release of model payloads from leukosomes (Fig. 4b–d) was attributed to the presence of the membrane proteins and the increased bilayer thickness.
Figure 4. Leukosomes retain drug loading and release properties similar to control liposomes.

a, Dexamethasone, caffeine and paclitaxel molecular formula and their water solubility are reported. They are representative of hydrophilic, amphiphilic, and hydrophobic drugs, respectively. b–d, Encapsulation efficiency and in vitro release profile of dexamethasone (b), caffeine (c), and paclitaxel (d)-loaded liposomes (GREEN) and leukosomes (RED). Leukosomes showed loading properties similar to conventional liposomes, while they delayed the release of their payload. Error bars represent the s.d. of 3 replicates.
Targeting and efficacy of leukosomes
Leukosomes displayed preferential targeting of inflamed endothelia, both in vitro and in vivo. For these studies we selected DXM, an anti-inflammatory glucocorticoid40, to be used as a proof-of-concept to demonstrate the therapeutic potential of leukosomes. DXM encapsulation did not affect the surface identity of leukosomes (Supplementary Fig. 8), indicating that the carrier’s surface properties were preserved after drug loading. A flow chamber assay was used to test the ability of liposomes and leukosomes to adhere to a reconstructed monolayer of activated human umbilical vein endothelial cells (HUVEC) under physiologically relevant shear stresses. Compared to conventional liposomes, leukosomes preferentially recognized the inflamed endothelium (Supplementary Fig. 9). To demonstrate successful treatment, we performed PCR analysis and observed that DXM-loaded leukosomes reduced the expression of pro-inflammatory markers (CCL2 and IL6) and endothelial adhesion molecules (ICAM-1 and VCAM-1) as well as increased levels of the anti-inflammatory gene MRC-1 (Supplementary Fig. 10).
To validate these results in an in vivo model of localized inflammation, we subcutaneously injected lipopolysaccharide (LPS) (10 μg) into the ears of mice. This treatment induced a confined inflammation, manifested by redness, edema, tissue thickening, and neutrophil infiltration, as confirmed by bioluminescence analysis (Supplementary Fig. 11). Being a unilateral inflammatory model, each mouse served as its own control41. Intravital microscopy (IVM) analysis displayed a significant increase in the accumulation of leukosomes in the inflamed ear (5-fold and 8.5-fold increase compared to control liposomes at 1h and 24h after injection, respectively (p<0.1, Fig. 5a).
Figure 5. Leukosomes preferentially adhere to inflamed vasculature in vivo and improve tissue healing by preserving its architecture and reducing neutrophil infiltration.

a, IVM images of inflamed-vasculature targeting relative to rhodamine-labeled liposomes and leukosomes. Compared to control liposomes, leukosomes showed a 5-fold and 8-fold increased accumulation into ear tissue at 1 and 24h after particles’ injection, respectively. Error bars represent the s.d. of measurements from at least 10 field of views taken from 3 mice. b, Histological analysis of not inflamed (control) and inflamed (free DXM, empty and DXM-loaded liposomes, empty and DXM-loaded leukosomes, and untreated) ear tissues shows an alteration of tissue architecture in the untreated group and in the ones treated with free DXM, empty and DXM-loaded liposomes. c, Inspection of ear cryo-sections revealed a significantly reduced thickness for the groups treated with leukosomes (empty and DXM-loaded) compared to the other groups. No statistically significant difference was observed among the control and the leukosomes-treated groups. d, Immunofluorescence analysis of ear sections at 24 hours reveals that both empty and DXM-loaded leukosomes exhibited a significant reduction in neutrophil infiltration in ear tissue compared to the other groups. e, Fluorescence intensity quantification of labeled neutrophils is reported. *p<0.1; **p<0.01; ***p<0.001. Scale bar = 50 μm. Error bars in c,e represent the s.d. obtained from at least 15 areas taken from 8 mice.
Inspection of IVM frames revealed an opposing behavior for liposomes and leukosomes at these two time points. Although leukosomes exhibited a 5 to 8-fold increase in accumulation (Supplementary Fig. 12a), liposomes were found more abundant into the extravascular space at 1h (Supplementary Fig. 12b), possibly as a result of the enhanced permeability and retention effect occurring at the vascular level following the LPS-induced inflammation42. On the other hand, leukosomes were found associated with the inflamed vasculature (Supplementary Fig. 12b), due to their bioinspired targeting properties. However, at 24h liposomes were in equilibrium between the two environments, while leukosomes continued to extravasate across the vascular barrier and were retained within the perivascular space (Supplementary Fig. 12c). These observations led us to believe that at early time points, the accumulation of leukosomes was mediated by the active adhesion to inflamed endothelium followed by the successful extravasation into the ear parenchyma at later time points. Conversely, liposomes passively distributed contingent on flow dynamics. To identify the key molecules and mechanisms responsible for leukosome targeting, we investigated, both in vitro and in vivo, particles’ adhesion to inflamed endothelium upon blocking two specific biomarkers. We selected LFA-1 and CD45, which have direct43 and indirect44 roles on leukocyte adhesion to the endothelium. A significant reduction of leukosome adhesion in vitro to inflamed endothelia was observed after LFA-1 (p<0.005, anti-LFA-1 leukosomes vs. leukosomes) and CD45 (p<0.001, anti-CD45 leukosomes vs. leukosomes) were blocked on the leukosome surface (Supplementary Fig. 13). Similarly, blocking LFA-1 or CD45 in vivo resulted in a significant decrease in the targeting of the inflamed ear (Supplementary Fig. 14; p<0.001). This result confirmed that LFA-1 was largely responsible for the vasculature adhesion of leukosomes and clarified the role of CD45 in leukocyte adhesion mediated by LFA-1 during an inflammatory response45, 44, 46.
The accumulation of leukosomes at the site of inflammation, as well as their biodistribution and pharmacokinetic profiles, were also assessed through spectrofluorometric analysis of homogenized explanted tissues (Supplementary Fig. 15). Compared to control liposomes, a significant reduction in the accumulation of leukosomes into MPS organs (2.6-fold decrease in spleen and 1.5-fold decrease in kidneys, liver, and lungs), as well as prolonged circulation (5-fold increase), was observed. In addition, leukosomes showed a 7-fold increase in accumulation into the ear compared to control liposomes (Supplementary Fig. 15), thus confirming IVM data.
We also evaluated leukosomes ability to reduce localized inflammation. The right ears of mice (n=8) were treated with PBS (as control), empty liposomes and leukosomes, DXM-loaded liposomes and leukosomes (5 mg/kg), and free DXM (5 mg/kg) after LPS injection. Macroscopic observations of the ear showed evident signs of improvement in the mice treated with empty and DXM loaded-leukosomes while the other groups continue to show signs of acute tissue inflammation (i.e., presence of prominent edema) (Supplementary Fig. 16). Similarly, H&E staining displayed normal ear tissue architecture for empty and DXM-loaded leukosomes groups while other treatments resulted in a substantial alteration of the ear’s architecture, increased neutrophil infiltration, and edematous transudate (Fig. 5b). A significant reduction in the thickening of the ear (Fig. 5c) and lower neutrophil infiltration (Fig. 5d,e) was also observed in the leukosome-treated groups. These findings further validated the H&E analysis and suggested empty nanoparticles may have an active role in the modulation of the inflammatory environment independent from their therapeutic payload.
Safety and immunogenicity of leukosomes
We next evaluated if the systemic administration of a high dose of leukosomes (1,000 mg/kg) would trigger an inflammatory response. Serum levels of cytokines (IL-6, TNFα, and IL-1β) were observed after 1 and 7 days with no significant difference between leukosomes and the control group (Fig. 6a). Furthermore, histological analysis on major organs demonstrated negligible long-term changes in tissue architecture (Fig. 6b). In addition, assessment of hepatic (aspartate aminotransferase [AST], alanine aminotransferase [ALT], and Alkaline phosphatase [ALP]) and renal (blood urea nitrogen [BUN]) function revealed minimal differences between treatment groups (Supplementary Fig. 17). Finally, flow cytometry profiles of IgM and IgG-positive liposomes and leukosomes, previously incubated with serum of untreated and treated mice, displayed no observable elevation of autologous antibody titers (Fig. 6c–e). In particular, compared to IgM-labeled particles, which reflect the amount of low affinity and less specific antibodies generated toward the nanoparticles (Fig. 6d), IgG-labeled particles resulted in a 10-fold decrease in abundance (Fig. 6e), indicating a minimal impact in the generation of high affinity antibodies following leukosome exposure. IgM and IgG labeled fewer than 3 and 0.3% of leukosomes in the host serum, similar to values observed with liposomes (Fig. 6d,e). These results indicate that leukosomes did not initiate a significant adaptive immune response or antibody production against leukosome membrane antigens. This suggests their safe use in immune-competent mice.
Figure 6. Immunogenicity and safety of leukosomes.

a, Serum levels of the main cytokines (IL-6, TNF-α, and IL-1β) in mice (n=5) treated with a high dosage of leukosomes (1000 mg/Kg). Blood samples were collected 1 and 7 days after leukosome i.v. administration. Error bars represent the s.d. of 5 replicates. b, Representative haematoxylin and eosin stained sections of indicated organs from mice 1 week after systemic injection of leukosomes, Liposomes, and PBS (CONTROL). c, Flow cytometry profiles of IgM and IgG-positive liposomes and leukosomes, previously incubated with serum of untreated (control) and treated mice. FACS analysis showed no significant elevation of autologous antibody titer compared with the control. Very limited leukosomes, fewer than 3 and 0.3% were labeled by host serum and secondary antibodies (anti-IgM and IgG, respectively) (d, e), and the same trend can be observed with control liposomes. This result suggests that leukosomes do not initiate any significant adaptive immune response and antibody production against membrane antigens related to the particles. Error bars represent the s.d. of 5 replicates.
Outlook
In the past decades, the development of bioinspired delivery systems was largely based on two strategies: bottom-up approaches, such as surface functionalization with antibodies that mimic original cell surface proteins14, 47; and top-down strategies, such as cell-derived nanovesicles and nano-ghosts8, 16. Compared to these methods, we developed a novel synthetic route to obtain biomimetic vesicles that possess: i) high surface complexity obtained through a facile one-step process that does not require chemical synthesis or complex purification steps, ii) the versatility in formulation and applications typical of liposomes, such as their capacity to load, retain, and release different payloads, iii) elevated and standardized yield of the manufacturing process, and iv) a stable, safe, and non-immunogenic final product. From a functional standpoint, leukosomes retained the physiological tropism of leukocytes toward inflamed vasculature and promoted the preferential accumulation into inflamed tissue, the reduction of neutrophils infiltration, and the prevention of tissue damage through the resolution of localized inflammation.
To the best of our knowledge, this approach represents the first time that a complex material such as the plasmalemma has been formulated into a lipid vesicle, using the established TLE method, to achieve the reproducible incorporation of membrane proteins into a lipid bilayer. We believe that in the future, by combining cell biology with nanotechnology, it will be possible to use the plasmalemma of virtually any cell type for the development of biomimetic particles. Furthermore, the combination of membrane proteins purified from different cellular types (leukocytes, red blood cells, platelets) will result in the creation of chimeric leukosomes that exploit the intrinsic properties of the original cells to exert more advanced drug delivery functions. Finally, the high versatility of this approach suggests that the leukosomes might be an effective delivery platform for the treatment of a broad range of disorders that have low therapeutic alternatives (e.g. rheumatoid arthritis, cancer, inflamed bowel diseases) but share the same inflammatory background.
Methods
Assembly and physical characterization of leukosomes
Phosphocholine-based phospholipids (DPPC, DSPC and DOPC) and cholesterol (Avanti Polar Lipids) were dissolved in a chloroform:methanol mixture (3:1 v/v) and the solvent was evaporated through a rotary evaporator (BÜCHI Labortechnik AG, Switzerland) to form a film according to the well-established TLE procedure. Films were hydrated with a PBS dispersion of membrane proteins (1:300 protein-to-lipid ratio) or PBS to respectively assemble leukosomes or liposomes (control). Lipid suspension was extruded ten times through 200 nm pore-size cellulose acetate membranes at 45°C. Physical characterization was performed with a Nanosizer ZS (Malvern Instruments). For Cryo-TEM analysis, liposomes and leukosomes were plunge-frozen on holey film grids (R2x2 Quantifoil®; Micro Tools GmbH, Jena, Germany) as previously reported48. Images were acquired on a JEOL 2100 electron microscope under low electron-dose conditions (<5 – 20 electrons/Å2) using a 4,096 × 4,096 pxl CCD camera (UltraScan 895, GATAN, Inc., nominal magnifications 20,000x). The proteomic profile was obtained via peptide-level LC/MSE analysis by in-solution trypsin digestion after reduction and alkylation of disulfide bridges and de-lipidation with methanol/chloroform extraction. Bradford (Bio-Rad) protein assay was employed to determine protein concentration, followed by trypsin digestion (overnight at 37°C with an enzyme:substrate = 1:50 molar ratio). AFM images of the liposomes and leukosomes were collected in Scan Asyst ®mode by Multimode (Bruker, CA, USA) using single-beam silicon cantilever probes (Bruker MLCT: resonance frequency 10 KHz, nominal tip radius of curvature 10 nm, force constant of 0.04N/m). Fourier Transform Infrared spectroscopy measurements in attenuated total reflection were performed using a single reflection diamond element. For the study, the FTIR spectrometer Nicolet was equipped with a nitrogen cooled mercury cadmium telluride detector. Leukosomes with different protein/lipid ratio (1:100, 1:300, 1:600) were prepared for DSC measurements using a Mettler-Toledo Star DSC (Mettler-Toledo, Switzerland), to evaluate the bilayer thermal transitions at increasing protein contents. Passive loading of leukosomes was obtained by hydrating the lipid film with a caffeine (1:10 caffeine to lipid ratio), or a dexamethasone (1:5 dexamethasone to lipid ration) solution, or by dissolving paclitaxel (1:30 paclitaxel to lipid ratio) in the chloroform:methanol mixture containing the lipids. See supplementary information for further details.
In vivo confocal imaging
All animal experiments were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals approved by The Houston Methodist Institutional Animal Care and Use Committee guidelines in accordance to protocols AUP-0714-0043 and AUP 0611-0032. IVM imaging was performed under anesthesia with isoflurane. Rhodamine-labeled particles (liposomes and leukosomes) were injected intravenously via retro orbital injection. 70 kDa fluorescein isothiocyanate (FITC)-labeled dextran dye (5 mg/ml; 50 μL in PBS) (Invitrogen, Carlsbad, CA) was used as vessel tracer as previously reported10. IVM studies of leukosomes’ dynamics were performed to determine effectiveness of tissue targeting and accumulation. Upon systemic administration, we monitored the dynamic flow and the real-time accumulation of liposomes and leukosomes for up to 60 min post-injection. Adhesion to the inflamed vasculature was monitored using an upright Nikon A1R laser scanning confocal microscope, equipped with resonance scanner, motorized and heated stage, and a Nikon long working distance 4x and 20x dry plan-apochromatic objectives. Images were obtained with a three-channel setup in which fluorescence was collected at 488/525 nm for FITC dextran, and at 561/579 nm for rhodamine-labeled particles. Image acquisition was performed over n=10 field of views (FOVs) at a resolution of 512 × 256 pixels with an optical slice thickness of 5 μm. To determine the extent of leukosomes and liposomes accumulation in the ear parenchyma, the animals were imaged 1 h, and 24 h after the i.v. injection (50 μL, 1 mg/mL). The images were analyzed using Nikon Elements.See Supplementary Information for further details.
Histology of ear tissue
Explanted mice ears were washed twice with PBS and embedded in a cryomold in O.C.T. (Tissue-Tek® O.C.T. Compound, Sakura® Finetek), and instantly frozen at −80 degree. Ten μm-thick slides were obtained cutting ears block with a cryostat at −20 degree. The slides were stored at −20 degree. For H/E staining, slides were thawed, hydrated, washed and stained with ematoxylin and eosin (Sigma-Aldrich®).
Immunofluorescence analysis of ear tissue
Once cryo-sections were obtained as described in the previous paragraph, immunofluorescence (IF) staining was performed as previously reported49. Briefly, slides were thawed and blocked with BSA 5% (Sigma-Aldrich®) PBS 1x solution. After washing, they were incubated overnight at 4 degrees with anti-neutrophil antibody (Alexa Fluor 647 anti-mouse Ly-6G/Ly-6C (Gr-1) Biolegend®). Excess of the anti-neutrophil antibody was washed out with PBS 1X. Cells nuclei were stained with DAPI. Slides were sealed with ProLong Gold antifade reagent (Life technologies™). Images were captured with a Nikon® Eclipse Ti Inverted Fluorescent Microscope equipped with Hamamatsu Digital Camera C11440 ORCA-Flash 2.8.
Immunogenicity and safety of leukosomes
8-week-old BALB/C mice (n=5) were intravenously injected with leukosomes and control liposomes once per week for one month. Then, the blood was collected from the mice at 6 weeks after the last injection and the serum isolated, as also reported by Copp and coworkers50. The sera were used as primary antibody and were incubated with the particles, which were first blocked with BSA and FBS at room temperature for 30 minutes. After one wash, anti-mouse IgM and IgG secondary antibodies labeled with different fluorochromes were incubated with particles at room temperature for 30 minutes. After washing, the particles were analyzed by FACS analysis. IgM or IgG-positive particles indicate that specific antibodies were generated in the host blood against them.
Supplementary Material
Acknowledgments
The authors would like to gratefully aknowledge Dr. Mauro Ferrari for the valuable and stimulating discussions about the study. The authors would like to thank Dr. Junping You for his help in the animal procedures. The authors acknowledge support from the National Institute of Health (1R21CA173579-01A1 and 5U54CA143837 PSOC Pilot project), the Department of Defense (W81XWH-12-10414 BCRP Innovator Expansion), George J. and Angelina P. Kostas Charitable Foundation, Brown Foundation Inc., William Randolph Hearst Foundation, and The Regenerative Medicine Program Cullen Trust for Health Care to E.T.; R.M was supported by grant RF-2010-2305526; C.C. and A.P. were supported by grant RF-2010-2318372 from Italian Ministry of Health. We thank Associazione Bianca Garavaglia, Via C. Cattaneo, 8, 21052 Busto Arsizio Varese, Italy and Project CREME “Campania Research in Experimental Medicine” POR Campania FSE 2007/2013. We ackowledge Dr. D.A. Engler and the Proteomics Core, Dr. D. Haviland and the Flow Cytometry Core, Dr. A.L. Rivera and the Research Pathology Core at HMRI. We thank M.G. Landry and M. Evangelopoulos for graphical assistance with the creation of the schematics. The authors also acknowledge the Sealy Center for Structural Biology and Molecular Biophysics at the University of Texas Medical Branch at Galveston for providing research resources.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Author contribution
E.T. conceived the Leukosome platform, wrote the paper and was the principal investigator of the major supporting grants. E.T. and R.M. designed the research project and defined the goals of the present study. R.M. developed and optimized the protocols for leukosome assembly, supervised all the experiments, and evaluated the therapeutic efficacy with contributions from J.O.M. and E.D.R.; C.C performed all the proteomic experiments and the interpretation of the data on protein enrichment; J.O.M. and E.D.R. performed the intravital microscopy experiments and analysis; M.E. performed flow cytometry and optimized the in vitro flow systems. J.O.M. carried out BLI analysis and revised the manuscript; F.T. performed FTIR and AFM analyses; S.M. performed DSC analysis; F.T., S.M., and A.D.V. performed H&E and immunofluorescence staining and optical and confocal laser microscopy imaging; M.B.S performed Cryo-TEM and assisted with analysis; I.K.Y. performed cytokine and organ functionality analyses; P.Z. performed the immunological analysis of leukosomes and gave his expert advice about the the study of the immunogenic response; N.E.T.F. performed dexamethasone loading and release experiments; X.W. performed the PCR analysis; A.P. assisted with the editing of the manuscript and mentored the authors during the development of the project.
References
- 1.Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13:813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nature Reviews Drug Discovery. 2014 doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasciotti E, et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nature nanotechnology. 2008;3:151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 4.Parodi A, et al. Bromelain Surface Modification Increases the Diffusion of Silica Nanoparticles in the Tumor Extracellular Matrix. ACS nano. 2014 doi: 10.1021/nn502807n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 6.Kudgus RA, et al. Tuning Pharmacokinetics and Biodistribution of a Targeted Drug Delivery System Through Incorporation of a Passive Targeting Component. Sci Rep. 2014;4 doi: 10.1038/srep05669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. Journal of Controlled Release. 2015 doi: 10.1016/j.jconrel.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu CMJ, et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu C-MJ, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015 doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parodi A, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nature nanotechnology. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammer DA, et al. Leuko-polymersomes. Faraday discussions. 2008;139:129–141. doi: 10.1039/b717821b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doshi N, et al. Platelet mimetic particles for targeting thrombi in flowing blood. Advanced Materials. 2012;24:3864–3869. doi: 10.1002/adma.201200607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature biotechnology. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbins GP, et al. Tunable leuko-polymersomes that adhere specifically to inflammatory markers. Langmuir. 2010;26:14089–14096. doi: 10.1021/la1017032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anselmo AC, et al. Platelet-like nanoparticles: mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS nano. 2014;8:11243–11253. doi: 10.1021/nn503732m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toledano Furman NE, et al. Reconstructed stem cell nanoghosts: a natural tumor targeting platform. Nano letters. 2013;13:3248–3255. doi: 10.1021/nl401376w. [DOI] [PubMed] [Google Scholar]
- 17.Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nature Reviews Drug Discovery. 2011;10:521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 18.Alvarez-Lorenzo C, Concheiro A. Bioinspired drug delivery systems. Current opinion in biotechnology. 2013;24:1167–1173. doi: 10.1016/j.copbio.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Gutiérrez Millán C, Colino Gandarillas CI, Sayalero Marinero ML, Lanao JM. Cell-based drug-delivery platforms. Therapeutic delivery. 2012;3:25–41. doi: 10.4155/tde.11.141. [DOI] [PubMed] [Google Scholar]
- 20.Millan CG, Marinero MaLS, Castaneda AZ, Lanao JM. Drug, enzyme and peptide delivery using erythrocytes as carriers. Journal of controlled release. 2004;95:27–49. doi: 10.1016/j.jconrel.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Bretscher MS. Asymmetrical lipid bilayer structure for biological membranes. Nature. 1972;236:11–12. doi: 10.1038/newbio236011a0. [DOI] [PubMed] [Google Scholar]
- 22.Demetzos C. Differential scanning calorimetry (DSC): a tool to study the thermal behavior of lipid bilayers and liposomal stability. Journal of liposome research. 2008;18:159–173. doi: 10.1080/08982100802310261. [DOI] [PubMed] [Google Scholar]
- 23.Manconi M, et al. Ex vivo skin delivery of diclofenac by transcutol containing liposomes and suggested mechanism of vesicle–skin interaction. European Journal of Pharmaceutics and Biopharmaceutics. 2011;78:27–35. doi: 10.1016/j.ejpb.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Mura S, Manconi M, Sinico C, Valenti D, Fadda AM. Penetration enhancer-containing vesicles (PEVs) as carriers for cutaneous delivery of minoxidil. International Journal of Pharmaceutics. 2009;380:72–79. doi: 10.1016/j.ijpharm.2009.06.040. [DOI] [PubMed] [Google Scholar]
- 25.Chow TS. Nanoscale surface roughness and particle adhesion on structured substrates. Nanotechnology. 2007;18:115713. [Google Scholar]
- 26.Schaap IA, Eghiaian F, des Georges A, Veigel C. Effect of envelope proteins on the mechanical properties of influenza virus. Journal of Biological Chemistry. 2012;287:41078–41088. doi: 10.1074/jbc.M112.412726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mereghetti P, et al. A Fourier transform infrared spectroscopy study of cell membrane domain modifications induced by docosahexaenoic acid. Biochimica et Biophysica Acta (BBA) - General Subjects. 2014;1840:3115–3122. doi: 10.1016/j.bbagen.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Lodish H, et al. Molecular cell biology. 2000 [Google Scholar]
- 29.Benmerah A, Scott M, Poupon V, Marullo S. Nuclear Functions for Plasma Membrane-Associated Proteins? Traffic. 2003;4:503–511. doi: 10.1034/j.1600-0854.2003.00102.x. [DOI] [PubMed] [Google Scholar]
- 30.Durr E, et al. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nature biotechnology. 2004;22:985–992. doi: 10.1038/nbt993. [DOI] [PubMed] [Google Scholar]
- 31.Lund R, Leth-Larsen R, Jensen ON, Ditzel HJ. Efficient isolation and quantitative proteomic analysis of cancer cell plasma membrane proteins for identification of metastasis-associated cell surface markers. Journal of proteome research. 2009;8:3078–3090. doi: 10.1021/pr801091k. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Zhang M, Go VLW, Hu S. Membrane proteomic analysis of pancreatic cancer cells. Journal of biomedical science. 2010;17:74. doi: 10.1186/1423-0127-17-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corbo C, et al. Proteomic profiling of a biomimetic drug delivery platform. Current drug targets. 2015;16:1540–1547. doi: 10.2174/1389450115666141109211413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118:6743–6751. doi: 10.1182/blood-2011-07-343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soto Pantoja DR, Kaur S, Miller TW, Isenberg JS, Roberts DD. Leukocyte surface antigen CD47. UCSD Molecule Pages. 2013;2 [Google Scholar]
- 36.Hu CMJ, et al. ‘Marker-of-self’ functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale. 2013;5:2664–2668. doi: 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen T. Liposomal Drug Formulations. Drugs. 1998;56:747–756. doi: 10.2165/00003495-199856050-00001. [DOI] [PubMed] [Google Scholar]
- 38.Cosco D, Paolino D, Cilurzo F, Casale F, Fresta M. Gemcitabine and tamoxifen-loaded liposomes as multidrug carriers for the treatment of breast cancer diseases. International Journal of Pharmaceutics. 2012;422:229–237. doi: 10.1016/j.ijpharm.2011.10.056. [DOI] [PubMed] [Google Scholar]
- 39.Bernsdorff C, Reszka R, Winter R. Interaction of the anticancer agent Taxol™ (paclitaxel) with phospholipid bilayers. Journal of Biomedical Materials Research. 1999;46:141–149. doi: 10.1002/(sici)1097-4636(199908)46:2<141::aid-jbm2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 40.Franchimont D, Kino T, Galon J, Meduri GU, Chrousos G. Glucocorticoids and inflammation revisited: the state of the art. Neuroimmunomodulation. 2002;10:247–260. doi: 10.1159/000069969. [DOI] [PubMed] [Google Scholar]
- 41.Gross S, et al. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat Med. 2009;15:455–461. doi: 10.1038/nm.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azzopardi EA, Ferguson EL, Thomas DW. The enhanced permeability retention effect: a new paradigm for drug targeting in infection. Journal of Antimicrobial Chemotherapy. 2013;68:257–274. doi: 10.1093/jac/dks379. [DOI] [PubMed] [Google Scholar]
- 43.Ishibashi M, et al. Integrin LFA-1 regulates cell adhesion via transient clutch formation. Biochemical and biophysical research communications. 2015;464:459–466. doi: 10.1016/j.bbrc.2015.06.155. [DOI] [PubMed] [Google Scholar]
- 44.Arroyo AG, et al. Induction of tyrosine phosphorylation during ICAM-3 and LFA-1-mediated intercellular adhesion, and its regulation by the CD45 tyrosine phosphatase. The Journal of cell biology. 1994;126:1277–1286. doi: 10.1083/jcb.126.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sigal A, et al. The LFA-1 Integrin Supports Rolling Adhesions on ICAM-1 Under Physiological Shear Flow in a Permissive Cellular Environment. The Journal of Immunology. 2000;165:442–452. doi: 10.4049/jimmunol.165.1.442. [DOI] [PubMed] [Google Scholar]
- 46.Lorenz HM, et al. CD45 mAb Induces Cell Adhesion in Peripheral Blood Mononuclear Cells via Lymphocyte Function-Associated Antigen-1 (LFA-1) and Intercellular Cell Adhesion Molecule 1 (ICAM-1) Cellular Immunology. 1993;147:110–128. doi: 10.1006/cimm.1993.1052. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, et al. Inflamed leukocyte-mimetic nanoparticles for molecular imaging of inflammation. Biomaterials. 2011;32:7651–7661. doi: 10.1016/j.biomaterials.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherman MB, et al. Removal of divalent cations induces structural transitions in red clover necrotic mosaic virus, revealing a potential mechanism for RNA release. Journal of virology. 2006;80:10395–10406. doi: 10.1128/JVI.01137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiappini C, et al. Biodegradable silicon nanoneedles delivering nucleic acids intracellularly induce localized in vivo neovascularization. Nature materials. 2015 doi: 10.1038/nmat4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Copp JA, et al. Clearance of pathological antibodies using biomimetic nanoparticles. Proceedings of the National Academy of Sciences. 2014;111:13481–13486. doi: 10.1073/pnas.1412420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.