

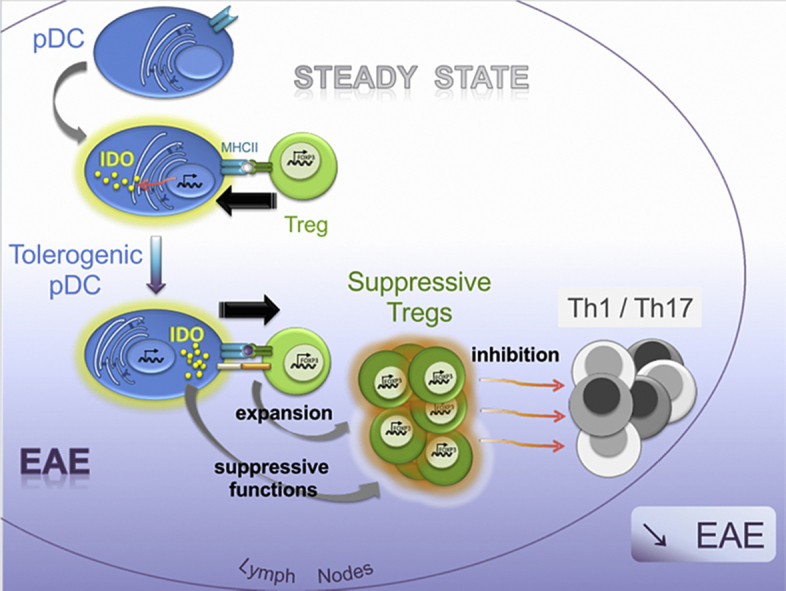
Abstract
Plasmacytoid dendritic cells (pDCs) have been shown to both mediate and prevent autoimmunity, and the regulation of their immunogenic versus tolerogenic functions remains incompletely understood. Here we demonstrate that, compared to other cells, pDCs are the major expressors of Indoleamine-2,3-dioxygenase (IDO) in steady-state lymph nodes (LNs). IDO expression by LN pDCs was closely dependent on MHCII-mediated, antigen-dependent, interactions with Treg. We further established that IDO production by pDCs was necessary to confer suppressive function to Tregs. During EAE development, IDO expression by pDCs was required for the generation of Tregs capable of dampening the priming of encephalitogenic T cell and disease severity. Thus, we describe a novel crosstalk between pDCs and Tregs: Tregs shape tolerogenic functions of pDCs prior to inflammation, such that pDCs in turn, promote Treg suppressive functions during autoimmunity.
Keywords: Regulatory T cells; Plasmacytoid dendritic cells; Indoleamine 2,3-dyoxygenase; Antigen presentation; Tolerance; Experimental autoimmune encephalomyelitis
Abbreviations used: Ag, antigen; cDCs, conventional dendritic cells; BM, bone marrow; CNS, central nervous system; cTECs, cortical thymic epithelial cells; dLN, draining lymph node; EAE, experimental autoimmune encephalomyelitis; IDO, indoleamine-2,3-dyoxygenase; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; pDCs, plasmacytoid dendritic cells
Graphical abstract
Highlights
-
•
IDO expression by LN pDCs is closely dependent on MHCII-mediated, antigen-dependent, interactions with Tregs.
-
•
pDCs are the predominant source of IDO in both steady-state and inflamed lymph nodes.
-
•
IDO production by pDCs is necessary to confer suppressive function to Tregs in EAE.
1. Introduction
Plasmacytoid dendritic cells (pDCs) are important sensors of non-self-nucleic acids derived from bacteria or viruses and are crucial mediators of innate anti-microbial responses through the production of inflammatory cytokines and type-I IFNs [1], [2]. In addition, pDCs have been implicated in the development of several autoimmune diseases, including lupus, psoriasis, multiple sclerosis (MS) and type-1 diabetes [3], [4], [5], [6], [7]. Following abnormal release of self-DNA during inflammatory processes, pDCs are activated through TLR signalling and subsequently produce type-I IFN [8]. Importantly, a few years ago, the notion emerged that pDCs act not only as innate sensors but can also function as bona fide antigen (Ag) presenting cells (APCs) and directly impact T cell responses [9]. It was shown that pDCs capture and process Ags [10], and load antigenic peptides onto MHC class I (MHCI) [11] and MHC class II (MHCII) molecules [12], [13], [14]. The modulation of Ag-presenting pDC functions led to important consequences on T cell immunity, the outcome being highly dependent on the cytokine microenvironment [15].
Many studies, including those investigating oral tolerance and allograft models, suggest that steady-state Ag-presenting pDCs exclusively promote T cell tolerance [16], [17], [18]. Although the nature of the factors controlling distinct pDC functions remains to be established, once activated, pDCs exhibit both immunogenic and tolerogenic functions. For example, using mice exhibiting a specific loss of MHCII expression by pDCs, we showed that CpG-B activated pDCs present Ag and promote effector Th17 cell differentiation, a property that can be exploited for anti-tumor vaccines [19]. Pro-pathogenic Ag-presenting pDCs were similarly described in a mouse model of atherosclerosis in which pDCs induced pathogenic Th1 cells [20]. In addition, BST-2 mediated specific Ag delivery to CpG-activated pDCs led to cytotoxic T lymphocyte (CTL) and Th1 cell differentiation and triggered protective immunity against viral infection and tumor growth [21]. In contrast, in the context of EAE, Ag targeting to pDCs via Siglec-H promoted CD4+ T cell anergy and inhibited CNS inflammation [22]. We previously demonstrated that in EAE, pDCs present myelin Ags on MHCII molecules to induce the expansion of suppressive Tregs, a phenomenon correlated with disease amelioration [23].
Indoleamine 2,3-dioxygenase (IDO) is an immunomodulatory enzyme involved in the initial and the rate-limiting step of tryptophan catabolism. Upon inflammation, IDO production has been shown to compromise T cell proliferation, promote T cell anergy and Tregs [24], [25], [26]. Depending on the experimental context, IDO can be induced either by IFN-γ, IFN-α/β, or TGF-β. CTLA-4 binding to cell-surface expressed costimulatory molecules promotes IDO production by pDCs through IFN-γ or IFN-α/β signalling [27], [28], [29], [30]. Furthermore CD200-Ig binding to his cognate receptor induces IDO in an IFN-α/β dependent signalling pathway [31]. Both IFN-γ and IFN-α/β pathways result in IDO+ immunosuppressive effects which are closely dependent on the catalytic activity of the enzyme.
CTLA-4-binding also promote IDO in tumor contexts, but the enzyme has reveal activity in only a minor DC subpopulation expressing the marker CD19, but none of the pDC classical markers [32], [33]. IDO enzymatic functions in tumor dLN-sorted pDCs have been correlated to in vitro Treg differentiation and suppressive functions [24], [34]. More recently, Pallotta and colleagues described that IDO+ pDCs induced long-lived Tregs by using a TGF-β-dependent pathway distinct from the catalytic activity of the enzyme. In mouse a model of skin delayed-type hypersensitivity, they shown that whereas IFN-γ-dependent IDO enzymatic activity in pDCs leads to T cell anergy, TGF-β induced IDO phosphorylation results in increased Treg frequencies [35].
It is so far unknown whether IDO expression in naïve pDCs pre-exists, and how it would be regulated in steady-state LNs. In contrast, recent work has implicated IDO expression in pDC immunoregulatory functions, including Treg induction, in inflamed LNs. Furthermore, IDO production by tumor-associated pDCs has been correlated to in vitro Treg-mediated suppression. However, the nature of the cells expressing IDO, as well as the impact on Treg functions in chronic inflammatory diseases, such as autoimmune disorders, remain undetermined.
Here we show that in steady-state lymph nodes (LNs), IDO is highly expressed by pDCs compared to other LN resident cells. We further established that IDO expression is positively regulated in steady-state pDCs following MHCII-mediated interactions with Tregs. During autoimmune disorders, such as EAE, IDO expression by MHCII competent pDCs is mandatory to confer suppressive functions to pDC-induced Tregs. IDO-competent Ag-presenting pDCs promote Tregs that inhibit autoimmune effector T cell responses in LNs, resulting in reduced disease severity. Therefore, we have identified a bidirectional interaction between pDCs and Tregs that favours self-tolerance.
2. Materials and methods
2.1. Mice
All mice had a pure C57BL/6 background and were bred and maintained under SPF conditions at Geneva medical school animal facility and under EOPS conditions at Charles River, France or at the National Institutes of Health, Bethesda, US. DEREG [36], Ubiquitin-eGFP [37], pIII + IV−/− [38], IDO−/− [39], BDCA2-DTR [40], MARILYN Rag2−/−, OTII Rag2−/− [41], AND Rag2−/− [42], SMARTA Rag1−/− [43], Rag2−/−, Scurfy [44], CD45.1 (Charles River, France), and 2D2 [45] mice have been previously described. WT C57BL/6 mice were purchased from Harlan laboratories (France) or Taconic (US). All procedures were approved by and performed in accordance with the guidelines of the animal research committee of Geneva or of the NIH.
2.2. Generation of BM chimeric mice
BM chimeric mice were generated as described [19]. Briefly, BM cells were recovered from tibia and femurs of donor mice. 5 to 7 × 106 cells were injected intravenously into sub-lethally irradiated recipient mice (two consecutive doses of 450 cGy). Reconstitution was assessed by analysing blood cells by flow cytometry after 6–8 weeks. For mixed BM chimeras, CD45.2 WT eGFP and CD45.2 pIII + IV−/− BM cells were simultaneously transferred into irradiated CD45.1 WT recipient mice in a 1:1 ratio.
2.3. EAE experiments
Active EAE was induced by immunizing mice, subcutaneously in both flanks, with 100 μg of MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK, Biotrend) emulsified in incomplete Freund's adjuvant (BD Diagnosis) supplemented with 500 μg/ml Mycobacterium tuberculosis H37Ra (BD Diagnosis). At the time of immunization and 48 h later, mice also received 300 ng of pertussis toxin (Sigma-Aldrich) into the tail vein. Mice were monitored daily for disease clinical symptoms, and blindly scored as follows. 1, flaccid tail; 2, impaired righting reflex and hind limb weakness; 3, complete hind limb paralysis; 4, complete hind limb paralysis with partial fore limb paralysis; 5, moribund.
For passive EAE induction, encephalitogenic CD4+ T cells were generated in vitro from LN and spleen cells of 2D2 mice as described [46]. 1–2 × 106 total cells were injected i.p. into recipient mice. Mice received 67 ng of pertussis toxin at the day of cell injection and 48 h later. Mice were monitored daily for disease clinical symptoms as described above.
Adoptive transfers of Treg cells were performed as follows. WT → WT, pIII + IV−/− → WT and IDO−/− → WT chimeric mice were immunized or not with MOG35–55 + CFA. CD4+ CD25hi T cells were harvested from total skin LNs (naïve) or dLNs (day 10 after EAE immunization) and 1–5 × 105 CD4+ CD25hi T cells were injected intravenously into tail vein of recipient mice. EAE was induced by active immunization the day after Treg transfer. In some experiments, CD45.1 mice were used as recipients, and Treg migration was assessed in dLNs at day 3 and in dLNs and SC at day 15 after EAE immunization.
Adoptive transfers of pDCs were performed as follows. 1.2–1.5 × 106 BM derived pDCs loaded with 10 μg/mL of MOG35–55 were injected intravenously into tail vein of recipient mice, and EAE was induced by active immunization the day after.
In some experiments, EAE mice were treated i.p. at indicated time points with DT (100 ng/mouse for BDCA2-DTR and 1 μg/mouse for DEREG).
2.4. Ex vivo cell isolation
Treg cells were isolated from total skin LNs of naïve mice or from dLNs of EAE mice (day 10 after immunization). LNs were scratched and LN cells were subjected to CD4+ T cell enrichment using CD4+ T cell isolation kit (Miltenyi biotec). CD4+ CD25hi Treg cells were next sorted using a MoFlowAstrios (Beckman Coulter).
For qPCR experiments, pDCs, cDCs, B cells and macrophages were recovered from LN after digestion with an enzymatic mix containing collagenase D (1 mg/mL) and DNAse I (10 μg/mL) (Roche) in HBSS. B cells were isolated using anti-CD19 beads (Miltenyi Biotech), macrophages, cDCs and pDCs were then purified, after CD3+ cells depletion (Miltenyi Biotech), by flow cytometry as CD11c−CD11b+Ly6C+ for macrophages, CD11c+PDCA1- for cDCs and CD11cintPDCA1+SiglecH+ for pDCs, using a MoFlowAstrios (Beckman Coulter) or BD FACSAria (BD Biosciences).
2.5. In vitro BM derived DC generation
pDCs were generated from BM of WT, pIII + IV−/− and IDO−/− mice as previously described [23]. Briefly, BM cells were recovered from tibia and femurs of mice and cultured, after red cell lysis, for 7 days in complete RPMI medium (10% heat-inactivated fetal bovine serum, 50 mM 2-βMercaptoethanol, 100 mM sodium Pyruvate and 100 μM of Penicillin/Streptomycin) supplemented with 100 ng/mL of murine Flt3L (PeproTech).
cDCs were generated as pDCs from BM of WT mice, by supplementing complete RPMI medium with 20 ng/ml granulocyte-macrophage colony-stimulating factor (PeproTech), and activated for 24 h with 5 ng/ml LPS (Enzo Life Sciences, Inc.).
2.6. Co-cultures
CD4+ T cells were recovered from scratched total skin LNs of 2D2 mice and purified with CD4+ T cell isolation kit (Miltenyi Biotec) according to manufacturer's instructions. Both cell types purity was assessed by flow cytometry using a Cyan™ ADP (Beckman Coulter) and exceeded 90%.
WT and Rag2−/− pDCs were recovered from total skin LN and spleen after digestion with the enzymatic mix described above. pDCs were isolated using the Plasmacytoid Dendritic Cell isolation kit II (Miltenyi Biotec) according to manufacturer's instructions. pDCs were loaded or not with MOG35–55 peptide (10 μg/mL). 300 000 pDCs were seeded in 48 well plates with 200 000 2D2 CD4+ T cells. Cells were co-cultured in complete RPMI medium for 16 h and pDCs were isolated again using the Plasmacytoid Dendritic Cell isolation kit II (Myltenyi Biotech) according to manufacturer's instructions.
WT BM-derived pDCs were generated in vitro for 7–8 days and purified using the Plasmacytoid Dendritic Cell isolation kit II (Miltenyi Biotec) according to manufacturer's instructions. Cell purity was assessed by flow cytometry using a Cyan™ ADP (Beckman Coulter) and exceeded 90%. pDCs were loaded or not with MOG35–55 peptide (10 μg/mL). 500 000 pDCs were seeded in 24 well plates with 300 000 2D2 CD4+ T cells or 300 000 pDCs were seeded in 48 well plates with 100 000 2D2 CD4+ T cells or 100 000 2D2 CD4+ CD25− cells or 100 000 2D2 CD4+ CD25+ cells. Cells were co-cultured in complete RPMI medium for 16 h and pDCs were isolated again using the Plasmacytoid Dendritic Cell isolation kit II (Miltenyi Biotec) according to manufacturer's instructions.
In vitro Treg suppressive assays were performed as follows. WT → WT and IDO−/− → WT chimeric mice were immunized or not with MOG35–55 + CFA. CD4+ CD25hi T cells were harvested from total skin LNs (naïve) or dLNs (day 10 after EAE immunization) and 17 000 cells were incubated with 50 000 proliferation dye-labeled 2D2 CD4+ T cells (Treg:2D2, ratio 1:3) and 50 000 LPS activated, MOG35–55 loaded, BM derived cDCs for 5 days. 2D2 T cell proliferation was assessed by flow cytometry. The percentage of Treg-mediated suppression was related to 0% of proliferation (relates to 100% of suppression) in absence of cognate MOG35–55 peptide.
2.7. Flow cytometry
Monoclonal antibodies used for flow cytometry were from: Biolegend; anti-CD11c (N418), anti-Ly6C (HK1.4), anti-Vβ11 (KT11), anti-CD45.2 (104); from eBioscience: anti-CD4 (GK1.5 and RM4-5), anti-CD69 (H1.2F3), anti-CD25 (PC61.5), anti-IL10 (JES5-16E3) anti-TER119 (TER-119), anti-Foxp3 (FJK-16s), anti–IL-17 (ebio17B7), anti-IFN-γ (XMG1.2) anti-Siglec-H (ebio440c), anti-Ki67 (SolA15), anti-CD45.1 (A20), anti-CD11b (M1/70), anti-CD11c (N418), anti-TCRβ (H57-597), anti-ICOS (C398.4A), anti-CD103 (2E7), anti CD16/32 (93), anti-CD5 (53–7.3); from BD: anti-CD25 (PC61), anti-CD19 (1D3), anti-CD3 (145-2C11), anti-CD4 (RM54-5), anti-Ki67 (B56) and anti–IFN-γ (XMG1.2). Cell proliferation was assessed using CellTrace™ Violet Cell Proliferation Kit (ThermoFisher Scientific). Gating on viable cells was performed using the Fixable Viability Dye eFluor® 780 (eBioscience).
For flow cytometry analysis, single cell suspensions were incubated with FcBlock (anti-CD16/32 FcγRII-RIII) for 10 min, at 4 °C and stained with antibodies. Intracellular cytokine stainings were done using the Intracellular Fixation & Permeabilisation buffer set (eBioscience) or with the Fix & Perm kit (BD Biosciences) for IL-10. Cell proliferation was assessed by flow cytometry using anti–mouse Ki67 and respective isotype control (Rat IgG2a, kappa). For IFN-γ, IL-17 and IL-10 staining, cells were re-stimulated in complete RPMI containing PMA/ionomycin, and incubated 4 h at 37 °C, 5% CO2. Golgi stop solution (BD Biosciences) was added to the last 2.5 h of culture. Data were acquired with a Cyan™ ADP or a Gallios (Beckman Coulter) and analysed using FlowJo software (FlowJo company). cDCs were defined as CD11chiPDCA-1- and pDCs as CD11cintPDCA-1+ or CD11cintSiglec-H+. B cells and macrophages were defined respectively as CD19+ and CD11b+Ly6C+.
2.8. Quantitative RT-PCR
Total RNA was isolated and prepared with TRIzol reagent (Invitrogen) and RT-PCR were performed as described [23]. cDNA was synthesized with random hexamers and M-MLV Reverse Transcriptase (Promega). PCR were performed with CFX Connect Real-time System (Bio rad) and iQ SYBR green Super-mix (Bio-Rad Labolatories). GAPDH mRNA was used for normalization. Primer sequences were as follows: IDO, forward, 5′-GGG ATG ACG ATG TTC GAA AG-3′ and reverse 5′-CAG GAC ACA GTC TGC ATA AG-3′; GAPDH, forward, 5′- CCC GTA GAC AAA ATG GTG AAG -3′ and reverse 5′- AGG TCA ATG AAG GGG TCG TTG -3′.
2.9. Statistics
Significance was assessed by two-tailed Mann-Whitney test or by one-way ANOVA with Bonferroni post Hoc test. EAE incidence was analysed using two-way ANOVA with Bonferroni post Hoc test. All statistical analyses were done using Prism 5.0 software (GraphPad Software). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS = Non significant.
3. Results
3.1. IDO expression by pDCs in steady-state LN is dependent on MHCII restricted Ag-specific interactions with Treg
We analysed IDO mRNA expression in different cell subtypes sorted from steady-state LNs. IDO mRNA was expressed to far greater levels (>5 fold) by pDCs (CD11cintPDCA-1+), than by cDCs (CD11chiPDCA-1−), B cells (CD11c−CD19+), and macrophages (CD11c−CD11b+LyC6int/+) (Fig. 1A). Consistently, IDO preferential expression by steady-state LN pDCs compared to cDCs was also observed in Balb/c mice, ruling out a mouse C57BL/6 strain restricted effect (Supplementary Fig. 1A). IDO expression by pDCs was shown to be induced by T-cell produced cytokines, such as IFN-γ and TGF-β [35], [47]. To determine whether, in steady state, T cells are involved in the regulation of IDO expression by pDCs, we measured IDO mRNA in Rag2−/− mice, which are devoid of T cells. Compared to WT mice, pDCs from steady-state LNs of Rag2−/− exhibited a significant reduction of IDO mRNA, which was in fine comparable to levels obtained in IDO−/− pDCs (Fig. 1B). We next tested whether interactions with T cell were required for IDO induction in pDCs of naïve mice. We examined IDO expression in pDCs from LNs of mice expressing a monoclonal population of CD4+ T cells specific to OVA peptide, OT-II Rag2−/− mice. IDO expression was significantly reduced and was comparable to what was seen in Rag2−/− mice (Fig. 1B). Thus, OT-II transgenic CD4+ T cells were insufficient at inducing IDO in pDCs.
Fig. 1.

IDO expression by pDCs is induced after Ag-specific interactions with Tregs. (A) IDO mRNA expression in B cells, cDCs, pDCs and macrophages (MØ) sorted from total skin LNs of naïve WT. (B) IDO mRNA in pDCs isolated from total skin LNs of naive WT, Rag2−/−, OTII Rag2−/− and IDO−/− mice. (C) CD5 expression levels by AND Rag2−/−, Smarta Rag1−/− (SMA) and Marilyn Rag2−/− (MAR) CD4+ TCR tg T cells (left), and IDO mRNA levels in pDCs isolated from skin LN of AND, SMA, MAR and WT mice (right). (C) Foxp3+CD4+ Treg frequencies in LNs of OTII Rag2−/− and female Marilyn Rag2−/− mice. (E) Foxp3+CD4+ Treg frequencies in LN cells of 2D2 TCR tg mice (left), and IDO mRNA levels in pDCs isolated from skin LN of Rag2−/− mice and co-cultured in vitro with 2D2 TCR tg CD4+ T cells and MOG35–55 peptide for 16 h (right). (F) IDO mRNA levels in ex-vivo pDCs sorted from skin LN of 3wk-old WT, pIII + IV−/− and Scurfy mice. (G) IDO mRNA levels of ex vivo WT pDCs and WT BM derived pDCs loaded or not with MOG35–55 and co-cultured in vitro with 2D2 CD4+ T cells for 16 h. (H) IDO mRNA levels of ex vivo WT pDCs and WT BM derived pDCs loaded with MOG35–55 and co-cultured in vitro for 16 h with 2D2 CD4+ CD25− cells, 2D2 CD4+ T cells or 2D2 CD4+ CD25+ Treg cells. (A–H) Results are representative of at least 2 independent experiments. Error bars depict mean ± SEM. (A, B, C, G, and H) One-way ANOVA with Bonferroni post Hoc test or (E and F) two-tailed Mann-Whitney test was used. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS = Non significant.
This result might be explained by a low TCR self-reactivity for MHCII molecules of the OTII transgenic CD4+ T cells [48]. To test the possibility that distinct TCR self-reactivity would exhibit differential ability to induce IDO expression in pDCs, we isolated pDCs from different CD4+ TCR transgenic Rag1−/− mice that express distinct levels of CD5, which reflects clone-specific strength self-reactivity [48]. Each TCR transgenic T cell population display a specific surface amount of CD5 that covered a range from low (Marilyn, TCR specific for HY), medium (Smarta, TCR specific for LCMV), to high (AND, TCR specific for Pigeon Cytochrome c) (Fig. 1C). IDO mRNA expression was reduced in LN pDCs isolated from all TCR transgenic Rag−/− mice compared to WT mice (Fig. 1C), showing that IDO induction in pDCs is independent of TCR self-reactivity. An alternative hypothesis which might explain why IDO expression in negligible is a potential specific requirement of Tregs in regulating IDO expression in pDCs, this population being almost absent in LNs of the TCR transgenic T cell mice tested (Fig. 1D). Consistently, MOG35–55-loaded Rag2−/− pDCs, exhibit a significant restoration of IDO expression when co-cultured with 2D2 TCR tg CD4+ T cells, which contain a low but significant frequency of Foxp3+ cells (Fig. 1E). Moreover, pDCs isolated from LNs of Scurfy mice, which are devoid of Foxp3+ Tregs [44], presented a significant reduction of IDO mRNA expression compared to WT pDCs (Fig. 1F). Thus, our data suggest that Tregs significantly contribute to induce IDO expression by pDCs in steady-state LN. We next evaluated whether Ag-specific pDC-T cell contacts were required to promote IDO expression in pDCs. To do so, cultures were done using in vitro generated BM-derived pDCs, for which IDO mRNA levels were found to be negligible, reinforcing the idea that a crosstalk with T cells is indeed required for pDCs to competently express IDO (Fig. 1G). When co-cultured with 2D2 TCR tg CD4+ T cells, a modest increase in IDO mRNA expression was observed in BM-derived pDCs (Fig. 1G). IDO expression was further increased by 2 fold when pDCs were previously loaded with MOG35–55 peptide, reaching similar levels of expression as pDCs isolated from WT LNs (Fig. 1G), Therefore, MHCII-restricted Ag-specific interactions with CD4+ T cells significantly contribute to the induction of IDO expression in pDCs. To test a role for Tregs in this process, we repeated co-culture experiments using 2D2 TCR tg CD4+ T cells that were separated into in CD25hi and CD25neg populations. While 2D2 TCR tg CD4+CD25neg cells were incompetent at inducing IDO, 2D2 TCR tg CD4+CD25hi cells significantly enhanced IDO mRNA levels in pDCs loaded with MOG35–55 (Fig. 1H), confirming that Tregs are mandatory at promoting IDO expression in pDCs. pDCs co-cultured with total 2D2 TCR tg CD4+ T cells exhibit a slight, but not significant increase in IDO mRNA levels compared to pDCs co-cultured with 2D2 TCR tg CD4+CD25hi cells, suggesting that although Tregs are required to promote IDO, non-Treg CD4+ T cells might also contribute to this process (Fig. 1H).
To clearly demonstrate that MHCII-restricted Ag specific interactions between pDCs and T cells are necessary to induce IDO in pDCs, we used genetically deficient mice that selectively lack MHCII expression by pDCs. These mice have been described before and are deficient for the promoters III and IV (pIII + IV) of CIITA, the master regulator for MHCII expression [38]. In mice, CIITA is under the control of cell specific promoters, pI, pIII and pIV [49], [50] (Supplementary Fig. 2). Absence of pIV leads to MHCII abrogation on cortical thymic epithelial cells (cTECs), resulting in the lack of CD4+ T cell positive selection. To restore CD4+ T cell thymic positive selection by MHCII competent cTECs, bone marrow (BM) cell precursors from pIII + IV−/− mice need to be injected into irradiated WT recipients (pIII + IV−/− → WT) and compared to WT → WT controls. pIII + IV−/− → WT mice exhibiting genetic deficiencies compared to WT → WT, it is possible that distinct immunological environments will affect IDO expression by pDCs. Therefore, to immerse MHCII competent and deficient pDCs in an identical milieu, we performed mixed BM chimeric mice using BM cells from Ubi-eGFP WT and pIII + IV−/− mice (ratio 1:1) that were co-injected into irradiated recipient mice expressing the congenic marker CD45.1 (Fig. 2A). LN cells were sorted 2 months later as pDCs and cDCs from donor BM cells (gated on CD45.2+) and further separated as WT (eGFP+) or pIII + IV−/− (eGFP−) cells (Fig. 2A and B). We confirmed that cDCs expressed little IDO mRNA (Fig. 2C). IDO expression by MHCII deficient pDCs was impaired compared to MHCII competent pDCs in mixed BM chimeric mice (Fig. 2C). Since cells were isolated from the same LNs, decreased IDO expression in absence of MHCII expression by pDCs was not related to different cytokine expression profiles, but linked to a defective MHCII expression by pDCs. An alternative explanation is that CIITA directly acts as a transcription factor regulating IDO gene expression. However, we observed a similar reduction of IDO mRNA in pDCs isolated from H2-Db−/− mice (not shown), ruling out this hypothesis. Altogether, our data demonstrated that MHCII-restricted Ag specific interactions with Tregs are required for the induction of IDO expression by pDCs in steady-state LNs.
Fig. 2.

IDO expression by pDCs is dependent of MHCII-TCR interactions. (A-C) Mixed BM chimeric mice were generated by co-transferring (1:1) CD45.2 GFP+ WT and CD45.2 pIII + IV−/− BM cells in lethally irradiated CD45.1 WT mice. (A) Experimental design. (B) WT GFP+ cDCs and pDCs, and pIII + IV−/− cDCs and pDCs were sorted from total skin LN of naïve mice based on CD45.2, CD11c, PDCA-1 and GFP markers. (C) IDO mRNA expression level in indicated cells is represented. Results are representative of at least 3 independent experiments. Error bars depict mean ± SEM. One-way ANOVA with Bonferroni post Hoc test was used. **P < 0.01, NS = Non significant.
3.2. IDO expression in LNs is restricted to pDCs during EAE, and is impaired in MHCII−/− pDCs
We next evaluated whether, as in steady-state LNs, pDCs remains the major expressors of IDO in LNs in a model of chronic inflammation. IDO has been shown to exert a protective role in EAE using either IDO−/− mice or IDO blocking antibodies [51], [52]. Therefore, we have quantified IDO mRNA expression in different cell types sorted from LNs draining the site of EAE immunization (day 10) in wild type (WT) mice. IDO was predominantly (>5 fold) expressed by pDCs compared to other LN cells (Fig. 3A). Levels of expression were comparable to steady-state LN pDCs (Supplementary Fig. 3A). Next, we analysed IDO expression by MHCII deficient pDCs during EAE. pDCs and cDCs were sorted from draining LNs of WT → WT and pIII + IV−/− → WT mice 10 days after EAE induction. Again, cDCs expressed little IDO mRNA (Fig. 3B). We observed a substantial reduction in IDO expression by pDCs sorted from pIII + IV−/− → WT compared to WT → WT chimeras (Fig. 3B). Differential IDO expression by MHCII competent and MHCII deficient pDCs might be explained by distinct inflammatory cytokinic environments in WT → WT and pIII + IV−/− → WT mice, as the latter developed more severe EAE [23]. Therefore, we performed mixed BM chimeric mice using as before BM cells from Ubi-eGFP WT and pIII + IV−/− mice (ratio 1:1) that were injected into irradiated CD45.1 recipient mice. EAE was induced in mixed BM chimeric mice, and LN cells were sorted 10 days after immunization as pDCs and cDCs from donor BM cells (gated on CD45.2+) and further separated as WT (eGFP+) or pIII + IV−/− (eGFP−) cells. IDO expression was significantly impaired in MHCII deficient compared to MHCII competent pDCs isolated from LN of EAE mice (Fig. 3C). Therefore, decreased IDO expression in absence of MHCII expression by pDCs was not related to different cytokine expression profiles between WT → WT and pIII + IV−/− → WT chimeras during EAE, but rather linked to a defective MHCII expression by pDCs. Altogether, our data suggest that IDO expression by LN pDCs is dependent on their expression of MHCII molecules, independent on the inflammatory status of the mice, and reflects a more general regulation of IDO protein expression at the mRNA level.
Fig. 3.

MHC-II sufficient pDC express IDO during EAE. (A) IDO mRNA expression in B cells, cDCs, pDCs and macrophages (MØ) sorted from dLNs of WT EAE mice 10 days after immunization. (B) EAE was induced in WT → WT and pIII + IV−/− → WT chimeric mice, and IDO mRNA was measured in cDCs and pDCs sorted from dLN 10 days after immunization. (C) Mixed BM chimeric mice were generated by co-transferring (1:1) CD45.2 GFP+ WT and CD45.2 pIII + IV−/− BM cells in lethally irradiated CD45.1 WT mice. WT GFP+ cDCs and pDCs, and pIII + IV−/− cDCs and pDCs were sorted from dLNs 10 days after EAE induction based on CD45.2, CD11c, PDCA1 and GFP markers. IDO mRNA expression level in indicated cells (A–C) Results are representative of at least 3 independent experiments. Error bars depict mean ± SEM. One-way ANOVA with Bonferroni post Hoc test was used. *P < 0.05, ****P < 0.0001, NS = Non significant.
3.3. IDO deficiency leads to exacerbated encephalitogenic T cell priming
To investigate the role of pDC-induced Treg in EAE pathogenesis, we have induced EAE in mice lacking Treg and MHCII expression by pDCs. We backcrossed DEREG mice, in which Foxp3+ Tregs can be selectively depleted using diphtheria toxin (DT) injection [36], with pIII + IV−/− mice and generated DEREGxpIII + IV−/− → WT chimeras (Supplementary Fig. 4 A–C). In untreated mice, as previously described [23], EAE was exacerbated (Supplementary Fig. 4D) when pDCs did not express MHCII. Moreover, EAE clinical course was similarly aggravated in DEREG → WT treated with DT and DEREGxpIII + IV−/− → WT chimeras injected or not with DT (Supplementary Fig. 4D). Encephalitogenic Th1 and Th17 CD4+ T cell frequencies were increased in DT-injected DEREG → WT and DEREGxpIII + IV−/− → WT mice compared to un-injected DEREG → WT mice (Supplementary Fig. 4E). This demonstrates that the depletion of Tregs and the absence of MHCII expression by pDCs similarly enhance pathogenic T cell priming and disease severity. Altogether, our data show that pDC-instructed Tregs inhibit the priming of encephalitogenic T cells in LNs during EAE.
We next evaluated whether IDO was implicated in the regulation of encephalitogenic T cell priming during EAE. Knowing that MHCII deficient pDCs exhibit impaired IDO expression both in steady-state and during EAE (Fig. 2, Fig. 3C), we analysed EAE development in WT → WT, pIII + IV−/− → WT and IDO−/− → WT chimeric mice. As before, disease was exacerbated in mice lacking MHCII on pDCs (pIII + IV−/− → WT) compared to control WT → WT animals (Fig. 4A). This could be consequent to either an absence of Ag-presentation by pDCs and/or a resulting defect in IDO expression by pDCs from pIII + IV−/− → WT mice. Consistently with previously published data [51], [52], mice deficient for IDO (IDO−/− → WT) developed aggravated EAE compared to WT controls. Importantly, clinical scores of IDO−/− → WT were comparable to mice lacking MHCII on pDCs (Fig. 4A). Notably, we did not notice any significant impact of IDO deficiency during EAE induced by transferring MOG35–55-specific 2D2 CD4+ effector T cells (Fig. S5), suggesting a critical role for IDO in the modulation of pathogenic T cell priming in SLOs during EAE. To confirm this hypothesis, we measured effector cytokine production by T cells before clinical symptom appearance (day 9) in IDO−/− → WT, pIII + IV−/− → WT and WT → WT chimeras. Elevated IFN-γ and IL-17 producing encephalitogenic CD4+ T cell frequencies were observed in LNs of IDO deficient mice (IDO−/− → WT) and mice lacking MHCII expression by pDCs (pIII + IV−/− → WT) compared to WT → WT controls, (Fig. 4B). Foxp3+CD25hi Treg proliferation was impaired in pIII + IV−/− → WT mice, whereas it was unaffected in IDO−/− → WT mice (Fig. 4C, left), showing that MHCII-mediated Ag presentation, but not IDO expression by pDCs, promoted Foxp3+ Treg proliferation. The proliferation of Foxp3+ Tregs exhibiting a suppressive phenotype (CD103+ICOS+) [53] (Fig. 4C middle), as well as the frequency of IL-10 expressing Foxp3+ Tregs (Fig. 4C right), were significantly decreased in LNs from both pIII + IV−/− → WT and IDO−/− → WT compared to WT → WT mice. Our results identify a new role for IDO in impacting the ability of Tregs to suppress encephalitogenic T cells in LNs.
Fig. 4.

IDO is required during EAE priming phase (A-C) EAE was induced in WT → WT (■), pIII + IV−/− → WT (○) and IDO−/− → WT ( ) BM chimeras. (A) Clinical scores were followed daily (two-way ANOVA with Bonferroni post Hoc test). Frequencies of (B) IFN-γ+ (left) and IL-17+ (right) CD4+ T cells, (C) Ki67+ (left), Ki67+CD103+ICOS+ (middle) and IL10+ (right) among CD25hiFoxp3+CD4+Tregs in dLN at d9 (one-way ANOVA with Bonferroni post Hoc test). (A–C) Results are representative of at least 3 independent experiments with 8 mice per group. Error bars depict mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, NS = Non significant.
) BM chimeras. (A) Clinical scores were followed daily (two-way ANOVA with Bonferroni post Hoc test). Frequencies of (B) IFN-γ+ (left) and IL-17+ (right) CD4+ T cells, (C) Ki67+ (left), Ki67+CD103+ICOS+ (middle) and IL10+ (right) among CD25hiFoxp3+CD4+Tregs in dLN at d9 (one-way ANOVA with Bonferroni post Hoc test). (A–C) Results are representative of at least 3 independent experiments with 8 mice per group. Error bars depict mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, NS = Non significant.
3.4. IDO deficiency in MOG35–55-presenting pDCs leads to EAE exacerbation
We have shown that IDO mRNA was preferentially expressed by pDCs compared to other LN cells both in steady-state and during EAE (Fig. 1, Fig. 3A). However, whether restricted IDO expression by pDCs is sufficient and required to inhibit disease development still need to be clarified. To address this, we used BDCA-2 DTR mice in which endogenous pDCs were depleted following DT injection [54]. It has been demonstrated that active immunization induces DT toxicity, and may therefore confound experiments in DTR transgenic mice for neuroinflammatory models, such as EAE [55]. To avoid this problem, we generated BM chimeric mice by injecting BM cells from BDCA-2 DTR into irradiated WT recipients. In these chimeric mice, no signs of toxicity or lethality in DT-treated mice upon EAE induction were observed. Moreover, efficient depletion of endogenous pDCs in the blood and LNs of BDCA-2 DTR → WT chimeras was achieved after DT injection (not shown). We transferred (i.v.) MOG35–55-loaded WT or IDO−/− pDCs into DT-treated BDCA-2 DTR → WT chimeras one day prior to EAE induction. In these settings, IDO was selectively supplied - or not - by adoptively transferred pDCs. Following EAE induction, only WT, but not IDO−/− pDCs significantly inhibited disease development (Fig. 5A). Foxp3+CD25+ Treg proliferation was increased in LN upon pDC transfer compared to control BDCA-2 DTR → WT EAE mice, whether transferred pDCs expressed IDO or not (Fig. 5B), confirming that IDO expression by pDCs is not involved in MHCII-dependent, pDC-mediated, Treg expansion. In contrast, the frequencies of suppressive Tregs co-expressing CD103 and ICOS (Fig. 5C), expressing high levels of CD25 (Fig. 5D), or upregulating the activation marker CD69 (Fig. 5E), were significantly reduced in LN from BDCA-2 DTR → WT EAE mice transferred with IDO−/− pDCs compared to WT pDCs. These results demonstrate that during EAE, IDO expression by pDCs is required to elicit pDC-mediated suppressive Tregs.
Fig. 5.

IDO+pDCs drive suppressive Tregs during EAE. (A–E) MOG35–55 loaded BM-derived pDCs from WT (■) and IDO−/− () were transferred (arrow) or not (○) into BDCA2-DTR → WT BM chimeras and EAE was induced 1 day after. Mice received 5 consecutive DT injections every 3–4 days. (A) Clinical scores were followed daily (two-way ANOVA with Bonferroni post Hoc test). Frequencies of (B) CD4+CD25+ Foxp3+ Treg cells (one-way ANOVA with Bonferroni post Hoc test), (C) CD103+ICOS+ Tregs, (D) CD25high Tregs and (E) CD69+ Tregs from dLNs are represented at d10 after EAE immunization (two-tailed Mann-Whitney test). (A–E) Results are representative of at least 2 independent experiments with 6–8 mice per group. Error bars depict mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
3.5. IDO is required for in vivo Treg-mediated EAE suppression
We next tested the in vivo suppressive activity of Tregs primed during EAE in pIII + IV−/− → WT, IDO−/− → WT and WT → WT chimeric mice. Purified CD4+CD25+ cells, which are predominantly Foxp3+ for all conditions (Fig. 6A), were transferred into WT hosts one day prior to EAE induction (Fig. 6A and B). Tregs primed in WT mice reduced EAE incidence, delayed disease onset, and conferred significant protection as we have previously reported [23]. In contrast, Tregs primed in mice lacking MHCII expression by pDCs did not exhibit any suppressive activity (Fig. 6B and Table 1). Strikingly, Tregs primed in absence of IDO did not confer any delay in disease onset, nor reduced EAE severity (Fig. 6B and Table 1). Frequencies of donor Tregs primed in WT and IDO−/− mice were similar in dLNs (day 3) and dLNs and SC (day 15) of recipient mice. These results suggest that the absence of EAE protection by Tregs primed in IDO deficient mice does not rely on an impaired migration of donor cells, but rather results from impaired intrinsic suppressive functions (Supplementary Fig. 6A and B). Accordingly, compared to Tregs isolated from WT EAE mice, Tregs primed in IDO deficient EAE mice were significantly less efficient at suppressing MOG35–55 loaded DC induced 2D2 CD4+ T cell proliferation in vitro (Fig. 6C). Furthermore, Tregs isolated from naïve WT and IDO−/− mice did not confer any suppression, further suggesting that Ag-specific priming in IDO+ context is required for the acquisition of Treg suppressive functions (Fig. 6C). In agreement, in vivo, only Tregs activated following EAE immunization in WT mice efficiently inhibited EAE incidence and severity (day 14), whereas neither Tregs isolated from naïve WT, nor Tregs isolated from naïve or immunized IDO−/− mice, conferred any protection upon transfer into EAE mice (Fig. 6D). Therefore, EAE-mediated activation is mandatory to confer IDO-dependent Treg suppressive functions.
Fig. 6.

Tregs primed by IDO+pDCs are suppressive in vivo and control EAE development. (A, B) CD4+CD25hi cells were purified from dLNs of WT → WT, pIII + IV−/− → WT and IDO−/− → WT BM chimeras 10 days after EAE induction, and transferred (arrow) into WT recipients further immunized for EAE the day after. (A) Experimental design is represented. Foxp3 expression in sorted cells. (B) Clinical scores were followed daily in control mice (⊡) and in mice transferred with WT Treg (■), IDO−/− Tregs () or pIII + IV−/− Tregs (○) (two-way ANOVA with Bonferroni post Hoc test). (A, B) Results are representative of at least 2 independent experiments. Error bars depict mean ± SEM. *P < 0.05, **P < 0.01. See also Table 1. (C, D) CD4+CD25hi cells were purified from total skin LNs of naïve WT → WT and IDO−/− → WT BM chimeras or from dLNs of WT → WT and IDO−/− → WT BM chimeras 10 days after EAE induction. (C) CD4+CD25hi cells were with proliferation dye-labeled 2D2 CD4+ T cells and LPS activated, MOG35–55 loaded, cDCs. 2D2 T cell proliferation was assessed after 5 days. Flow cytometry histograms represent 2D2 T cell proliferation for indicated conditions. Histograms represent the percentages of Treg-mediated suppression (two-tailed Mann-Whitney test). Results are representative of 2 independent experiments. Error bars depict mean ± SEM. **P < 0.01, NS = Non significant. (D) CD4+CD25hi cells were transferred into WT recipients further immunized for EAE the day after. Clinical scores and incidence (d15) are depicted. Data are representative of 2 experiments. Error bars represent mean ± SEM. One-way ANOVA with Bonferroni post Hoc test was used. *P < 0.05.
Table 1.
Tregs primed by IDO+pDCs delay and dampen clinical EAE. CD4+CD25hi cells were purified from dLNs of WT → WT (WT Tregs), pIII + IV−/− → WT (pIII + IV−/− Tregs) and IDO−/− → WT (IDO−/− Tregs) BM chimeras 10 days after EAE induction, and transferred into WT recipients further immunized for EAE the day after. Mean and cumulative clinical scores, mean day of onset and disease incidence are indicated (two-way ANOVA with Bonferroni post Hoc test). Results are representative of 2 independent experiments. ***P < 0.001.
| Mean clinical score | Cumulative clinical score | Mean day of onset | Incidence | |
|---|---|---|---|---|
| Ctrl EAE | 1.32 ± 0.32 | 25.13 | 10 | 100% |
| EAE + WT Tregs | ***0.57 ± 0.17 | 10.80 | 16 | 80% |
| EAE + IDO−/− Tregs | 1.70 ± 0.39 | 32.25 | 11 | 100% |
| EAE + pIII + IV−/− Tregs | 1.36 ± 0.36 | 25.88 | 12 | 100% |
Altogether, our data demonstrate that IDO provided by pDCs is critical for the acquisition of Ag-specific Treg in vivo suppressive functions.
4. Discussion
Whether pDCs exhibit tolerogenic or immunogenic APC functions in different immunogical context is an important matter of debate, and might depend on their own ability to produce inflammatory cytokines, as suggested by previous studies [9]. pDC tolerogenicity has been correlated to their expression of IDO in different settings [24], [31], [35]. In a mouse model of skin inflammation, IDO was induced in pDCs either after IFN-γ or TGF-β stimulation, the first pathway promoting IDO enzymatic activity-dependent tryptophan depletion, the second one implicating IDO phosphorylation and subsequent Treg induction [35]. Here we show that already in steady-state, pDCs are the major expressors of IDO in LNs. We further provide evidence that IDO expression in naïve pDCs is regulated through Ag-specific MHCII-restricted interaction with Foxp3+ Tregs. First, pDCs isolated from TCR tg mice in which CD4+ T cells all recognize a non-expressed antigenic peptide, exhibited substantially reduced IDO expression, independently of the self-reactivity of the TCR. Second, in absence of an existing Treg population in those TCR tg mice, LN pDCs exhibited a dramatic reduction in IDO expression. In addition, pDCs isolated from LN of Treg-deficient Scurfy mice expressed very little IDO mRNA. We cannot exclude that in Scurfy mice, the non-Treg CD4+ T cell population, which is strongly biased towards a pro-inflammatory autoimmune repertoire compared to WT mice, might impact IDO expression. Nevertheless, IDO expression by pDCs from T cell deficient mice was induced after co-culture with a CD4+ T cells only when the later contained a descent Treg population. In addition, IDO restoration was further dependent on the presence of the cognate antigenic peptide. The contribution of Treg in inducing IDO in pDCs has been suggested before [56], however without real evidence for a requirement of MHCII-restricted, Ag-specific interactions between pDCs and Tregs. Whether a particular Treg subpopulation would be specifically involved in this crosstalk with pDCs to induce IDO expression, and possibly the expression of other genes as well, remains to be determined. A role for IL-10 has been demonstrated in IDO expression stabilization [57]. Therefore, it is possible that IL-10 producing Tregs might be more competent to induce IDO up-regulation in pDCs. Our results reinforce the idea that Treg exhibit their Ag-specific immunomodulatory roles by impacting different cellular targets, not only by inhibiting effector T cells [58], inducing tumor-associated DC death [59] but also by promoting pDC tolerogenic functions. Future investigations will determine whether other pDC-specific tolerogenic features similarly depend on interactions with Tregs.
IDO expression by DCs has also been correlated with high levels of ICOS [60] and CTLA-4 expression by T cells, and might depend, or not, on IFN-γ production [28], [31], [32], [33], [61]. In mixed BM chimeric mice containing both MHCII sufficient and deficient pDCs, IDO is selectively abolished in MHCII deficient pDCs, whereas the same Treg populations expressing a certain level of ICOS, CTLA-4 or other molecules are present. Therefore, although ICOS and CTLA-4 might be required to promote IDO in pDCs, cells nevertheless need to express MHCII molecules.
During EAE, mice lacking MHCII on pDCs developed exacerbated disease, with increased IFN-γ and reduced TGF-β production in LNs compared to WT mice [23]. MHCII-deficient pDCs nevertheless still exhibit a strong impairment in IDO expression upon EAE. Therefore, neither IFN-γ, nor TGF-β, was sufficient to induce IDO in absence of MHCII expression by pDCs. In addition, we show that pDCs tolerogenicity in EAE context is dependent on their expression of IDO. First, in IDO deficient mice, encephalitogenic T cell frequency was increased, and as a consequence, disease was exacerbated. Second, adoptively transferred Tregs inhibited EAE only when isolated from actively MOG35–55 immunized WT, but not IDO−/− mice, suggesting that Tregs need to be primed in an IDO sufficient microenvironment to acquire their suppressive functions. Third, in LNs, IDO was predominantly expressed by pDCs, in both naïve and EAE mice. Finally, in an experimental setting in which we controlled IDO expression exclusively in pDCs, we observed an inhibition of EAE, as well as an increase in suppressive Tregs, only when pDCs expressed IDO. Notably, Treg induced after Ag-specific interaction with IDO+ pDCs are necessary to inhibit pathogenic T cells in LNs. Altogether, our results demonstrate that IDO expression by pDCs is required for the generation of suppressive Tregs that, upon Ag-specific activation, competently inhibit encephalitogenic T cells during EAE priming phase. In conclusion, we showed that naïve pDCs are high IDO expressors in the LN, a property acquired after a cross-talk with Foxp3+ Tregs. By expressing IDO, pDCs become competent to confer suppressive functions to Tregs in the context of EAE, where Ag-presenting IDO+ pDCs promote suppressive Tregs that inhibit the priming of encephalitogenic Th1 and Th17 cells and dampen CNS autoimmunity. Future investigations will determine whether other pDC features and functions, apart from IDO expression, are impacted following interactions with Tregs. Together, our data have identified a new regulatory role for Tregs in shaping Ag-presenting pDC functions towards tolerogenicity in autoimmunity.
Competing interests
The authors have no competing financial interests.
Acknowledgements
The authors thank C. Gameiro for excellent assistance in flow cytometry, the E. Shevach lab for providing us with Scurfy mice and T. Sparwasser for providing us with DEREG mice. We also thank R. Germain, Leslie Guery and I. Dunand-Sauthier for valuable discussions, D. Merkler and M. Gannagé for reviewing the manuscript, and C. Orabona, M. Benkhoucha and G. Schneiter for technical help. This work was supported by the Swiss National Science Foundation (PP00P3_152951 to S.H.), the European Research Council (281365 to S.H.) and the Swiss Multiple Sclerosis Society (to S.H), and by the Intramural Research Program of NIAID, NIH (to J.N.M.).
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jaut.2016.07.004.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Cervantes-Barragan L., Zust R., Weber F., Spiegel M., Lang K.S., Akira S., Thiel V., Ludewig B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colonna M., Trinchieri G., Liu Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 3.Chan V.S., Nie Y.J., Shen N., Yan S., Mok M.Y., Lau C.S. Distinct roles of myeloid and plasmacytoid dendritic cells in systemic lupus erythematosus. Autoimmun. Rev. 2012;11:890–897. doi: 10.1016/j.autrev.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Kared H., Masson A., Adle-Biassette H., Bach J.F., Chatenoud L., Zavala F. Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4(+)CD25(+) regulatory T-cells. Diabetes. 2005;54:78–84. doi: 10.2337/diabetes.54.1.78. [DOI] [PubMed] [Google Scholar]
- 5.Longhini A.L., von Glehn F., Brandao C.O., de Paula R.F., Pradella F., Moraes A.S., Farias A.S., Oliveira E.C., Quispe-Cabanillas J.G., Abreu C.H. Plasmacytoid dendritic cells are increased in cerebrospinal fluid of untreated patients during multiple sclerosis relapse. J. Neuroinflammation. 2011;8:2. doi: 10.1186/1742-2094-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nestle F.O., Conrad C., Tun-Kyi A., Homey B., Gombert M., Boyman O., Burg G., Liu Y.J., Gilliet M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005;202:135–143. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reder A.T., Feng X. Aberrant type I interferon regulation in autoimmunity: opposite directions in MS and SLE, shaped by evolution and body ecology. Front. Immunol. 2013;4:281. doi: 10.3389/fimmu.2013.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilliet M., Cao W., Liu Y.J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 9.Swiecki M., Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015;15:471–485. doi: 10.1038/nri3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villadangos J.A., Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29:352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Di Pucchio T., Chatterjee B., Smed-Sorensen A., Clayton S., Palazzo A., Montes M., Xue Y., Mellman I., Banchereau J., Connolly J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoeffel G., Ripoche A.C., Matheoud D., Nascimbeni M., Escriou N., Lebon P., Heshmati F., Guillet J.G., Gannage M., Caillat-Zucman S. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity. 2007;27:481–492. doi: 10.1016/j.immuni.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 13.Sapoznikov A., Fischer J.A., Zaft T., Krauthgamer R., Dzionek A., Jung S. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J. Exp. Med. 2007;204:1923–1933. doi: 10.1084/jem.20062373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young L.J., Wilson N.S., Schnorrer P., Proietto A., ten Broeke T., Matsuki Y., Mount A.M., Belz G.T., O'Keeffe M., Ohmura-Hoshino M. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat. Immunol. 2008;9:1244–1252. doi: 10.1038/ni.1665. [DOI] [PubMed] [Google Scholar]
- 15.Guery L., Hugues S. Tolerogenic and activatory plasmacytoid dendritic cells in autoimmunity. Front. Immunol. 2013;4:59. doi: 10.3389/fimmu.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goubier A., Dubois B., Gheit H., Joubert G., Villard-Truc F., Asselin-Paturel C., Trinchieri G., Kaiserlian D. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. 2008;29:464–475. doi: 10.1016/j.immuni.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X., Mishra P., Yu S., Beckmann J., Wendland M., Kocks J., Seth S., Hoffmann K., Hoffmann M., Kremmer E. Tolerance induction towards cardiac allografts under costimulation blockade is impaired in CCR7-deficient animals but can be restored by adoptive transfer of syngeneic plasmacytoid dendritic cells. Eur. J. Immunol. 2011;41:611–623. doi: 10.1002/eji.201040877. [DOI] [PubMed] [Google Scholar]
- 18.Ochando J.C., Homma C., Yang Y., Hidalgo A., Garin A., Tacke F., Angeli V., Li Y., Boros P., Ding Y. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat. Immunol. 2006;7:652–662. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 19.Guery L., Dubrot J., Lippens C., Brighouse D., Malinge P., Irla M., Pot C., Reith W., Waldburger J.M., Hugues S. Ag-presenting CpG-activated pDCs prime Th17 cells that induce tumor regression. Cancer Res. 2014;74:6430–6440. doi: 10.1158/0008-5472.CAN-14-1149. [DOI] [PubMed] [Google Scholar]
- 20.Sage A.P., Murphy D., Maffia P., Masters L.M., Sabir S.R., Baker L.L., Cambrook H., Finigan A.J., Ait-Oufella H., Grassia G. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation. 2014;130:1363–1373. doi: 10.1161/CIRCULATIONAHA.114.011090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loschko J., Schlitzer A., Dudziak D., Drexler I., Sandholzer N., Bourquin C., Reindl W., Krug A.B. Antigen delivery to plasmacytoid dendritic cells via BST2 induces protective T cell-mediated immunity. J. Immunol. 2011;186:6718–6725. doi: 10.4049/jimmunol.1004029. [DOI] [PubMed] [Google Scholar]
- 22.Loschko J., Heink S., Hackl D., Dudziak D., Reindl W., Korn T., Krug A.B. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J. Immunol. 2011;187:6346–6356. doi: 10.4049/jimmunol.1102307. [DOI] [PubMed] [Google Scholar]
- 23.Irla M., Kupfer N., Suter T., Lissilaa R., Benkhoucha M., Skupsky J., Lalive P.H., Fontana A., Reith W., Hugues S. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells inhibits T cell-mediated autoimmunity. J. Exp. Med. 2010;207:1891–1905. doi: 10.1084/jem.20092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baban B., Chandler P.R., Sharma M.D., Pihkala J., Koni P.A., Munn D.H., Mellor A.L. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J. Immunol. 2009;183:2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W., Liang X., Peterson A.J., Munn D.H., Blazar B.R. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 2008;181:5396–5404. doi: 10.4049/jimmunol.181.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J., Yang Y., Fan H., Zou H. Tolerogenic splenic IDO (+) dendritic cells from the mice treated with induced-Treg cells suppress collagen-induced arthritis. J. Immunol. Res. 2014;2014:831054. doi: 10.1155/2014/831054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fallarino F., Grohmann U., Hwang K.W., Orabona C., Vacca C., Bianchi R., Belladonna M.L., Fioretti M.C., Alegre M.L., Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 28.Grohmann U., Orabona C., Fallarino F., Vacca C., Calcinaro F., Falorni A., Candeloro P., Belladonna M.L., Bianchi R., Fioretti M.C. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 29.Manlapat A.K., Kahler D.J., Chandler P.R., Munn D.H., Mellor A.L. Cell-autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19+ dendritic cells. Eur. J. Immunol. 2007;37:1064–1071. doi: 10.1002/eji.200636690. [DOI] [PubMed] [Google Scholar]
- 30.Mellor A.L., Baban B., Chandler P., Marshall B., Jhaver K., Hansen A., Koni P.A., Iwashima M., Munn D.H. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 31.Fallarino F., Asselin-Paturel C., Vacca C., Bianchi R., Gizzi S., Fioretti M.C., Trinchieri G., Grohmann U., Puccetti P. Murine plasmacytoid dendritic cells initiate the immunosuppressive pathway of tryptophan catabolism in response to CD200 receptor engagement. J. Immunol. 2004;173:3748–3754. doi: 10.4049/jimmunol.173.6.3748. [DOI] [PubMed] [Google Scholar]
- 32.Baban B., Hansen A.M., Chandler P.R., Manlapat A., Bingaman A., Kahler D.J., Munn D.H., Mellor A.L. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int. Immunol. 2005;17:909–919. doi: 10.1093/intimm/dxh271. [DOI] [PubMed] [Google Scholar]
- 33.Munn D.H., Sharma M.D., Mellor A.L. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J. Immunol. 2004;172:4100–4110. doi: 10.4049/jimmunol.172.7.4100. [DOI] [PubMed] [Google Scholar]
- 34.Sharma M.D., Hou D.Y., Liu Y., Koni P.A., Metz R., Chandler P., Mellor A.L., He Y., Munn D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113:6102–6111. doi: 10.1182/blood-2008-12-195354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pallotta M.T., Orabona C., Volpi C., Vacca C., Belladonna M.L., Bianchi R., Servillo G., Brunacci C., Calvitti M., Bicciato S. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 36.Lahl K., Loddenkemper C., Drouin C., Freyer J., Arnason J., Eberl G., Hamann A., Wagner H., Huehn J., Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaefer B.C., Schaefer M.L., Kappler J.W., Marrack P., Kedl R.M. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 38.Reith W., LeibundGut-Landmann S., Waldburger J.M. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 2005;5:793–806. doi: 10.1038/nri1708. [DOI] [PubMed] [Google Scholar]
- 39.Baban B., Chandler P., McCool D., Marshall B., Munn D.H., Mellor A.L. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J. Reprod. Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Swiecki M., Gilfillan S., Vermi W., Wang Y., Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33:955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnden M.J., Allison J., Heath W.R., Carbone F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 42.Kaye J., Hsu M.L., Sauron M.E., Jameson S.C., Gascoigne N.R., Hedrick S.M. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 1989;341:746–749. doi: 10.1038/341746a0. [DOI] [PubMed] [Google Scholar]
- 43.Oxenius A., Bachmann M.F., Zinkernagel R.M., Hengartner H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 1998;28:390–400. doi: 10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 44.Means G.D., Toy D.Y., Baum P.R., Derry J.M. A transcript map of a 2-Mb BAC contig in the proximal portion of the mouse X chromosome and regional mapping of the scurfy mutation. Genomics. 2000;65:213–223. doi: 10.1006/geno.2000.6173. [DOI] [PubMed] [Google Scholar]
- 45.Bettelli E., Pagany M., Weiner H.L., Linington C., Sobel R.A., Kuchroo V.K. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domingues H.S., Mues M., Lassmann H., Wekerle H., Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS One. 2010;5:e15531. doi: 10.1371/journal.pone.0015531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fallarino F., Gizzi S., Mosci P., Grohmann U., Puccetti P. Tryptophan catabolism in IDO+ plasmacytoid dendritic cells. Curr. Drug Metab. 2007;8:209–216. doi: 10.2174/138920007780362581. [DOI] [PubMed] [Google Scholar]
- 48.Mandl J.N., Monteiro J.P., Vrisekoop N., Germain R.N. T cell-positive selection uses self-ligand binding strength to optimize repertoire recognition of foreign antigens. Immunity. 2013;38:263–274. doi: 10.1016/j.immuni.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LeibundGut-Landmann S., Waldburger J.M., Krawczyk M., Otten L.A., Suter T., Fontana A., Acha-Orbea H., Reith W. Mini-review: specificity and expression of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol. 2004;34:1513–1525. doi: 10.1002/eji.200424964. [DOI] [PubMed] [Google Scholar]
- 50.LeibundGut-Landmann S., Waldburger J.M., Reis e Sousa C., Acha-Orbea H., Reith W. MHC class II expression is differentially regulated in plasmacytoid and conventional dendritic cells. Nat. Immunol. 2004;5:899–908. doi: 10.1038/ni1109. [DOI] [PubMed] [Google Scholar]
- 51.Kwidzinski E., Bunse J., Aktas O., Richter D., Mutlu L., Zipp F., Nitsch R., Bechmann I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005;19:1347–1349. doi: 10.1096/fj.04-3228fje. [DOI] [PubMed] [Google Scholar]
- 52.Sakurai K., Zou J.P., Tschetter J.R., Ward J.M., Shearer G.M. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002;129:186–196. doi: 10.1016/s0165-5728(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 53.Barthlott T., Bosch A.J., Berkemeier C., Nogales-Cadenas R., Jeker L.T., Keller M.P., Pascual-Montano A., Hollander G.A. A subpopulation of CD103(pos) ICOS(pos) Treg cells occurs at high frequency in lymphopenic mice and represents a lymph node specific differentiation stage. Eur. J. Immunol. 2015;45:1760–1771. doi: 10.1002/eji.201445235. [DOI] [PubMed] [Google Scholar]
- 54.Rowland S.L., Riggs J.M., Gilfillan S., Bugatti M., Vermi W., Kolbeck R., Unanue E.R., Sanjuan M.A., Colonna M. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med. 2014;211:1977–1991. doi: 10.1084/jem.20132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer Zu Horste G., Zozulya A.L., El-Haddad H., Lehmann H.C., Hartung H.P., Wiendl H., Kieseier B.C. Active immunization induces toxicity of diphtheria toxin in diphtheria resistant mice–implications for neuroinflammatory models. J. Immunol. Methods. 2010;354:80–84. doi: 10.1016/j.jim.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 56.Mellor A.L., Munn D.H. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 57.Munn D.H., Sharma M.D., Lee J.R., Jhaver K.G., Johnson T.S., Keskin D.B., Marshall B., Chandler P., Antonia S.J., Burgess R. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 58.Grant C.R., Liberal R., Mieli-Vergani G., Vergani D., Longhi M.S. Regulatory T-cells in autoimmune diseases: challenges, controversies and–yet–unanswered questions. Autoimmun. Rev. 2015;14:105–116. doi: 10.1016/j.autrev.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 59.Boissonnas A., Scholer-Dahirel A., Simon-Blancal V., Pace L., Valet F., Kissenpfennig A., Sparwasser T., Malissen B., Fetler L., Amigorena S. Foxp3+ T cells induce perforin-dependent dendritic cell death in tumor-draining lymph nodes. Immunity. 2010;32:266–278. doi: 10.1016/j.immuni.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 60.Coquerelle C., Oldenhove G., Acolty V., Denoeud J., Vansanten G., Verdebout J.M., Mellor A., Bluestone J.A., Moser M. Anti-CTLA-4 treatment induces IL-10-producing ICOS+ regulatory T cells displaying IDO-dependent anti-inflammatory properties in a mouse model of colitis. Gut. 2009;58:1363–1373. doi: 10.1136/gut.2008.162842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mellor A.L., Chandler P., Baban B., Hansen A.M., Marshall B., Pihkala J., Waldmann H., Cobbold S., Adams E., Munn D.H. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int. Immunol. 2004;16:1391–1401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.