

Abstract
Therapeutic protein kinase inhibitors are designed on the basis of kinase structures. Here, we define intrinsically disordered regions (IDRs) in structurally hybrid kinases. We reveal that 65% of kinases have an IDR adjacent to their kinase domain (KD). These IDRs are evolutionarily more conserved than IDRs distant to KDs. Strikingly, 36 kinases have adjacent IDRs extending into their KDs, defining a unique structural and functional subset of the kinome. Functional network analysis of this subset of the kinome uncovered FAK1 as topologically the most connected hub kinase. We identify that KD-flanking IDR of FAK1 is more conserved and undergoes more post-translational modifications than other IDRs. It preferentially interacts with proteins regulating scaffolding and kinase activity, which contribute to cytoskeletal remodeling. In summary, spatially and evolutionarily conserved IDRs in kinases may influence their functions, which can be exploited for targeted therapies in diseases including those that involve aberrant cytoskeletal remodeling.
Keywords: Intrinsic disorder in kinases, evolution of kinases, FAK1 signaling, hub proteins, protein-protein interaction networks, network medicine
Graphical Abstract
1. INTRODUCTION
The human kinome, consisting of 518 protein kinases distributed in 10 distinct groups, phosphorylates more than 90% of the proteome at least once in a lifetime of a cell, modulating critical cellular processes1,2. Consequently, aberrant activation of kinases drives pathogenesis of many human diseases. Thus, inhibiting pathogenic kinases is a major focus of targeted therapeutics3. Since diseases like organ fibrosis and cancer are characterized by aberrant activation of kinases, use of kinase inhibitors (KIs) is wide-spread in treating these pathologies. However, KIs often have off-target effects and become less effective due to chemoresistance4–8. While various classes of KIs exist, majority of them are designed to target structurally conserved catalytic kinase domains (KDs). Compensatory mutations at key gatekeeper residues in the catalytic cleft or at other regions within the KD confer drug-resistance9,10. Highly specific allosteric kinase inhibitors, which target functionally important regions outside the kinase domains, represent an exciting avenue to develop specific kinase inhibitors11. However, despite these advances, only a small subset of the human kinome is therapeutically targetable. Therefore, it is imperative to identify novel regions outside the catalytic kinase domain that may serve as potential drug target sites for the development of more specific allosteric kinase inhibitors. Herein, we postulate that identification of evolutionarily divergent and functionally important regions in protein kinases provide novel opportunities to target specific kinases.
To define regulatory regions that may influence the catalytic activity of kinases independent of the enzymatically active KDs, we focused on intrinsically disordered regions (IDRs), which are emerging as potential molecular targets12,13. IDRs participate in various disease pathogenesis and exist as highly dynamic conformational ensembles14,15. Some of these regions acquire stable structures by engaging in protein-protein interactions (PPIs). IDRs can interact with multiple unrelated protein partners and fold differently while interacting with different binding partners16. This disorder-based binding promiscuity of IDRs, acquired via faster rate of evolution at the amino acid level, allow them to play central roles in multiple cellular processes15,17–19. Our analysis reveal that 83% of kinases in the human kinome are hybrid proteins20; i.e., proteins containing both ordered domains and IDRs21. These hybrid kinases are involved in extensive networks of kinase-kinase interactions, potentially driving disease pathogenesis20. Although kinases possess highly structured and conserved KDs, the preponderance and functional consequences of IDRs and their rate of evolution remain unexplored. Most importantly, the regulation of IDRs adjacent to KDs has not been investigated. Therefore, herein, we sought to analyze the human kinome to define spatial and functional relationship between IDRs in kinases with their KD.
First, we predicted the prevalence of IDRs in the kinome and revealed differential degrees of disorder amongst different kinase groups. Second, we calculated the rate of evolution of all kinases, their KDs, and IDRs to identify that IDRs indeed evolve at a much higher rate compared to the KDs and the rest of the proteins. Third, we found that IDR evolution is spatially restricted; IDRs adjacent to KDs evolve slowly as compared to the IDRs away from KDs. Surprisingly, we uncovered 36 kinases that have IDRs within the KD that form elaborate PPI networks, of which FAK1 is topologically the most significant hub kinase. FAK1 IDRs engage in a large number of PPIs, a majority of which are cytoskeletal remodeling proteins with high IDR content. Taken together, our study has uncovered differentially evolving IDRs adjacent to the KDs and highlighted IDR-driven FAK1 pathway involved in cytoskeletal remodeling. Our detailed analysis of regulatory regions in kinases, which are independent of the KD provides a novel approach to understanding the role of IDR in modulating the cytoskeleton. These IDR sites are likely therapeutic targets for treating diseases like cancer and fibrosis which are driven by aberrant cytoskeletal changes associated with pathogenic cell proliferation and invasion.
2. MATERIALS AND METHODS
2.1 Derivation of PPIs and network analysis
Experimentally validated protein-protein interaction (PPI) data of the 36 kinases and that of FAK1 interacting proteins was assembled using manual data curation and various softwares including as described previously20. PPI network was constructed and visualized using Cytoscape22. Network analysis was performed to identify topologically significant hubs from the PPI networks using Network Analyzer23 and CentiScaPe plug in tools24.
2.2 Gene Ontology analysis
Gene ontology analysis was performed with BiNGO25. BiNGO was used to create two networks of molecular functions of proteins that either interacted with ordered regions or IDRs of FAK1. The two networks were merged using Cytoscape option “Merge” (Tools → Merge → Networks). In order to identify unique molecular functions present in the two networks, “difference” operation was selected and a new network was created with molecular functions exclusively present in either of the networks. p-value was calculated using Hypergeometric test and corrected with Benjamini & Hochberg False Discovery Rate. Molecular function terms with p < 0.05 were considered to be significant.
2.3 Defining KD range
Protein kinase domains were manually curated from Uniprot database, which defines the kinase domains either through manual curation of published articles or based on functional and sequence similarity using PROSITE ProRule PRU00159. We also validated the kinase domains of 36 kinases with IDRs extending into their KDs using Interpro, a database of protein sequence analysis and classification. The secondary analysis confirmed the range of the kinase domains found in Uniprot, which provided a smaller range of kinase domains compared to the Interpro database, which increases the confidence of overlapping IDRs and KDs and reduces instances of false positives.
2.4 Rate of evolution of kinases, KDs, and IDRs
The relative rates of evolution for the proteins and their domains were calculated as described earlier26. Ortholog groups were obtain using the OMA Browser27 and alignments were obtained with MAFFT28. The orthologs alignment for the kinases was subdivided into each domain or IDR using the positions in the Human as a reference. An evolutionary distance matrix was obtained for each of these section of alignment (using protdist from PHYLIP 3.69 (ref. 29)) and the distance from any ortholog to the human sequence was averaged. This number is then divided by the average in a matrix that considers the average distances to the orthologs for the same species of all proteins in the human proteome. The resulting rate ratio thus indicates the relative rate of evolution of the protein (or protein domain) to the average human proteins.
2.5 Correlation and significance studies
Rates of evolution of multiple IDRs from a single kinase were averaged to avoid oversampling kinases with multiple IDRs. Rates of evolution of WPs, their KDs, and their IDRs were then compared with each other to determine correlation. Correlations between KD-WP, IDR-WP, and IDR-KD were determined by calculating Pearson correlation coefficient (r) for 414 kinases with IDRs using PEARSON function in MS-EXCEL. Level of significance was assumed to be at or below 0.01 for two-tailed t-test with degree of freedom (df) = 412.
2.6 FAK1 interactome mapping and PPI distribution calculations
Original research articles that had reported all of the FAK1 PPIs were identified and by manual literature search, the protein-regions participating in interaction with FAK1 were identified. Protein interactions involving a structured region of FAK1 were considered to be “Ordered-region interactions (O)”, whereas, interactions involving the IDRs of FAK1 were considered to be “IDR interactions (DO)”. Further, per residue intrinsic disorder prediction of the FAK1-interacting partners was performed to identify whether the regions of interacting partners involved in interaction with FAK1 were disordered. Following calculations were used to identify per residue PPI distribution.
2.7 Disease and function enrichment
Proteins were analyzed using the “Core Analysis” option in the Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, www.ingenuity.com). The default Human proteome from IPA was used as the background dataset. Disease enrichment data was derived by selecting “Diseases and Disorders” from “Customize Chart” functions. Similarly, functional enrichment data was derived by selecting “Molecular and Cellular Functions” and “Physiological Functions” from “Customize Chart” functions. The raw data was used to generate bar graphs using p-values.
2.8 Network Analysis
Core analysis of 36 kinases was performed in IPA using the entire human kinome as a reference dataset. The top two functional networks were merged and the centrality of FAK1 was calculated by determining the total number of edges emanating from FAK1 node. Each edge represents an experimentally validated functional relationship between FAK1 and another protein.
2.9 Multiple sequence Alignment of FAK1
FAK1 protein sequences from different species were retrieved from Ensembl. Alignment feature in Macvector program (MacVector Inc., Apex, North Carolina) was used. We used ClustalW Alignment to perform multiple sequence alignment. Gonnet series matrix was used with open gap penalty of 10.0, extend gap penalty of 0.2, and delay divergent of 30%. We allowed gap separation distance of 4 with residue-specific penalties.
3. RESULTS
3.1 Development of a bioinformatics pipeline that identifies FAK1 as a critical hub-kinase
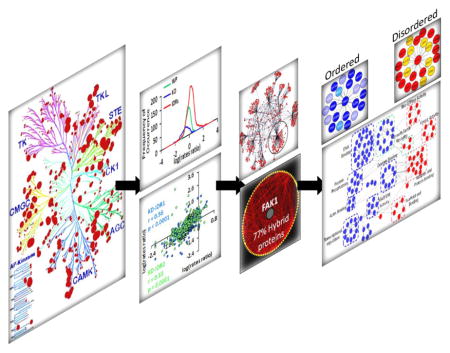
We developed a strategy that first analyzed the predicted disorder in the human kinome to reveal disorder variability between different kinase groups (Fig. 1A). Second, we analyzed the evolution of kinases, KDs, and IDRs to reveal spatial regulation of IDR evolution (Fig. 1B). Third, we identified conserved IDRs flanking the KD as an important feature of kinases, uncovering 36 kinases with IDRs extending into their KDs (Fig. 1C). Fourth, FAK1 emerged as the topologically most central kinase, which engages in multiple PPIs via its IDRs contributing to activation of processes involved in cytoskeletal remodeling (Fig. 1C–F).
Figure 1. Flow chart depicting the approach leading to identification of intrinsically disordered FAK1 and its interacting partners modulating cytoskeletal pathway.

(A) Analysis of disorder prediction of 504 human kinases20 identified prevalence of IDRs adjacent to KDs in 65% of the kinases. (B) Calculation of evolutionary conservation of IDRs revealed that although IDRs evolve at a much faster rate than the whole protein and the KD, the IDRs adjacent to KDs evolve at a significantly slower rate than other IDRs. (C) 36 kinases were identified to have IDRs within the kinase domains. Network analysis of the interactome of 36 kinases identified FAK1 as a topologically most central hub kinase. (D) FAK1 has three IDRs, which are divergent in their sequence, but conserved in their spatial arrangement. (E) 77% of the FAK1 primary interactome is comprised of hybrid proteins. (F) Functional network analysis highlighted cytoskeletal remodeling pathway to be regulated by intrinsically disordered regions present in FAK1 and its interacting partners.
3.2 Differential evolution of IDRs in the kinome highlights correlative evolution of KDs and adjacent IDRs
We analyzed 504 kinases to identify the prevalence of intrinsic disorder in their structure. Moreover, our previous analysis identified that 83% of the kinases (414 out of 504) have at least one IDR20, making the kinome a family of hybrid proteins with a varying degree of disordered residues (Fig. 2A; Table 1 in Ref [30]). Each kinase group is classified by its differential evolution that includes sequence and functional similarity as observed in the kinome dendrogram31. While the evolutionary rate of IDRs is faster than the ordered regions in various protein families32, the relative evolutionary rates of IDRs in the kinome has not been determined. Our analysis of the IDRs, KDs, and the whole protein (WP) revealed that the IDRs evolve at a much higher rate, while the KDs evolve at a much slower rate in comparison to the whole protein (Fig. 2B) and (Fig. 1 and Tables 2, 3 in Ref [30]). Interestingly, we also observed a number of kinases with conserved IDRs, which evolved at lower rates (negative rate ratio). Therefore, we plotted the average evolutionary rates of IDRs with respect to the evolutionary rates of their KDs and WPs, which revealed that highly conserved kinases had relatively conserved IDRs (Fig. 2C, Kinases 1–150; Fig. 1 in Ref[30]) and kinases with relatively neutral evolution (rates approaching zero) had IDRs that were evolving at a rapid rate (Fig. 2D, kinases 251–414; Fig. 1 in Ref[30]).
Figure 2. Differential evolution of IDRs in the kinome highlights 36 kinases with IDRs in their KD.

(A) PONDR-FIT analysis predicts that 25% of the amino acids in the kinome are disordered. An amino is considered disordered if its disorder value is >0.5. Percent disorderliness is overlaid on the kinome dendrogram (cell signaling, Danvers, MA) using KINOMErender34, a visualization tool for overlapping annotations on a phylogenic tree of protein kinases. Each circle represents a kinase. Size of the circle represents percent disorderliness in the kinase. (B) Distribution of the rate of evolution of IDRs, KDs, and WPs reveals that IDRs evolve at a higher rate compared to the KDs and whole kinases. (C) Kinases along with their IDRs and KDs have a varying degree of evolution. Heat-map represents evolutionary rates of 414 kinases (WPs: Whole Proteins), their corresponding KDs (Kinase Domains), and the average rates of their corresponding IDRs. The bar graph demonstrates the average evolutionary rates of IDRs, KDs, and WPs in a window of 50 kinases each. The kinases are sorted from slowest evolving IDRs to fastest evolving IDRs from left to right on the X-axis, respectively.
The landscape of the heat-map (Fig. 2C) suggests that as the average rates of evolution of IDRs in a given kinase increases, the overall rates of evolution for the kinase also increases. To verify this phenomenon, we defined the relationship between evolution of kinases, their KDs, and their IDRs. Our studies revealed that there is a significant correlation between (1) the rate of evolution of an IDR and the KD, (2) the rate of evolution of an IDR and the whole protein (WP), and (3) the rate of evolution of a KD and the WP. This observation suggests that KDs and IDRs in a given kinase evolve in a correlative manner, albeit at different individual rates (Fig. 3A). Therefore, it is likely that a highly conserved protein will have a relatively lower rate of evolution for its IDRs and KDs compared to a highly divergent protein, which will have a higher rate of evolution for its IDRs and KDs.
Figure 3. IDRs, KDs, and WPs evolve in a correlative manner.

(A) Scatter plot of the relationships between the rate of evolution of each kinase (WP) and its KD (KD-WP; blue), each kinase and average rate of all of its IDRs (IDR-WP; green), and KD and average rate of all the IDRs of that kinase (IDR-KD; red). Pearson’s coefficient (r) reveals that there is a significant positive relationship between rate of evolution of WPs and their KDs (blue circles), WPs and their IDRs (green circles), and KDs and corresponding IDRs (red circles) (p < 0.0001). Kinases with at least one IDR were analyzed (n = 414). (B) IDRs adjacent to KDs evolve at a slower rate than IDRs away from the KDs. KD = Kinase Domain; IDR1 = Closest IDR to the KD; IDR2 = second closest IDR to the KD. (C) Analysis of evolutionary rates of KD with IDR1 (Blue) and KD with IDR2 (green) reveal higher correlation between the KDs and IDR1 (r= 0.56) vs KDs and IDR2 (r = 0.33) within the same kinase. 211 kinases containing at least two IDRs were analyzed.
Since our data suggested that IDRs and KDs had correlative evolutionary rates, we tested whether this correlative evolutionary pressure had any preference for IDRs that are found adjacent to the KDs. We analyzed the kinome for spatial arrangement of IDRs within 100 amino acids on either N-terminus or C-terminus of the KD, since the predicted optimal number of residues corresponding to the maximum free energy of unfolding is 100 amino acids33. We find that 65% of the kinases (325 of 504 kinases) have at least one IDR present within 100 amino acids of the KD (Table 4 in Ref [30]). Functional importance of these IDRs can be explained, in part, by their evolution in relation to other IDRs and the KDs. To test the hypothesis that IDRs adjacent to KDs evolve in correlation to the KDs, we analyzed kinases that had at least two IDRs and compared the evolution of IDRs closest to KDs (IDR1) with the evolution of second closest IDRs (IDR2). We identified a total of 211 kinases with at least two IDRs. Interestingly, we found that although both IDR1 and IDR2 evolved at a significantly faster rate than the KD, IDR1 was under greater evolutionary selection pressure and evolved at approximately 6-fold slower rate than IDR2 (Fig. 3B; Tables 2, 3 in Ref [30]). Further, we also identified a correlation between the rates of evolution of KDs and IDR1 compared to the KDs and IDR2 (Fig. 3C). In summary, our analysis reveals that IDRs evolve at a significantly higher rate than the KD and the WP. Further, IDRs evolve at a differential rate based on their spatial arrangement with respect to the KDs.
3.3 Functional network analysis reveals FAK1 as topologically most significant hub
Since 325 kinases were identified to contain IDR1 adjacent to the KD, we further analyzed the spatial arrangement of these IDR1s with respect to their KDs. Strikingly, a unique subset of 36 kinases emerged with IDR1 that were not only adjacent to their KD but also extended into their KD (Fig. 2A and Table 5 in Ref [30]). We hypothesized that these 36 kinases, with peculiar arrangement of IDRs, may participate in unique set of cellular functions. To test this hypothesis, first, we derived experimentally validated PPIs, thereby defining the primary interactome of each of the 36 kinases. A total of 959 proteins were identified to be interacting with these 36 kinases. Considering that IDRs facilitate PPIs, these interactions may be influenced by either direct binding of IDRs or allosteric influence of IDRs facilitating these interactions (Fig. 4A; Table 6 in Ref [30]). Our network analysis identified Focal Adhesion Kinase (FAK1) as topologically the most significant protein in this network using degree centrality as a measure35 (Fig. 4A). In order to confirm the importance of FAK1, in addition to the statistical network analysis, we analyzed functional enrichment of the 36 kinases against the entire kinome. Upon functional network analysis (see Methods), we identified that FAK1 had the highest number of functional relationships with other proteins in the network (Fig. 4A, B, blue edges). Thus, FAK1 was identified as a central hub of the network of 36 kinases using independent statistical and functional approaches.
Figure 4. Functional network analysis of the interactome of 36 Kinases reveals FAK1 as topologically most significant hub.

(A) A primary interactome was derived from 959 experimentally validated PPIs of 36 kinases. The PPI network was visualized using Cytoscape22. Topologically significant hub proteins were determined by calculating degree centrality as a measure (yellow circles). FAK1 was determined to be topologically most significant kinase. (B) In order to identify whether the topologically most significant kinases are functionally important, top two functional networks enriched by 36 kinases when compared to the entire kinome were merged using IPA (Background dataset used: Human Kinome – 504 kinases). Red colored symbols represent proteins that are part of the 36 kinases. The four topologically significant kinases are highlighted with their functional relationships with other proteins in the network, which are depicted as blue lines. FAK1, the most central kinase from (A) had the highest number of relationships within the network. (C) Functional and disease enrichment of 36 kinases using IPA revealed involvement of these kinases in critical cellular and molecular functions (Background dataset: Human Kinome – 503 kinases). Cellular processes related to cytoskeletal remodeling such as cellular morphology and movement were highlighted. Infectious diseases were enriched as the most significant disease. Red dotted line represents the threshold for significance (p<0.05).
Functional enrichment of this subset of the kinome identified various cellular processes such as cell signaling, cell growth and proliferation, and other cytoskeletal remodeling-related functions such as cellular movement and morphology (Fig. 4C). These results support our concept that cells may preferentially utilize these 36 kinases within the kinome to drive a unique set of functions. Importantly, we observe that FAK1 is a central hub kinase amongst the 36 kinases, facilitating cytoskeletal remodeling-related functions such as cellular movement (Fig. 2 in Ref [30]). In summary, our topological and functional analyses highlight FAK1 as a functionally important hub kinase.
3.4 FAK1 IDRs are spatially conserved in evolution
Since we identified FAK1 as a topological and functional hub kinase of the 36 kinases, we sought to determine how FAK1 IDRs can be regulated, which, in turn, can regulate function of FAK1. FAK1 protein structure consists of three domains: FERM domain, KD, and FAT domain. We identified three large IDRs flanking each of the three domains using eight different disorder predictors (Fig. 5A). The middle IDR (IDR1) overlapped with the KD at its C-terminal end (Fig. 5A). Since IDRs adjacent to KDs evolve slowly, we wanted to know whether this was the case for FAK1. To determine the evolutionary sequence conservation of FAK1 IDRs, we performed multiple sequence alignment of FAK1 orthologs from 11 different species ranging from invertebrates to human. Majority of the protein is largely conserved across the nine species from H. sapiens to D. rerio, while C. elegans and L. variegates (invertebrate species) had poor alignment and conservation of the IDR sequences (Fig. 5B). Evolutionary analysis of FAK1, its KDs, and its IDRs reveal that IDR1 is indeed more conserved than the other two IDRs (Fig. 5C), suggesting differential sequence adaptation of IDRs depending on their spatial arrangement within FAK1 protein structure.
Figure 5. FAK1 IDRs are spatially conserved in evolution.

(A) Eight distinct disorder predictors were used to calculate disorder regions with high confidence for the human FAK1 protein. The average disorder score of all predictors is plotted (red line) ±SE as error bars (orange lines). Blue bars represent structured regions of FAK1 and red bars represent IDRs. Broken red bars represent low agreement of disorder between eight predictors. (B) Multiple sequence alignment (MSA) was performed between FAK1 protein sequences from 11 species, ranging from humans to lower invertebrates including sea urchins (echinoderms). MSA scores were calculated using ClustalW in MacVector, a sequence analysis program. Green represents highly conserved amino acid residues while red represents highly divergent amino acid residues. A high degree of disagreement is observed in IDRs, while structured domains are highly conserved. (C) Relative evolution rates of FAK1 domains were calculated to reveal distinct rate of evolution amongst IDRs based on their spatial arrangement with respect to the KD. Whole protein (grey), KD (blue), and each of the three IDRs (red). (D) Disorder scores of FAK1 protein from 11 species ranging from humans to lower invertebrates such as sea urchins were calculated using the eight disorder predictors. Average disorder was calculated for FAK1 protein from each species and plotted as line graph.
Higher evolutionary rate, implied by low conservation, is often associated with lack of conserved function for a given region within the protein. The lack of evolutionary selection pressure allows such regions to evolve at a rapid rate36. However, to test the possibility that FAK1 IDRs are conserved at the spatial level and maintain intrinsic disorder, we hypothesized that despite the poor conservation at the amino acid level, IDRs are spatially conserved across evolution. To test this hypothesis, we predicted intrinsic disorder in all eleven orthologs using the same 8 disorder predictors used earlier in Fig. 5A. Consistent with our hypothesis, we find that IDRs in FAK1 are present in all of the eleven species, including invertebrate species that showed poor alignment of IDR sequences (Fig. 5D). In addition, the positions of IDRs are also relatively conserved, reaffirming the requirement for spatial conservation of IDRs. This spatial conservation of IDRs may enhance structural pliability adjacent to the FAK1 ordered domains, including its KD. Taken together, we reveal that FAK1 IDRs, although less conserved at the sequence level, are spatially conserved.
3.5 FAK1 IDR1 is more enriched in protein binding motifs
Since IDRs can adopt complimentary conformations to lower free energy and enhance PPI repertoire of a protein37, we sought to identify PPI-facilitating motifs within FAK1 IDRs. We used D2P2 database comprising of disordered protein predictions38. Our analysis revealed that FAK1 IDRs are rich in protein binding motifs such as ANCHOR-identified sites and MoRFs (Molecular Recognition Features), which facilitate PPIs through disorder-to-order transition39 (Fig. 6A, B). Of particular interest is the presence of a MoRF between amino acids 397–414, which immediately precedes the KD in the locally disordered linker region. Autophosphorylation of FAK1 at Y397 residue is critical for its activation, leading to interaction with SRC40. Thus, our results further confirm the importance of intrinsic disorder in FAK1 activity. Additionally, our prediction of PTM sites in FAK1 also indicates that FAK1 is selectively modified (via phosphorylation, acetylation, and ubiquitination) within its IDRs by at least 2.80 fold as compared to the structured regions (Fig. 6C; Fig. 2C in Ref [30]), suggesting that FAK1 activity may be modulated via PTMs-enriched IDRs. More importantly, our analysis suggests that KD-flanking IDR1 has a higher propensity to be post translationally modified in comparison to the IDR2 (Fig. 6C).
Figure 6. FAK1 IDRs are enriched in protein binding motifs.

(A) An overlap of ANCHOR and Post Translational Modification (PTM) sites with IDRs present in FAK1 protein revealed that they occur more frequently in IDRs. FAK1 protein sequence was analyzed using D2P2 database, a database of disordered protein predictions. The database predicted intrinsic disorder in FAK1. (B) Prediction of Molecular Recognition Features (MoRFs) in FAK1 protein using MoRFPred revealed enrichment of MoRFs in IDRs. (C) We further analyzed the regulation of IDRs via predicted post translational modifications (PTMs; see methods in Ref. [30]). PTMs were normalized to total of number of amino acids and % PTMs was calculated as described in Ref [30]. 33% of the residues in IDR1, 19% of the residues in IDR2, and 9% of the residues in remaining structured regions are predicted to be post-translationally modified.
3.7 FAK1-interacting proteins are enriched in cytoskeletal remodeling function
Kinases relay their specific cellular functions by interacting with multiple effector proteins. Since IDRs facilitate PPIs, we hypothesized that FAK1 IDRs may play critical roles in FAK1 functions. In order to determine the utility of intrinsic disorder in FAK1 functions, we analyzed its primary interactome of 111 proteins derived from known interactions, of which 86 proteins were identified as hybrid proteins (Fig. 7A; Table 7 in Ref [30]). Functional enrichment utilizing these 86 FAK1 interacting hybrid proteins revealed that they play a major role in influencing cytoskeletal pathway as evident by enriched functions for cellular movement and cell morphology, consistent with the notion that FAK1 is a modulator of cytoskeletal pathway41 (Fig. 3A in Ref [30]). Interestingly, role of FAK1 in mediating cytoskeletal reorganization involves 11 FAK1-interacting hybrid proteins, many of which serve as biomarkers in various cancer types (Fig. 3B in Ref [30]).
Figure 7. IDR-interacting protein partners of FAK1 participate in distinct molecular functions and drive FAK1-mediated cytoskeletal remodeling.

(A) FAK1 primary interactome was visualized using Cytoscape. 86 of 111 protein partners (77% of the interactome) were identified as hybrid proteins. (B) (i) Bar structure of FAK1 shows IDRs and three structured domains (figure not to scale). A total of 45 FAK1-interacting proteins were mapped to different regions of FAK1. (ii) Enrichment of PPIs in IDRs (3.12-fold) was observed. Number of PPIs were normalized to the length of ordered (RO) or disordered regions (RDO). (iii) Interactomes comprising of proteins interacting with ordered regions (Interactome A) or disordered regions (Interactome B) were identified. The color coding of the proteins corresponds to the region of FAK1 each protein interacts with, as depicted in the bar structure. (C) Gene ontology analyses on Interactome A and Interactome B was performed using BiNGO App in Cytoscape and unique molecular functions of Interactomes A and B were identified. Blue circles: molecular functions terms unique to Interactome A; Red circles: molecular functions terms unique to Interactome B. Molecular functions highlighted by green dotted lines represent common functions between the two interactomes. (D) Canonical pathways enriched by Interactome A and Interactome B were used for comparison analysis in IPA. Heat map depicting the −log(p-value) reveals signaling pathways that are specifically enriched via proteins in Interactome A or B. Top 10 canonical pathways are depicted here (Full list: Fig. 6 in Ref [30]). (E) Comparison of the molecular and cellular functions enriched by the proteins in Interactome A or B was performed using IPA. Processes involved in cytoskeletal remodeling had the highest difference between Interactome A and B (denoted by black stars). Red dotted line represents the threshold for significance (p<0.05).
To determine whether the interactomes of these 11 hybrid proteins are also enriched with IDRs, which may contribute to cytoskeletal remodeling, we analyzed the primary interactomes of these 11 hybrid proteins. Our analysis revealed that 76% of the proteins (882 of 1154 proteins) are hybrid proteins (Fig. 4A and Table 8 in Ref [30]). Furthermore, consistent with the role of IDRs in enhancing hub protein functions, there was a high degree of crosstalk within the secondary interactome highlighting multiple hub proteins (Fig. 4B in Ref [30]). In summary, these findings further support the concept that FAK1-mediated cytoskeletal remodeling pathway is replete with hybrid proteins.
3.8 IDR-interacting protein partners of FAK1 participate in distinct molecular functions
The role of intrinsic disorder in scaffolding is well known42 and our previous analyses identified a higher number of hybrid proteins in cytoskeletal remodeling. Therefore, we hypothesized that IDRs play an important role in mediating PPIs between FAK1 and its interacting partners, many of which function as scaffolding proteins in the formation and maintenance of intracellular focal adhesions and subsequent cytoskeletal remodeling. Through literature mining, we mapped a total of 45 proteins to different domains of FAK1. Consistent with our hypothesis, FAK1 utilizes its IDRs to interact with 23 proteins, a 3-fold enrichment when normalized to the total number of disordered and ordered residues of FAK1 (Fig. 7B(ii)). We hypothesized that IDR-interacting proteins may drive a distinct set of molecular functions when compared to the proteins interacting with the ordered regions. Therefore, we analyzed the gene-ontology (GO) terms associated with the proteins interacting with either ordered regions (Interactome A) or IDRs (Interactome B) to derive two functional networks (Fig. 7B(iii), Fig. 5 in Ref [30]).
Comparison of the two networks using Cytoscape22 identified clusters of unique functions driven by each set of proteins (Fig. 7C). Interestingly, IDR-interacting proteins participate in important molecular functions such as kinase activity, scaffolding, adaptor- and protein-binding, and Rho-GTPase activity. To further validate this result, we compared the signaling pathways enriched by proteins of either Interactome A or Interactome B. We revealed that canonical integrin signaling and FAK signaling pathways are primarily driven by proteins of Interactome B (Fig. 7D; Fig. 6 in Ref [30]). Further, when comparing the molecular and cellular functions, cytoskeletal remodeling-related functions such as cellular movement and morphology are preferentially enriched in the subset of proteins participating in IDR-driven interactions (Fig. 7E; Table 9 in Ref [30]). Taken together, these analyses implicate FAK1 IDRs in modulating interactions with proteins that preferentially participate in cytoskeletal remodeling.
4. DISCUSSION and CONCLUSION
The present study reveals that some degree of intrinsic disorder exists across all families of human kinases and that a large number of kinases have IDRs adjacent to their KDs. Interestingly, IDRs adjacent to their KDs evolve at a slower rate, while IDRs further away from KDs evolve at a faster rate. IDRs allow expansion of functional and regulatory repertoire of proteins43. Thus, rapidly evolving IDRs likely help facilitate acquisition of novel kinase functions including newer protein binding partner and substrates, while those adjacent to the KD may likely help fine-tune the catalytic roles of the KD and conserve their overall function. Our studies on evolutionary rates of IDRs with respect to KDs suggest a high correlation between the IDRs and KDs within each kinase. More specifically, there was higher correlation between the evolutionary rates of KDs and adjacent IDRs than the KDs and the spatially distant IDRs. The differential rates of evolution of IDRs based on their spatial arrangement with respect to the KD may have functional consequences. The relatively more conserved IDRs adjacent to the KDs help preserve the high level of substrate specificity of the catalytic domain, while the less conserved spatially distant IDRs may be involved in molecular recognition via their scaffolding activity, improving their biochemical and functional repertoire. This correlative of IDRs and KDs may contribute to enhancing the repertoire and optimization of kinase function, a possibility that should be further tested in future studies.
We identified 36 human kinases as a structurally unique subset of the kinome that have IDRs extending into their KDs. As our analysis suggests, the 36 kinases are part of seven different kinase groups. The “Other” kinase group has the most number of kinases with IDR1 extending into their KDs. The “Other” kinase group is classified as a group of kinases that do not fit into any of the other kinase groups, suggesting their functional dissimilarities with rest of the human kinome. Therefore, we believe that the functional enrichment of the 36 kinases is likely a function of their structural uniqueness.
Our results suggest that the 36 kinases, highlighted by their IDRs extending into the KDs, enrich a subset of functions when compared to the rest of the human kinome. Previous studies have suggested that hybrid proteins preferentially participate in cellular signaling by virtue of having higher degree of post-translational modifications44. It is possible that IDRs extending into the KDs provide a functional advantage to the kinases due to ability of IDRs to allosterically modulate the catalytic activity of a kinase13, thereby positively influencing phosphorylation of the kinase substrates. Consistent with our data, it has also been suggested that intrinsic disorder plays an important role in cytoskeletal remodeling45. Our analyses identify FAK1 as a topological and functional hub in the network of the 36 kinases, consistent with the known role of FAK1 as a master regulator of cytoskeletal remodeling-associated processes such as cellular movement and morphology41. Thus, the 36 kinases highly enrich the cellular signaling and cytoskeletal remodeling-associated functions, which highlights the functional role of the topological hub FAK1 in this network.
Despite the exhaustive protein signaling diversity found in nature, many of the signaling pathways are largely conserved in their functions across evolution. For example, core components of integrin-mediated adhesion pathway have been found in the genome of apusozoan protist Amastigomonas sp., that predates the divergence of metazoans and fungi46. However, it is equally bewildering that Integrin signaling pathway is most significantly regulated by rapidly evolving IDR-interacting proteins in the FAK1 interactome. It is possible that the IDRs, despite high evolutionary divergence, have functional conservation via maintained PPIs across evolution. The PPI motifs of IDRs such as MoRFs are highly locally conserved and dictate conservation of amino acid composition and physicochemical properties in the flanking regions47. Therefore, it is likely that IDR-interacting proteins in the FAK1 interactome can continue to relay highly conserved Integrin signaling pathway through conserved MoRFs while simultaneously adapting to novel PPIs through highly variable amino acids. Although more studies are warranted to further decipher the roles of IDRs in FAK1 signaling, our data provides preliminary evidence that correlative evolution of IDRs with the KD and WP serves two distinct functions: One – IDRs acquire novel functions for the kinase owing to their structural promiscuity; and two – IDRs maintain the kinase function through conservation of sequence motifs utilized for conserved PPIs.
Our analysis points towards unique roles of IDRs based on their spatial arrangement within the kinases. Our findings support the recently emerging notion that IDRs near structured catalytic domains in enzymes provide structural pliability that enhances their catalytic activity48. Here, our analysis identified IDR-mediated regulation of FAK1 activity. Indeed, future studies focusing on IDRs in regulation of catalytic activities of the kinome will help decipher its dynamic modulation in response to kinase inhibitors49. This insight will allow us to ascertain the underlying mechanisms of the acquired resistance to therapeutics, which may be driven by inherent signaling adaptability of the kinome.
While the current method of drug designing requires structural features to target functional regions on kinases, recent advances made in IDR-targeting therapeutics are encouraging. Further, although IDRs naturally do not form structures, they often undergo a disorder-to-order transition to facilitate interaction with a binding partner through conserved MoRFs and ANCHOR sites. As discussed above, these regions represent evolutionarily conserved local motifs and their structures can be potentially elucidated through conventional crystallography techniques when they are bound with their target protein partners. Therefore, targeting these interaction interfaces of kinases will likely interfere with binding partner and abrogate their pathogenic activities. Indeed, such an approach has been taken to interfere with Myc-MAX interaction where small molecules have been designed to specifically disrupt dimerization by targeting their interaction surface within the c-Myc IDR50,51. Likewise, a similar strategy was employed in developing FAK1 inhibitor, Y15. This inhibitor targets a structured region near an IDR causing interference with FAK1 autophosphorylation at Y39752, which lies within a disordered linker region40. In addition, designer peptidomimetics can be used to interfere with the IDR interface or inhibit vital PTMs on IDRs, thereby reducing their ability to engage in a large number of PPIs14. Thus, although IDR-based drug designing remains challenging, the present study provides the impetus to the concept of targeting functionally conserved IDRs. It is hoped that these studies will serve as a platform to develop better therapies taking IDRs into consideration in the future.
Highlights.
Human Kinome has a variable presence of intrinsically disordered regions (IDRs)
IDRs adjacent to the kinase domains observe variable evolutionary pressure
FAK1 is a central hub of a network of Kinases with IDRs adjacent to kinase domain
FAK1 IDRs are spatially conserved across evolution
FAK1 relays cytoskeletal remodeling preferentially through IDR-interacting proteins
Acknowledgments
FUNDING: This research was supported by USF-COM startup funds and the following grants to VD (AHA-SDG-0830101N and 1R21AG047473-01A1).
Footnotes
CONTRIBUTIONS: J.J.K, R.R.P., A.B., and B.X performed all the experiments. A.B. and E.R.M.T. performed evolutionary analysis of the IDRs and kinases. The project was conceived and designed by J.J.K. and V.D. and written by J.J.K. and V.D. with inputs from V.N.U.
COMPETING INTERESTS: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Petretti C, Prigent C. The Protein Kinase Resource: everything you always wanted to know about protein kinases but were afraid to ask. Biology of the cell/under the auspices of the European Cell Biology Organization. 2005;97:113–118. doi: 10.1042/BC20040077. [DOI] [PubMed] [Google Scholar]
- 3.Fabbro D, Cowan-Jacob SW, Moebitz H. “10 things you should know about protein kinases” IUPHAR Review 14. Br J Pharmacol. 2015 doi: 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10:111–126. doi: 10.1038/nrd3252. [DOI] [PubMed] [Google Scholar]
- 5.Dy GK, Adjei AA. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J Clin. 2013;63:249–279. doi: 10.3322/caac.21184. [DOI] [PubMed] [Google Scholar]
- 6.Pal R, Berlow N. A kinase inhibition map approach for tumor sensitivity prediction and combination therapy design for targeted drugs. Pac Symp Biocomput. 2012:351–362. [PubMed] [Google Scholar]
- 7.Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J Clin Oncol. 2011;2:80–93. doi: 10.5306/wjco.v2.i2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Manstein V, et al. Resistance of Cancer Cells to Targeted Therapies Through the Activation of Compensating Signaling Loops. Curr Signal Transduct Ther. 2013;8:193–202. doi: 10.2174/1574362409666140206221931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nature structural & molecular biology. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandarlapaty S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer discovery. 2012;2:311–319. doi: 10.1158/2159-8290.CD-12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller S, Chaikuad A, Gray NS, Knapp S. The ins and outs of selective kinase inhibitor development. Nature chemical biology. 2015;11:818–821. doi: 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- 12.Metallo SJ. Intrinsically disordered proteins are potential drug targets. Current opinion in chemical biology. 2010;14:481–488. doi: 10.1016/j.cbpa.2010.06.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babu MM, Kriwacki RW, Pappu RV. Structural biology. Versatility from protein disorder. Science. 2012;337:1460–1461. doi: 10.1126/science.1228775. [DOI] [PubMed] [Google Scholar]
- 14.Babu MM, van der Lee R, de Groot NS, Gsponer J. Intrinsically disordered proteins: regulation and disease. Current opinion in structural biology. 2011;21:432–440. doi: 10.1016/j.sbi.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Peng Z, et al. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cellular and molecular life sciences : CMLS. 2014 doi: 10.1007/s00018-014-1661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu WL, et al. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein science : a publication of the Protein Society. 2013;22:258–273. doi: 10.1002/pro.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. Journal of biomolecular structure & dynamics. 2012;30:137–149. doi: 10.1080/07391102.2012.675145. [DOI] [PubMed] [Google Scholar]
- 18.Light S, Sagit R, Sachenkova O, Ekman D, Elofsson A. Protein expansion is primarily due to indels in intrinsically disordered regions. Molecular biology and evolution. 2013;30:2645–2653. doi: 10.1093/molbev/mst157. [DOI] [PubMed] [Google Scholar]
- 19.Brown CJ, Johnson AK, Dunker AK, Daughdrill GW. Evolution and disorder. Current opinion in structural biology. 2011;21:441–446. doi: 10.1016/j.sbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kathiriya JJ, et al. Presence and utility of intrinsically disordered regions in kinases. Molecular bioSystems. 2014 doi: 10.1039/c4mb00224e. [DOI] [PubMed] [Google Scholar]
- 21.Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein science : a publication of the Protein Society. 2013;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 24.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25:2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 26.Bezginov A, Clark GW, Charlebois RL, Dar VU, Tillier ER. Coevolution reveals a network of human proteins originating with multicellularity. Molecular biology and evolution. 2013;30:332–346. doi: 10.1093/molbev/mss218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic acids research. 2011;39:D289–294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Briefings in bioinformatics. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 29.Felsenstein J. Mathematics vs. Evolution: Mathematical Evolutionary Theory. Science. 1989;246:941–942. doi: 10.1126/science.246.4932.941. [DOI] [PubMed] [Google Scholar]
- 30.Kathiriya JJ, et al. Data on Evolution of Intrinsically Disordered Regions of the human kinome and Contribution of FAK1 IDRs to Cytoskeletal Remodeling. Data in Brief. 2016 doi: 10.1016/j.dib.2016.11.099. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends in biochemical sciences. 2002;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- 32.Brown CJ, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. Journal of molecular evolution. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 33.Xu D, Nussinov R. Favorable domain size in proteins. Fold Des. 1998;3:11–17. doi: 10.1016/S1359-0278(98)00004-2. [DOI] [PubMed] [Google Scholar]
- 34.Chartier M, Chenard T, Barker J, Najmanovich R. Kinome Render: a stand-alone and web-accessible tool to annotate the human protein kinome tree. PeerJ. 2013;1:e126. doi: 10.7717/peerj.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maslov S, Sneppen K. Specificity and stability in topology of protein networks. Science. 2002;296:910–913. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]
- 36.Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 37.Kim PM, Sboner A, Xia Y, Gerstein M. The role of disorder in interaction networks: a structural analysis. Molecular systems biology. 2008;4:179. doi: 10.1038/msb.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oates ME, et al. D(2)P(2): database of disordered protein predictions. Nucleic acids research. 2013;41:D508–516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Disfani FM, et al. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics. 2012;28:i75–83. doi: 10.1093/bioinformatics/bts209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nature reviews. Molecular cell biology. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 42.Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in scaffold proteins: getting more from less. Progress in biophysics and molecular biology. 2008;98:85–106. doi: 10.1016/j.pbiomolbio.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Latysheva NS, Flock T, Weatheritt RJ, Chavali S, Babu MM. How do disordered regions achieve comparable functions to structured domains? Protein science : a publication of the Protein Society. 2015 doi: 10.1002/pro.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nature reviews. Molecular cell biology. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guharoy M, Szabo B, Contreras Martos S, Kosol S, Tompa P. Intrinsic structural disorder in cytoskeletal proteins. Cytoskeleton. 2013;70:550–571. doi: 10.1002/cm.21118. [DOI] [PubMed] [Google Scholar]
- 46.Sebe-Pedros A, Roger AJ, Lang FB, King N, Ruiz-Trillo I. Ancient origin of the integrin-mediated adhesion and signaling machinery. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10142–10147. doi: 10.1073/pnas.1002257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang C, Noguchi T, Tominaga D, Yamana H. MFSPSSMpred: identifying short disorder-to-order binding regions in disordered proteins based on contextual local evolutionary conservation. BMC bioinformatics. 2013;14:300. doi: 10.1186/1471-2105-14-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larion M, Miller B, Brüschweiler R. Conformational heterogeneity and intrinsic disorder in enzyme regulation: Glucokinase as a case study. Intrinsically Disordered Proteins. 2015;3:1–4. doi: 10.1080/21690707.2015.1011008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graves LM, Duncan JS, Whittle MC, Johnson GL. The dynamic nature of the kinome. Biochem J. 2013;450:1–8. doi: 10.1042/BJ20121456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chemistry & biology. 2008;15:1149–1155. doi: 10.1016/j.chembiol.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Yu C, et al. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Scientific reports. 2016;6:22298. doi: 10.1038/srep22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golubovskaya VM, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. Journal of medicinal chemistry. 2008;51:7405–7416. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]