

Abstract
A multiplexed quantitative method for the analysis of three major unconjugated steroids in human serum by stable isotope dilution liquid chromatography-high resolution mass spectrometry (LC-HRMS) was developed and validated on a Q. Exactive Plus hybrid quadrupole/Orbitrap mass spectrometer. This quantification utilized isotope dilution and Girard P derivatization on the keto-groups of testosterone (T), androstenedione (AD) and dehydroepiandrosterone (DHEA) to improve ionization efficiency using electrospray ionization. Major isomeric compounds to T and DHEA; the inactive epimer of testosterone (epiT), and the metabolite of AD, 5α-androstanedione (5α-AD) were completely resolved on a biphenyl column within an 18 min method. Inter- and intra-day method validation using LC-HRMS with qualifying product ions was performed and acceptable analytical performance was achieved. The method was further validated by comparing steroid levels from 100 μL of serum from young vs older subjects. Since this approach provides high-dimensional HRMS data, untargeted analysis by age group was performed. DHEA and T were detected among the top analytes most significantly different across the two groups after untargeted LC-HRMS analysis, as well as a number of other still unknown metabolites, indicating the potential for combined targeted/untargeted analysis in steroid analysis.
Keywords: Mass spectrometry, Steroid, Girard P, Testosterone, High resolution/accurate mass, Liquid chromatography
Graphical abstract
1. Introduction
Dehydroepiandrosterone (DHEA), 4-androstene-3,17-dione (AD), testosterone (T) are androgenic 19-carbon steroids that serve as precursors for other male and female sex hormones and influence sex specific and sexually dimorphic processes such as spermatogenesis, muscle growth and neural development[1]. DHEA in humans is primarily produced by the adrenal glands, whereas both the adrenals and the gonads produce AD. T is produced in a sexually dimorphic manner, with around 95% produced by the testis in males, versus 50% by the ovaries and through adrenal precursor conversion in females, with the remainder produced by peripheral metabolism of AD. Biosynthesis of DHEA is accomplished primarily by cytochrome P450 17A1 from the 21-carbon substrate of 17-hydroxypregneolone, conversion of DHEA to AD is controlled by 3β-hydroxysteroid dehydrogenase (3β-HSD), and conversion of AD to T by 17β-HSD [2]. There is also an alternative pathway involving 17β-HSD- or aldo-keto reductase (AKR) 1C3-mediated conversion of DHEA to androst-5-ene-3β, 17β-diol followed by 3β-HSD-mediated conversion to T [3]. Disorders of metabolism and signaling of these steroids results in a range of morbidities, and the changes in the levels of these metabolites over the life course affect both development and age-related conditions. Diagnosis of diseases of steroid metabolism, as well as research into their more subtle variation as a driver or modifier of complex disease mandates their analysis in both clinical [4] and epidemiological [5] settings.
Quantification of steroid hormones from biological matrices is challenging due to strict requirement for sensitivity and specificity inherent in the analysis of relatively low abundance endogenous analytes [6]. This is further complicated by the change in concentration of steroids by developmental stage, with pre-pubertal children, post-menopausal women and older men having low levels of androgens [7] and estrogens [8]. Longitudinal studies and other large research studies are often limited in the amount of biospecimens collected. Thus, limitations on sample availability make highly sensitive and multiplexed sample sparing assays extremely valuable. Ligand binding assays (LBAs) were the most commonly used methods for the measurement of steroids in biological matrices attributed to their small sample consumption, high throughput, and femtomole to attomole level sensitivity. However, LBAs, especially for small molecules [9], are difficult or impossible to directly multiplex, are known to be prone to analytical interference including inter-assay variability due to cross-reactivity, and suffer a lack of specificity for the structurally related steroid metabolites [10]. Further, the inter-assay variability of immunoassay when different antibodies were employed has caused significant problems when applying LBA to longitudinal studies and epidemiologic studies. Liquid chromatography–mass spectrometry (LC–MS) based methods of steroid analysis have become increasingly popular due to sensitivity, specificity, robustness, and multiplexing ability, enabling robust quantification of multiple steroids in a single analytical run [11,12].
Unlike the epitope recognition underlying LBAs, the basis of specificity in LC–MS is chromatographic separation and spectral resolution. LC-tandem MS (MS/MS) further increases sensitivity and selectivity by monitoring one or more product ions after fragmentation of a selected precursor ion. Monitoring the precursor to product transitions in single or multiple reaction monitoring (SRM or MRM) mode results in gains in sensitivity from reduction in the noise level especially for triple quadrupole (QQ.Q.) based instruments [13]. However, in complex biological matrices, the selectivity of unit resolution mass spectrometer is limited by the resolving power of its mass analyzers, and interferences may bias results. Increasingly powerful high-resolution accurate mass (HR/AM) mass spectrometers derives specificity from high mass resolution. This may provide orthogonal specificity to fragmentation from a QQQ, e.g. in the case of the resolution of the M + 2 isotopologues from an analyte containing a double bond from the saturated analog (e.g. AD and 5α-AD). The utility of HR/AM MS also includes a considerably higher dimensionality of the data acquired from the analyzed sample, allowing a high degree of multiplexing, retrospective analysis, improved qualitative evaluation through isotope pattern analysis, untargeted analysis, and the ability to putatively assign chemical formula by accurate mass. Instruments combining HR/AM MS capabilities with more traditional isolation and fragmentation capabilities, including the quadrupole/Orbitrap or quadrupole/time-of-flight (TOF) hybrid instruments, can provide sensitive, specific quantification through multiple modes of mass spectrometry at scan speeds increasingly compatible with liquid chromatography. In hybrid instruments equipped with a mass filter to select precursor ions, a fragmentation cell, and then a high resolution mass analyzer, MS/HRMS analysis can also be performed [14]. The product ion chromatograms can be extracted from the MS/high-resolution MS data using a narrow mass tolerance (<5 ppm) commensurate with the mass accuracy of the analyzer, the resolution of the instrument and the complexity of the matrix. However, methods using hybrid instruments are sparse in the literature and none to our knowledge have yet been evaluated for quantitative performance for androgen analysis. This study was therefore designed to test the quantitative performance of a LC-HR/AM MS based assay of major androgens for simultaneous targeted and untargeted quantitation.
2. Experimental
2.1. Reagents and materials
Water, methanol, hexanes, dichloromethane, methyl tert-butyl ether (MTBE), acetonitrile and acetic acid were Optima LC–MS grade solvents from Fisher Scientific (Pittsburg, PA). Hydrochloric acid and acetic acid were from Fisher Scientific. Girard P reagent was from Tokyo Chemical Industry Company, LTD (Tokyo, Japan). Unlabeled androgens of ≥98% purity (T, AD, DHEA, 5α-AD and EpiT) were from Sigma-Aldrich (St Louis, MO). Stable isotope-labeled androgens ([13C3]-T 98% purity, [13C3]-AD 99% purity, and [2H5]-DHEA 97% purity) were from Cambridge Isotope Labs (And-over, MA). The internal standard eluting most closely to other analytes, [13C3]-T, was used for quantitation of epiT and 5α-AD. No detectable cross contamination of standards or internal standards was found when each standard and internal standard was analyzed alone (data not shown). Double charcoal stripped human serum used as a surrogate matrix was from Golden West Biologicals, Inc. and contained no detectable levels of the steroids measured (Temecula, CA, USA).
2.2. Sample preparation and Girard P derivatization
The internal standard solution containing 10 pg/μL [13C3]-T, 10 pg/μL [13C3]-AD, and 50 pg/μL [2H5]-DHEA in methanol (20 μL) was added to each sample of double-charcoal stripped serum or serum (100 μL), then the sample was extracted by liquid-liquid extraction (LLE) with MTBE. The serum containing internal standards was diluted with 400 μL of water, acidified with 5 μL 1 M HCl, and then 50 μL of saturated NaCl in water was added followed by 1.4 mL of MTBE. Samples were vortexed briefly, followed by shaking for 10 min, then centrifuged at 3500g for 5 min to separate the upper organic layer which was then transferred to a new glass tube. Extracted samples were evaporated to dryness under nitrogen, then re-suspended in 200 μL of 10% acetic acid in methanol for derivatization as previously described for derivatization of estrone [15]. Importantly, the mono-derivatization is rapid, going to essential competition within 1 min. 20 μL of Girard's reagent P (1 mg/mL in water) was added prior to a 10-min incubation at 60 °C to ensure complete reaction. Finally, samples were again evaporated to dryness under nitrogen before resuspension in 100 μL 50:50 methanol:water. 10 μL of the sample was then used for LC–MS/HRMS analysis, such that 10 pg of [13C3]-T and [13C3]-AD as well as 50 pg of [2H5]-DHEA were injected on column.
2.3. LC–MS/HRMS analysis of Girard P-derivatized steroids
LC–MS/HRMS analysis was conducted on an Ultimate 3000 quaternary UPLC equipped with a refrigerated autosampler (6 °C) and a column heater (60 °C) coupled to a Thermo QExactive Plus HRMS. LC separations were conducted on a Phenomenex Kinetex biphenyl column (2.6 μm, 100 A, 100 × 2.1 mm). A multi-step gradient at 0.2 mL/min flow with solvent A (water 1% acetic acid) and solvent B (acetonitrile 1% acetic acid) was as follows; 20% B from 0 to 1 min, increasing to 25% B from 1 to 5 min, increasing to 100% B from 5 to 8 min then holding 100% B until 12 min, then the column was returned to starting conditions and re-equilibrated at 20% B from 13 to 17 min. Column effluent was diverted to waste before 1 and after 14 min. The mass spectrometer was operated in positive ion mode alternating between full scan (200–800 m/z) at a resolution of 70,000 and parallel reaction monitoring at 17,500 resolution with a precursor isolation window of 0.7 m/z. Molecular (M+) precursor and the most intense product ions (m/z) were as follows; T, DHEA, EpiT, and 5α-AD (422.2802–343.2380), AD (420.2646–341.2224), [13C3]-T (425.2903–346.2481), [13C3]-AD (423.2746–344.2324), and [2H5]-DHEA (428.3179–349.2757). AD as well as [13C3]-AD were also detected as a doubly-charged product of a bis-Girard P derivatization, 2GP-M2+ (AD, 277.1697 – 237.6461, [13C3]-AD, 278.6729–239.1511).
2.4. Method validation
We conducted analytical method validation including inter-and intra-day studies of precision and accuracy, as well as testing for specificity and stability [16]. Calibration curves and quality controls were prepared by spiking individual standards in double-charcoal stripped human serum with a new batch of calibrators and quality controls prepared across 4 different days (inter-day) and with 4 different batches in one day (intra-day). Inter- and intra-day comparisons of accuracy and precision were conducted with standard curves from 19.5 to 1250 pg/mL (with constituent points at 19.53, 39.06, 78.13, 156.3, 312.5, 625.0, and 1250 pg/mL), and quality control samples prepared separately at LQC (50 pg/mL), MQC (200 pg/mL) and HQC (1000 pg/mL). Standard curve and QC points for DHEA were 10 times higher to reflect natural abundance. This range was chosen to cover the expected distribution of steroid levels from serum. QC and selected clinical samples were re-analyzed after storage in the autosampler for approximately 24 and 48 h to mimic storage conditions during analysis. Since a major capability of LC-HRMS is that retrospective analysis of the data is much more data rich, in validation we included epiT in our calibrations, using [13C3]-T as the internal standard to mimic the situation of examining retrospective analysis from an already analyzed set of samples. Further validation was conducted by UV-vis absorbance of the T calibrator stock, using the published co-extinction coefficient of 15,100 L/mol * cm at 241 nm [17]. Finally, validation to an external method was conducted by analysis of the National Institutes of Standards and Technology (NIST) Standard Reference Material (SRM) 971 – Hormones in Frozen Human Serum.
2.5. Clinical study
The blood collection protocol for serum collected as part of this study was approved by the University of Pennsylvania Review Board (Protocol # 800924). After the blood was collected, it was allowed to clot for 1 h at room temperature, serum was separated and aliquots were stored at −80 °C. Serum samples were allowed to thaw at room temperature and aliquots of 100 μL were used for the extraction and analyses.
2.6. Data analysis
LC-MS data was analyzed in XCalibur v2.6 and Tracefinder v3.2 (Thermo). Statistical analysis was performed in Microsoft Excel or Prism v6 (GraphPad Software Inc. La Jolla, CA). Quantification was conducted as indicated using HR/AM-MS1 with a 5 ppm window, with the qualifying product ion analyzed with a 5 ppm product ion window.
3. Results
3.1. Reaction of keto- containing steroids with Girard P reagent
Derivatization of keto-containing steroids has been previously used for estrone and its metabolites [15], as well as neutral steroids in MALDI-MS analysis. We successfully applied the derivatization strategy to the quantitation of major androgens, and the reaction scheme is shown in (Fig. 1) for T. DHEA and T formed mono-GP products and AD, containing two ketones, derivatized as both the mono-GP and bis-GP derivatives. Although sensitivity for AD was assumed to be reduced by the combination of mono- and bis-derivatives, AD is not sufficiently low in humans that this was an issue for method development or application. Furthermore, the use of a [13C]-labeled stable isotope analog compensates for the partial derivatization.
Fig. 1.
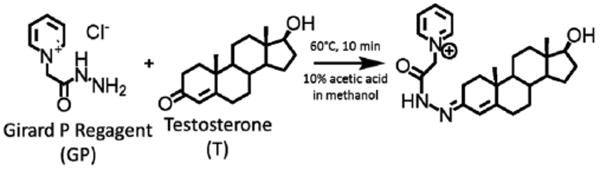
Derivitization of testosterone (T) by Girard's reagent P (GP) as a pre-ionized derivative containing a quaternary nitrogen.
3.2. Optimization and method validation of LC-HRMS analysis of Girard P derivatives
Chromatography was optimized using the intense [M]+ ion for each androgen-GP derivative to produce reliable separation with approximately 20–30 s peak widths. Previous methods for androgen analysis have reported that C18 stationary phases at higher flow rates are sub-optimal for resolving androgens [7]. Therefore, we examined the ability of a Phenomenex biphenyl column to chromatographically separate the androgens. All analyzed GP-androgens were baseline resolved in 17min, including a 4-min re-equilibration of the column (Fig. 2). DHEA and T were completely resolved, as evidenced by separation of their [M]+ ions at 6.9 min and 8.7 min respectively (Fig. 2A and B). The inactive epimer of T, epi-testosterone (epi-T), also formed a GP-derivative with an intense [M]+ ion that eluted at 9.8 min, baseline resolved from T. Another isobaric steroid metabolite, 5α-AD, was closely baseline resolved from all of the above steroids and eluted at 10.0 min (Fig. 2C), but with remarkably reduced signal intensity. The mono-GP of AD, eluted at 9.9 min, with shouldering peaks likely corresponding to the other possible mono-GP derivative resulting from cis- and trans-isomers at 9.7 min. The doubly-charged bis-GP derivative of AD eluted at 2.2 min, which was after the void volume of the LC-system (less than 1.5 min) (Fig. 2D). Each GP-androgen co-eluted exactly with their matched [13C3]-stable isotope labeled internal standard, except for DHEA, where [2H5]-DHEA eluted 0.1 min earlier at 6.7 min.
Fig. 2.
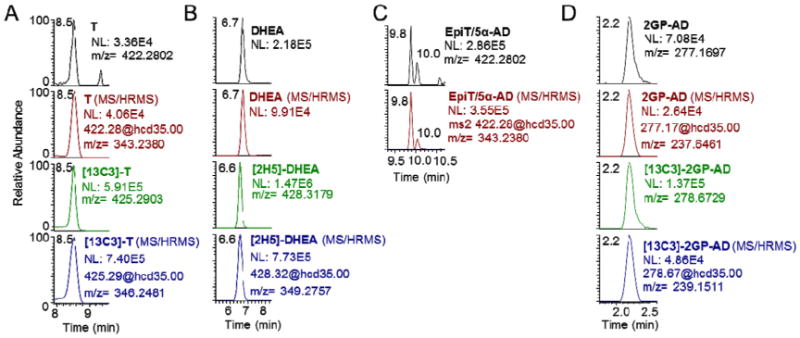
LC-HRMS and LC–MS/HRMS of GP derivatives of (A) T, (B) DHEA, (C) epiT (9.8) and 5α-AD (10.0), and (D) AD as the more intense bis-GP with co-elution of their corresponding stable isotope labeled internal standards for T, DHEA, and AD.
All androgen-GPs analyzed produced an intense product ion still containing the intact steroid ring with a loss derived from the GP derivative (the loss of a pyridinium ion [M-79]+) after higher-energy collision induced dissociation (HCD) with fragmentation that was similar to that previously observed for the estrone-GP derivative under CID fragmentation in a QQQ [15]. Thus, the major fragment ions as indicated in the methods section were used as qualifying transitions in quantitative analysis, and were consistently superimposable on the LC-HRMS analysis of the respective precursor [M]+ ion from patient samples (Fig. 2).
Quantitation for method validation was performed from the HR full scan data with qualifying peaks from the MS/HRMS using a 5 ppm (±2.5 ppm) window. Analysis of LQC (500 pg/mL DHEA, 50 pg/mL AD, T, epiT), MQC (2200 pg/mL, 200 pg/mL AD, T, epiT), HQC (10,000 pg/mL DHEA, 1000 pg/mL AD, T, epiT) was performed over 4 separate days from separate batches of calibration curves and quality controls, with one day analyzing 4 separately prepared sets of QCs. Typical calibration curves were linear in the form of y = mx + b for; T, y = 0.004582× –0.004553, r2 = 0.9994, AD, y = 0.006193× + 0.001915, r2 = 0.9977, DHEA y = 0.01198× + 0.0005762, r2 = 0.9995. Summary values for precision and accuracy are shown in Table 1. Accuracy and precision were below 20% CV for all but one value, and below 15% for most. Re-injection of samples after storage in the autosampler for 24 and 48 h resulted in calculated steroid amounts and signal intensity within 5% CV indicating sufficient stability for analysis (data not shown). Interestingly, other groups have reported difficulty with precision in analysis of AD, thus we believe this may represent instability of the AD analyte, more so than higher imprecision with LC-HRMS [7].
Table 1.
Method validation of inter- and intra-day precision and accuracy for DHEA T, AD, and epiT at the low, medium and high quality controls (LQC, MQC and HQCs of 50 pg/mL, 220 pg/mL and 1000 pg/mL with levels of DHEA 10 times higher to reflect natural abundance).
| LQC | MQC | HQC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||
| Mean | SD | Precision % | Accuracy % | Mean | SD | Precision % | Accuracy % | Mean | SD | Precision % | Accuracy % | |
| Inter-day (N = 4) | ||||||||||||
| DHEA | 577.6 | 81.4 | 14.1% | 115.5% | 2323.4 | 104.4 | 4.5% | 116.2% | 10031.7 | 422.2 | 4.2% | 100.3% |
| AD | 53.9 | 6.5 | 12.0% | 107.8% | 189.8 | 14.4 | 7.6% | 94.9% | 1046.4 | 102.5 | 9.8% | 104.6% |
| T | 53.4 | 10.1 | 19.0% | 106.7% | 237.2 | 14.9 | 6.3% | 118.6% | 1001.1 | 36.0 | 3.6% | 100.1% |
| EpiT | 56.3 | 8.4 | 14.9% | 112.5% | 223.8 | 14.0 | 6.3% | 111.9% | 1014.9 | 70.1 | 6.9% | 101.5% |
| Intra-day (N = 4) | ||||||||||||
| DHEA | 528.3 | 54.6 | 10.3% | 105.7% | 2295.6 | 98.0 | 4.3% | 114.8% | 9721.4 | 436.5 | 4.5% | 97.2% |
| AD | 47.3 | 1.2 | 2.6% | 94.7% | 196.8 | 43.0 | 21.8% | 98.4% | 1199.0 | 281.8 | 23.5% | 119.9% |
| T | 45.8 | 4.2 | 9.2% | 91.5% | 236.2 | 12.5 | 5.3% | 118.1% | 976.6 | 56.1 | 5.7% | 97.7% |
| EpiT | 51.7 | 3.9 | 7.5% | 103.4% | 213.6 | 11.7 | 5.5% | 106.8% | 939.1 | 57.7 | 6.1% | 93.9% |
NIST SRM 971 [18] was analyzed for T in triplicate using 100 μL aliquots to compare our method to the reference method used for certified values in the SRM [19]. The experimentally determined value of 6655.12 pg/mL for the male and 280.02 pg/mL for female serum using our assay is in close agreement, with 3.43% and 1.03% difference for male and female serum respectively, to the SRM values of 6434.204 pg/mL (male) and 277.17 pg/mL (female).
3.3. Analysis of steroids from patient samples
To demonstrate the utility of the method in untargeted analysis, simultaneously acquired with the targeted analysis, we analyzed steroids from 19 subjects, including 9 younger men and 1 younger woman (age 28–49, mean 38.6) and 8 older women and 1 older man (age 61–81, mean 74.6). We dichotomized the sample population to examine differences in levels of steroids by age group. Targeted analysis revealed the expected decrease in T given the age difference and sex distribution of the two sets (327.3 ± 68.3 vs 127.6 ± 54.2 pg/mL) as well as a decrease in DHEA (3311.1 ± 613.0 vs 1368.0 ±336.1 pg/mL) (p-val <0.05 for both).
Simultaneous untargeted analysis was conducted using the LC-HRMS data and SIEVE, a proprietary untargeted analysis software (Thermo). After LC alignment, frames were constructed using default settings with a minimum signal intensity of 500,000, yielding 20,805 frames after removal of duplicates. Data was then analyzed in Metaboanalyst 3.0 [20], a server based platform for metabolomics analysis. Non-informative variables were removed by filtering for inter-quartile range, and peaks detected in the blanks were excluded from analysis. Hierarchical clustering by Euclidean distance with the Ward clustering algorithm and heat-map visualization revealed the similarity of the blanks and QCs within their groups, but poor overall grouping by young vs old metabolome (Fig. 3).
Fig. 3.
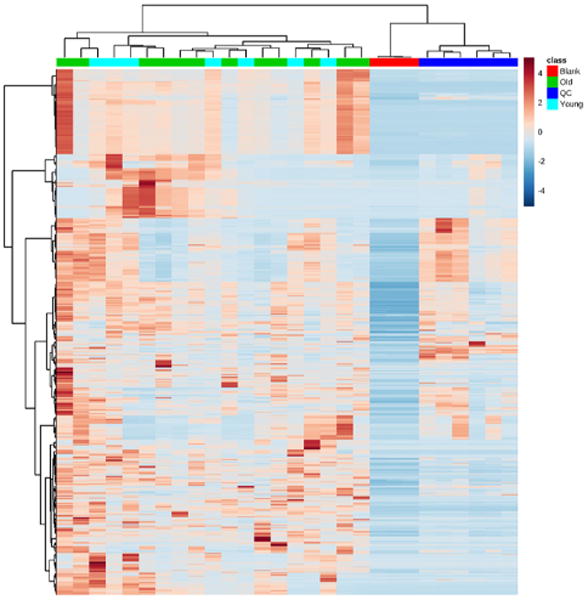
Hierarchical clustering of blank, old, young, and quality control (QC) samples.
The most highly variable metabolites by age group were displayed using a volcano plot using a cut-off fold-change of 1.5 and p-value of 0.05 using a conservative non-parametric t-test assuming unequal variance due to our limited sample size and prior knowledge of steroid levels (Fig. 4). The top 100 metabolites were chosen for putative identification (Supplemental Table 1), since this represents a reasonable number of metabolites for this time consuming step of metabolomics analysis. The Human Metabolome Database [21] was queried using the adjusted accurate mass to reflect the underivatized [MH]+ after adjustment of the mass for a mono-derivitization by GP. A feature (m/z 422.2795, Rt 6.89 min) corresponding to DHEA was identified as the top 25th feature, and a feature (m/z 422.2802, Rt 8.71 min) corresponding to T was identified as the top 88th feature. Further inspection indicated isotope (m/z 423.2836, Rt 8.71) and partially mis-aligned frames (m/z 422.2799, Rt 7.16) also corresponding to T and DHEA among the top ranked frames. Appropriate age-matching or stratification by sex would likely improve the ability to discriminate age-related steroid level changes, but the proof-of-principle for untargeted steroid analysis is still reflected with these findings.
Fig. 4.
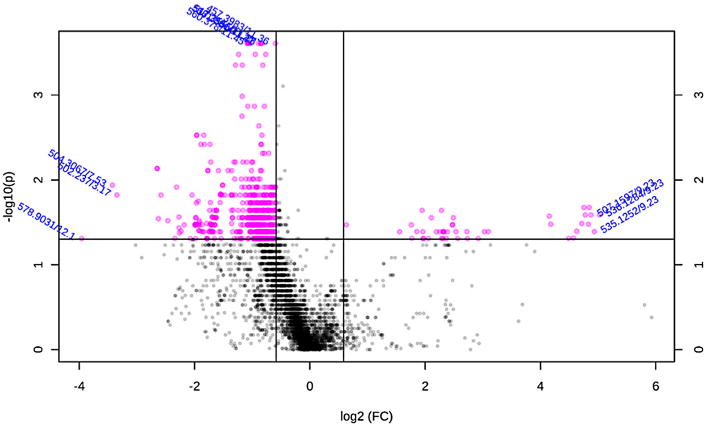
Volcano plot of LC-HRMS features (m/z/retention time) comparing serum of young vs old subjects. Features displayed as pink dots had a fold change (FC) > 1.5 and a p-value <0.05 by a t-test assuming unequal variance and non-parametric distribution.
4. Discussion
The main challenge of steroid analysis in special populations remains sensitivity, to allow quantitation of low abundance hormones within the constraint of feasible biospecimen consumption [22]. Limits of detection at the sub-pg on column range with a sample sparing method using only 100 μL of serum should provide adequate sensitivity for analysis of unconjugated steroids in even special populations with low levels of androgens. This is critical in both the clinical and epidemiological setting, where precise accurate and reproducible measurements are necessary [23]. The impetus for such steroid measurement can include diagnosis of steroid biochemical disorders [24] or more general studies of developmental changes [25]. A number of other processes and disorders, including aging, autism spectrum disorder [26] as well as various cancers [27], are hypothesized to be driven by sex steroid hormones and studies are warranted on these topics. Due to the clinical and research relevance of steroid bioanalysis, methods for steroid analysis are an active topic of research still evolving due to increasing understanding of the complexities of steroid biochemistry, as well as evolving analytical technology.
Derivatization has been a common strategy for modern LC–MS based methods of sterol metabolites, to overcome the limited ionization efficiency of steroids under ESI conditions at the source of the mass spectrometer [28]. Methods of direct analysis (without derivatization) have been reported for major steroids, but for adult patients and requiring relatively large amounts of blood product (e.g. 500 μL of serum) that would not be feasible in an epidemiological setting or for clinical samples requiring an extensive battery of tests from one sample [23]. For some steroids, advances in instrument sensitivity have allowed underivatized analysis on similarly limited sample volumes [29], but none of these method have to our knowledge have been on high resolution platforms. Increasing instrument sensitivity will likely allow underivatized methods of steroid analysis in populations with higher steroid levels, as methods without derivatization [29] have reported lower limits of quantitation at 0.1 nmol/L (28.8 pg/mL) for T and AD, which is lower than the LQC investigated here. Beyond the scope of investigation in this report, but consistent with comparison of this report to the existing literature, comparisons of LC–MS/MS and LC-HRMS have yielded relatively similar limits of quantitation for vitamin D metabolites [30]. Reviews on derivatization reagents and strategies, as well as the varied benefits in sensitivity, specificity, sample preparation, are available [31,32]. Pre-ionized derivatives, especially, have been adapted for high sensitivity analysis reserved for sample sparing methods, special populations, and low abundance metabolites of steroids [33]. The Girard P derivatization used here has been previously employed in estrone analysis from postmenopausal women, and MALDI/ToF-MS to examine oxysterols in the brain sections of rats, where the characteristic fragmentation of the derivative were crucial in dealing with the complex ion spectra detected from the tissue [34]. Girard P derivatization was also used in an excellent analysis of T/epiT ratios in urine for sports doping investigations, but the authors did not examine the inclusion of other steroids into quantitation [35], used sub-optimally pure deuterated internal standards, and did not pursue untargeted analysis from their samples.
Untargeted analysis of steroids offers potential opportunities, significant benefits, and some challenges. The complexity of steroid biochemistry, and the change of steroid metabolism through development and disease offers opportunities for metabolite discovery. The benefits of sterol panels in basic, clinical and epidemiological research have been previously identified, and this method provides a balanced approach between the ability to validate a panel of analytes of high interest, while simultaneously profiling a large number of other metabolites. A major benefit of steroid metabolomics is the availability of a wide range of analytical standards for steroid analysis, to provide robust identification of features that remains a bottleneck in metabolomics. Major challenges in steroid analysis, such as their low abundance and poor ionization efficiency in ESI are, as this report demonstrates, surmountable.
It is highly likely that our prior optimization of LC conditions contributed to the results regarding specificity. Thus, the generalizability of using LC-HRMS for untargeted approaches is likely to be analyte and method dependent. To this point, HRMS by direct infusion has been proposed for a number of lipid classes. However, steroids as a class, contain a large number of isobaric species as well as non-specific fragmentation derived from regio- and stereo-isomers with distinct biological roles. Direct infusion with sufficiently high resolution would be able to resolve near isobars, such as those derived from the overlap of the M + 2 isotopologues of major steroids (e.g. M + 2 of AD, [M + H]+ m/z 289.2072 vs 5α-AD, [M + H]+ m/z 289.2162). Even separation by C18 column chemistries has been reported to be insufficient to resolve major steroids including DHEA and T with short chromatographic methods, and overlapping fragmentation of major and minor product ions between DHEA and T was also reported by that same group [7]. The biphenyl column was selected after less ideal separation was found from analysis on a C18 column, but other reported successful stationary phase chemistries do include C18 and phenyl-hexyl columns.
Finally, the ability to multiplex steroid analysis within one assay is a critical parameter considering the diversity of steroid metabolism. In our study, we optimized quantitation of 3 major androgens, while ensuring the separation of major isobaric species. Quantitation of the major isobars, using internal standards already spiked into the samples for quantitation of the initial 3-steroid panel, proved adequate for quantitation for epiT. However, a limitation of this approach is that without the ideal stable isotope labeled internal standard in the samples of interest, the difference between surrogate matrix (double charcoal stripped serum in our case) and true matrix, or the inter-individual variation in suppressing compounds may confound measurement. Ultimately, validation with an appropriate stable isotope labeled analog and LC–MS/MS should be conducted for rigor. Another benefit of the analytic approach used here is that, in theory, LC-HRMS should allow nearly unlimited multiplexing within the dynamic range of the detector. Thus, derivatization may be a useful technique to increase the ion current derived from any given analyte to allow higher multiplexing. Combined with MS/HRMS, the upper bound of dynamic range may also be extended by using less efficient fragmentation to reduce any potential saturation of the detector. This may be especially important when assays are required to quantify analytes across 2–3 orders of magnitude as can be seen in some steroid and bile acid metabolites. This study should spur further examination of the increased specificity and other analytical benefits that may be gained by small molecule analysis by HRMS, especially on hybrid instruments with higher order fragmentation. It should also spur investigation of the potential drawbacks on quantitation by LC-HRMS alone, since parallel comparisons of quantitative results obtained by multiple scan modes (e.g. LC–MS/MS at different resolutions and mass windows for analysis) on the same instrument with the same sample are now possible.
Highlights.
Keto-steroids, including testosterone, are analytes of biological interest.
LC-high resolution MS offers unique potential for steroid assays.
Quantitative validation of a Girard P based keto-steroid assay was conducted.
Simultaneous untargeted steroid profiling and targeted quantitation is possible.
Acknowledgments
This project was supported by a Pennsylvania Department of Health CURE Grant to NWS, as well as NIH Grants K22ES26235, R21HD087866 (NWS), P42ES023720, P30ES013508 (IAB), and T32ES019851 (LB).
Footnotes
Conflict of interest disclosure: The authors declare no competing financial interest.
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.steroids.2016.10. 003.
Contributor Information
Alexander J. Frey, Email: ajf354@drexel.edu.
Qingqing Wang, Email: qingqing@mail.med.upenn.edu.
Christine Busch, Email: cbusch@mail.med.upenn.edu.
Daniel Feldman, Email: drf49@drexel.edu.
Lisa Bottalico, Email: bottlisa@mail.med.upenn.edu.
Clementina A. Mesaros, Email: mesaros@upenn.edu.
Ian A. Blair, Email: ianblair@exchange.upenn.edu.
Anil Vachani, Email: avachani@mail.med.upenn.edu.
Nathaniel W. Snyder, Email: nws28@drexel.edu.
References
- 1.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25(6):947–970. doi: 10.1210/er.2003-0030. [DOI] [PubMed] [Google Scholar]
- 3.Mitamura K, Nakagawa T, Shimada K, Namiki M, Koh E, Mizokami A, Honma S. Identification of dehydroepiandrosterone metabolites formed from human prostate homogenate using liquid chromatography–mass spectrometry and gas chromatography–mass spectrometry. J Chromatogr A. 2002;961(1):97–105. doi: 10.1016/s0021-9673(02)00134-6. [DOI] [PubMed] [Google Scholar]
- 4.Kushnir MM, Rockwood AL, Roberts WL, Yue B, Bergquist J, Meikle AW. Liquid chromatography tandem mass spectrometry for analysis of steroids in clinical laboratories. Clin Biochem. 2011;44(1):77–88. doi: 10.1016/j.clinbiochem.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Barrett-Connor E, Garland C, McPhillips JB, Khaw KT, Wingard DL. A prospective, population-based study of androstenedione, estrogens, and prostatic cancer. Cancer Res. 1990;50(1):169–173. [PubMed] [Google Scholar]
- 6.Mesaros C, Wang Q, Blair IA. What are the main considerations for bioanalysis of estrogens and androgens in plasma and serum samples from postmenopausal women? Bioanalysis. 2014;6(23):3073–3075. doi: 10.4155/bio.14.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kushnir MM, Blamires T, Rockwood AL, Roberts WL, Yue B, Erdogan E, Bunker AM, Meikle AW. Liquid chromatography-tandem mass spectrometry assay for androstenedione, dehydroepiandrosterone, and testosterone with pediatric and adult reference intervals. Clin Chem. 2010;56(7):1138–1147. doi: 10.1373/clinchem.2010.143222. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Bottalico L, Mesaros C. I.A. Blair, Analysis of estrogens and androgens in postmenopausal serum and plasma by liquid chromatography–mass spectrometry. Steroids. 2015;99(Pt A):76–83. doi: 10.1016/j.steroids.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fanelli F, Belluomo I, Di Lallo VD, Cuomo G, De Iasio R, Baccini M, Casadio E, Casetta B, Vicennati V, Gambineri A, Grossi G, Pasquali R, Pagotto U. Serum steroid profiling by isotopic dilution-liquid chromatography–mass spectrometry: comparison with current immunoassays and reference intervals in healthy adults. Steroids. 2011;76(3):244–253. doi: 10.1016/j.steroids.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Taieb J, Mathian B, Millot F, Patricot MC, Mathieu E, Queyrel N, Lacroix I, Somma-Delpero C, Boudou P. Testosterone measured by 10 immunoassays and by isotope-dilution gas chromatography-mass spectrometry in sera from 116 men, women, and children. Clin Chem. 2003;49(8):1381–1395. doi: 10.1373/49.8.1381. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Catlin DH, Demers LM, Starcevic B, Swerdloff RS. Measurement of total serum testosterone in adult men: comparison of current laboratory methods versus liquid chromatography-tandem mass spectrometry. J Clin Endocrinol Metab. 2004;89(2):534–543. doi: 10.1210/jc.2003-031287. [DOI] [PubMed] [Google Scholar]
- 12.Ackermans MT, Endert E. LC–MS/MS in endocrinology: what is the profit of the last 5 years? Bioanalysis. 2014;6(1):43–57. doi: 10.4155/bio.13.300. [DOI] [PubMed] [Google Scholar]
- 13.McLafferty FW. Tandem mass spectrometry. Science. 1981;214(4518):280–287. doi: 10.1126/science.7280693. [DOI] [PubMed] [Google Scholar]
- 14.Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, Snyder NW. LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and (13)C-isotopic labeling of acyl-coenzyme A thioesters. Anal Bioanal Chem. 2016;408(13):3651–3658. doi: 10.1007/s00216-016-9448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rangiah K, Shah SJ, Vachani A, Ciccimaro E. I.A. Blair, Liquid chromatography/mass spectrometry of pre-ionized Girard P derivatives for quantifying estrone and its metabolites in serum from postmenopausal women. Rapid Commun Mass Spectrom. 2011;25(9):1297–1307. doi: 10.1002/rcm.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.U. Fda. Draft Guidance for Industry: Bioanalytical Method Validation. US Department of Health and Human Services, US FDA; 2013. [Google Scholar]
- 17.Yasuda K. Synthesis of 2-Alpha-Halo-4-En-3-Oxo-Steroids. Chem Pharm Bull (Tokyo) 1964;12:1217–1224. doi: 10.1248/cpb.12.1217. [DOI] [PubMed] [Google Scholar]
- 18.https://www-s.nist.gov/srmors/certificates/971.pdf (accessed 09/16).
- 19.Tai SS, Xu B, Welch MJ, Phinney KW. Development and evaluation of a candidate reference measurement procedure for the determination of testosterone in human serum using isotope dilution liquid chromatography/tandem mass spectrometry. Anal Bioanal Chem. 2007;388(5-6):1087–1094. doi: 10.1007/s00216-007-1355-3. [DOI] [PubMed] [Google Scholar]
- 20.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0–making metabolomics more meaningful. Nucl Acids Res. 2015;43(W1):W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, Scalbert A. HMDB 3.0-the human metabolome database in 2013. Nucl Acids Res. 2013;41(Database issue):D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Mesaros C. I.A. Blair, Ultra-high sensitivity analysis of estrogens for special populations in serum and plasma by liquid chromatography-mass spectrometry: assay considerations and suggested practices. J Steroid Biochem Mol Biol. 2016 doi: 10.1016/j.jsbmb.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couchman L, Vincent RP, Ghataore L, Moniz CF, Taylor NF. Challenges and benefits of endogenous steroid analysis by LC–MS/MS. Bioanalysis. 2011;3(22):2549–2572. doi: 10.4155/bio.11.254. [DOI] [PubMed] [Google Scholar]
- 24.Rossi C, Calton L, Hammond G, Brown HA, Wallace AM, Sacchetta P, Morris M. Serum steroid profiling for congenital adrenal hyperplasia using liquid chromatography-tandem mass spectrometry. Clin Chim Acta. 2010;411(3-4):222–228. doi: 10.1016/j.cca.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Kulin HE, Finkelstein JW, D'Arcangelo MR, Susman EJ, Chinchilli V, Kunselman S, Schwab J, Demers L, Lookingbill G. Diversity of pubertal testosterone changes in boys with constitutional delay in growth and/or adolescence. J Pediatr Endocrinol Metab. 1997;10(4):395–400. doi: 10.1515/jpem.1997.10.4.395. [DOI] [PubMed] [Google Scholar]
- 26.Baron-Cohen S. The extreme male brain theory of autism. Trends Cognit Sci. 2002;6(6):248–254. doi: 10.1016/s1364-6613(02)01904-6. [DOI] [PubMed] [Google Scholar]
- 27.Santen RJ, Yue W, Wang JP. Estrogen metabolites and breast cancer. Steroids. 2015;99(Pt A):61–66. doi: 10.1016/j.steroids.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Griffiths WJ, Sjovall J. Analytical strategies for characterization of bile acid and oxysterol metabolomes. Biochem Biophys Res Commun. 2010;396(1):80–84. doi: 10.1016/j.bbrc.2010.02.149. [DOI] [PubMed] [Google Scholar]
- 29.Buttler RM, Martens F, Kushnir MM, Ackermans MT, Blankenstein MA, Heijboer AC. Simultaneous measurement of testosterone, androstenedione and dehydroepiandrosterone (DHEA) in serum and plasma using isotope-dilution 2-dimension ultra high performance liquid-chromatography tandem mass spectrometry (ID-LC–MS/MS) Clin Chim Acta. 2015;438 (:157–159. doi: 10.1016/j.cca.2014.08.023. [DOI] [PubMed] [Google Scholar]
- 30.Bruce SJ, Rochat B, Beguin A, Pesse B, Guessous I, Boulat O, Henry H. Analysis and quantification of vitamin D metabolites in serum by ultra-performance liquid chromatography coupled to tandem mass spectrometry and high-resolution mass spectrometry – a method comparison and validation. Rapid Commun Mass Spectrom. 2013;27(1):200–206. doi: 10.1002/rcm.6439. [DOI] [PubMed] [Google Scholar]
- 31.Niwa M, Watanabe M, Watanabe N. Chemical derivatization in LC–MS bioanalysis: current & future challenges. Bioanalysis. 2015;7(19):2443–2449. doi: 10.4155/bio.15.177. [DOI] [PubMed] [Google Scholar]
- 32.Marcos J, Pozo OJ. Derivatization of steroids in biological samples for GC–MS and LC–MS analyses, Bioanalysis. 2015;7(19):2515–2536. doi: 10.4155/bio.15.176. [DOI] [PubMed] [Google Scholar]
- 33.Higashi T, Ogawa S. Chemical derivatization for enhancing sensitivity during LC/ESI-MS/MS quantification of steroids in biological samples: a review. J Steroid Biochem Mol Biol ( 2015 doi: 10.1016/j.jsbmb.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Hornshaw M, Alvelius G, Bodin K, Liu S, Sjovall J, Griffiths WJ. Matrix-assisted laser desorption/ionization high-energy collision-induced dissociation of steroids: analysis of oxysterols in rat brain. Anal Chem. 2006;78(1):164–173. doi: 10.1021/ac051461b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danaceau JP, Scott Morrison M, Slawson MH. Quantitative confirmation of testosterone and epitestosterone in human urine by LC/Q-ToF mass spectrometry for doping control. J Mass Spectrom. 2008;43(7):993–1000. doi: 10.1002/jms.1443. [DOI] [PubMed] [Google Scholar]