

Abstract
Imaging findings of adult-onset mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome is poorly documented. The authors present a 48-year-old woman with subacute onset of word-finding difficulties and right arm stiffness. Magnetic resonance imaging performed 2 weeks prior revealed left temporal lobe diffusion and fluid-attenuated inversion recovery hyperintensity predominantly involving the cortex. The apparent diffusion coefficient map showed preserved signal in the temporal cortex. Subsequent magnetic resonance imagings demonstrated a new diffusion signal abnormality extending to the left parietal cortex and occipital cortex with resolving diffusion hyperintensity in the temporal lobe. MR spectroscopy showed scattered areas of lactate deposition. Diagnosis of MELAS syndrome was confirmed by genetic analysis. Fluctuating, migratory stroke-like lesions with a predilection for the parietal, temporal, and occipital cortex that do not conform to a vascular territory and a lactate spike at 1.3 ppm on MR spectroscopy are characteristic of MELAS syndrome. Preserved signal intensity on apparent diffusion coefficient is useful to distinguish MELAS syndrome from ischemic infarction where the signal is typically reduced.
Keywords: MELAS syndrome, Migratory stroke-like lesions, Lactate
Case report
A 48-year-old woman of Palestine origin presented to a local hospital with subacute onset of confusion and word-finding difficulties. Her symptoms had started 2 weeks earlier with a headache, nausea, and dizziness. Her medical history was significant for essential hypertension, poorly controlled type 2 diabetes mellitus, and bilateral hearing loss of unknown etiology requiring hearing aids since age 46 years. Computed tomography (CT) revealed a hypodense lesion within the left temporal lobe (edema) involving gray matter and white matter (Fig. 1). Magnetic resonance imaging (MRI) demonstrated left temporal lobe diffusion signal abnormality and fluid-attenuated inversion recovery (FLAIR) hyperintensity predominantly involving the cortex, with cortical and leptomeningeal contrast enhancement. The apparent diffusion coefficient (ADC) map showed preserved, isointense signal in the temporal cortex (Fig. 2). The ventricular system and remaining parenchyma were grossly normal. Normal patent vessels without stenosis were seen on MR angiogram (Fig. 3). Clinically, her symptoms initially stabilized, but in the following week, the patient's confusion worsened, and she developed clumsiness and stiffness of her right arm. At that point, she was referred to our institution for further evaluation.
Fig. 1.

CT performed at t = 0 revealed hypodensity (edema) in the left temporal gray matter and white matter suggestive of infarction.
Fig. 2.

MRI examinations at t = 0 and week 2 showing the migratory lesions of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). At t = 0, there is cortical diffusion weighted imaging (arrowhead) and FLAIR hyperintensity with isointense signal on the ADC map. This lesion shows partial resolution with new lesions in the temporal lobe, both anterior and posterior, to the original lesion with extension into occipital cortex (arrows) on the week 2 MRI.
Fig. 3.
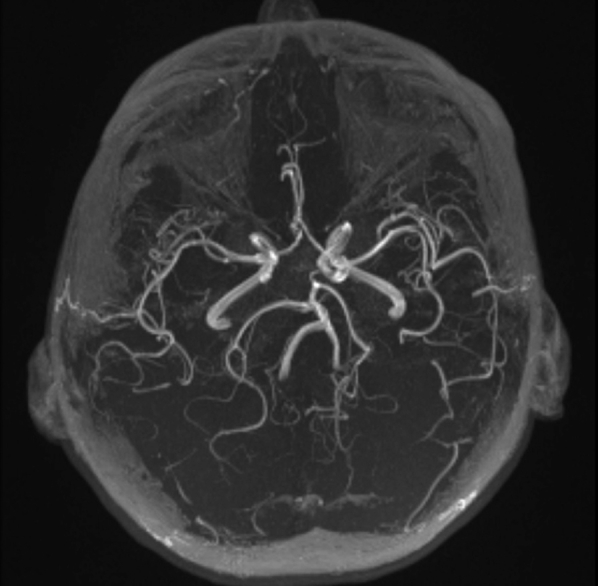
Normal patent vessels seen on MR angiogram at t = 0.
On admission, she had moderate expressive aphasia, mild dysarthria, and apraxia with paratonia of her right upper extremity. Imaging showed partial resolution of the left temporal lobe lesion, and new cortical diffusion weighted imaging abnormality in the left temporal and occipital lobes with corresponding FLAIR hyperintensity (Fig. 2). Normal ADC signal and postcontrast leptomeningeal enhancement were similarly seen in these regions. Positron emission tomography (PET) showed no evidence of malignancy but demonstrated reduced metabolic activity in the regions of signal abnormality with adjacent metabolic hyperactivity involving the left superior parietal and medial occipital lobes in a gyriform distribution. Lumbar puncture ruled out infection but was significant for elevated lactate to 5.1 mmol/L. Serum lactate was elevated to 4.6 mmol/L. Serum inflammatory markers (dsDNA, anti-Hu, anti-Ri, anti-Yo, SS-A, SS-B, C3, C4) were normal. On day 4 of hospitalization, the patient reported feeling better, and her family took her home against medical advice.
She then suffered 2 generalized tonic–clonic seizures associated with severe headache and blurred vision, and she was readmitted to our institution the same night. Continuous electroencephalogram showed left hemispheric slowing and frequent seizures originating from the left occipital lobe. Subsequent MRI demonstrated extension of the occipital lesion to involve more of the parietal and occipital lobes (Fig. 4). MR spectroscopy showed elevated lactate peak at 1.3 ppm diffusely throughout the brain (Fig. 5). A clinical diagnosis of MELAS syndrome was made.
Fig. 4.

MRI examinations showing interval progression from week 2 to 3. Week 3 shows a resolving temporal lobe lesion, and new diffusion weighted imaging signal abnormality within the occipital lobe (arrow) demonstrating the migratory pattern of MELAS.
Fig. 5.
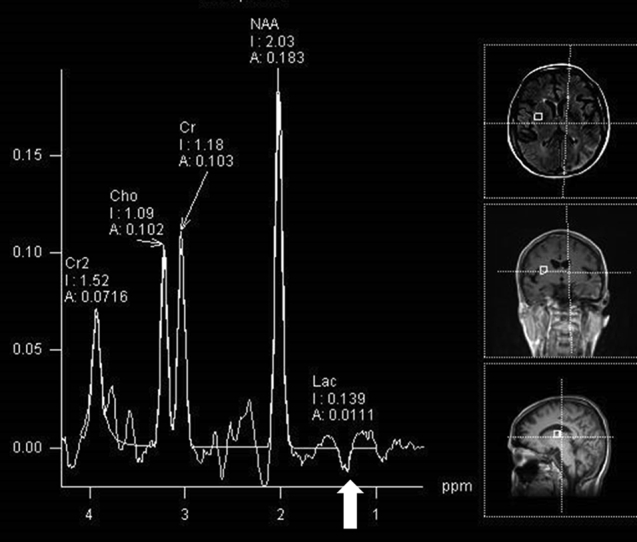
MR spectroscopy demonstrated elevation of the lactate peak, seen as an inverse doublet at 1.3 (arrow), within normal and abnormal regions of the brain.
The patient recovered without further complications on anticonvulsants and high-dose intravenous arginine then oral citrulline at 0.5 mg/kg. At 2-month follow-up, her cognition including language and activities of daily living had greatly improved. Genetic testing of patient's serum confirmed m.3243 A→G mutation (heteroplasmy 22%) in the MT-TL1 gene that encodes leucine transfer RNA, consistent with MELAS syndrome. A detailed 3-generation family history revealed no known neurologic, muscular, cardiac, or vision problems. Genetic counseling is ongoing.
Discussion
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome are maternally inherited multisystem disorder caused by mitochondrial DNA mutation [1], [2], [3]. Several point mutations in the mitochondrial DNA have been identified. In particular, an A to G transition at nucleotide position 3243 of the MT-TL1 gene that encodes transfer RNALeu(UUR) is present in 80% of the patients with MELAS syndrome [3]. Heteroplasmy within individual cells can result in unpredictable distribution of abnormal mitochondria in tissues and family members often inherited different percentage of mutant mitochondrial DNA. As such, constellation of symptoms and severity of symptoms are highly variable [4]. Review of our patient's family history suggests that she is only one who became symptomatic.
Pathophysiology of the disease is poorly understood. Current theories include inadequate energy production, microvascular angiopathy, and nitric oxide (NO) deficiency [5], [6], [7], [8]. Tissues with high energy demand are particularly susceptible to damage, such as the brain and skeletal muscles. Neurologic symptoms of MELAS syndrome typically present as stroke-like episodes and commonly include severe headache, sensorineural hearing loss, vision changes, expressive or receptive aphasia, rapid cognitive decline, and seizures. Exercise intolerance and muscle weakness are often present, along with lactic acidosis. While rare, systemic manifestations include hypertrophic cardiomyopathy, ophthalmoplegia, focal segmental glomerulosclerosis, and diabetes mellitus [9], [10], [11]. Treatment of MELAS encephalopathy is based on the assumption that energy deficit in the vascular endothelial cells lead to impaired cerebral blood flow and reduced NO production. Administration of NO precursors, specifically high-dose arginine and citrulline, has been shown to improve clinical symptoms of stroke-like episodes and to decrease the frequency and severity of these symptoms [3], [12], [13]. Recent evidence suggests that citrulline, a downstream and more immediate precursor of NO, to be superior to arginine in improving NO production [14]. The immediate symptomatic relief our patient experienced on initiation of treatment, and the excellent recovery and absence of new symptoms since then support the NO deficiency hypothesis and the use of NO precursors in the treatment of MELAS encephalopathy.
The combination of clinical suspicion, imaging patterns, and genetic analysis has become the mainstay of diagnosis and largely replaced the more invasive tissue biopsy. However, diagnosis remains challenging. Approximately 75% of MELAS syndrome cases present before 20 years of age; disease onset after the age of 40 years is rare and radiographic pattern of this population is poorly documented [15]. To complicate matters further, the focal neurologic deficits of abrupt onset MELAS syndrome are clinically indistinguishable from an ischemic stroke. Therefore, imaging is an essential component of the work-up and diagnosis.
Focal hypodensities with atrophy are commonly seen on CT scans of childhood-onset MELAS syndrome [16], [17], [18]. Global atrophy and calcification of the basal ganglia are sometimes present [16]. MRI characteristically shows hyperintensity on diffusion weighted imaging and FLAIR with a predilection for the parietal, temporal, and occipital cortex, without conforming to a single vascular territory, a hallmark finding of MELAS [19], [20], [21]. Blood vessels typically are normal on angiographic evaluation, although cases of vasoconstriction have been reported [22]. These diffusion signal abnormalities are likely to resolve over time, paralleling clinical improvement, while new lesions may develop with or without new symptoms. This migratory, waxing and waning pattern of stroke-like lesions on imaging is a cardinal feature of MELAS syndrome [16], [18], [23]. Pattern of ADC mapping is variable. Recent findings suggest the initial energy insufficiency causes cytotoxic edema and reduced ADC signal within the first 24 hours of a stroke-like episode. Development of vasogenic edema then follows and ADC signal increases within days to weeks [24]. Cortical contrast enhancement, as a result of breakdown of the blood-brain barrier or reperfusion hyperemia, and the subcortical hypermetabolic activity on PET/CT, indicate that significant regional hyperemia is present surrounding the stroke-like lesions. MR spectroscopy allows interrogation of tissue metabolites and has proven useful in some hereditary and acquired brain metabolic disorders [25], [26], [27]. The accumulation of lactate, represented by a peak or an inverse peak at 1.3 ppm, is sensitive for MELAS encephalopathy if found in areas that appear normal on imaging, as was seen in our patient [23], [28]. MR spectroscopy is also a potentially useful tool in assessing treatment response in patients with MELAS syndrome [29].
In conclusion, our case report suggests that the radiographic pattern of adult-onset MELAS syndrome is not significantly different from the pattern in patients who present in childhood. Migrating stroke-like lesions that do not conform to a vascular territory is highly indicative of MELAS syndrome. MR spectroscopy can help with the diagnosis.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
References
- 1.Hirano M., Ricci E., Koenigsberger M.R., Defendini R., Pavlakis S.G., DeVivo D.C. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125–135. doi: 10.1016/0960-8966(92)90045-8. [DOI] [PubMed] [Google Scholar]
- 2.Ciafaloni E., Ricci E., Shanske S., Moraes C.T., Silvestri G., Hirano M. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31:391–398. doi: 10.1002/ana.410310408. [DOI] [PubMed] [Google Scholar]
- 3.El-Hattab A.W., Adesina A.M., Jones J., Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015;116:4–12. doi: 10.1016/j.ymgme.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Goto Y., Horai S., Matsuoka T., Koga Y., Nihei K., Kobayashi M. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42:545–550. doi: 10.1212/wnl.42.3.545. [DOI] [PubMed] [Google Scholar]
- 5.King M.P., Koga Y., Davidson M., Schon E.A. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992;12:480–490. doi: 10.1128/mcb.12.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chomyn A., Enriquez J.A., Micol V., Fernandez-Silva P., Attardi G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J Biol Chem. 2000;275:19198–19209. doi: 10.1074/jbc.M908734199. [DOI] [PubMed] [Google Scholar]
- 7.El-Hattab A.W., Emrick L.T., Craigen W.J., Scaglia F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab. 2012;107:247–252. doi: 10.1016/j.ymgme.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Müller-Höcker J., Hübner G., Bise K., Förster C., Hauck S., Paetzke I. Generalized mitochondrial microangiopathy and vascular cytochrome c oxidase deficiency. Occurrence in a case of MELAS syndrome with mitochondrial cardiomyopathy-myopathy and combined complex I/IV deficiency. Arch Pathol Lab Med. 1993;117:202–210. [PubMed] [Google Scholar]
- 9.Fang W., Huang C.C., Lee C.C., Cheng S.Y., Pang C.Y., Wei Y.H. Ophthalmologic manifestations in MELAS syndrome. Arch Neurol. 1993;50:977–980. doi: 10.1001/archneur.1993.00540090074013. [DOI] [PubMed] [Google Scholar]
- 10.Sue C.M., Lipsett L.J., Crimmins D.S., Tsang C.S., Boyages S.C., Presgrave C.M. Cochlear origin of hearing loss in MELAS syndrome. Ann Neurol. 1998;43:350–359. doi: 10.1002/ana.410430313. [DOI] [PubMed] [Google Scholar]
- 11.Onishi H., Inoue K., Osaka H., Kimura S., Nagatomo H., Hanihara T. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and diabetes mellitus: molecular genetic analysis and family study. J Neurol Sci. 1993;114:205–208. doi: 10.1016/0022-510x(93)90299-e. [DOI] [PubMed] [Google Scholar]
- 12.Scaglia F., Northrop J.L. The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: a review of treatment options. CNS Drugs. 2006;20:443–464. doi: 10.2165/00023210-200620060-00002. [DOI] [PubMed] [Google Scholar]
- 13.Koga Y., Akita Y., Nishioka J., Yatsuga S., Povalko N., Tanabe Y. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- 14.El-Hattab A.W., Hsu J.W., Emrick L.T., Wong L.-J.C., Craigen W.J., Jahoor F. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab. 2012;105:607–614. doi: 10.1016/j.ymgme.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yatsuga S., Povalko N., Nishioka J., Katayama K., Kakimoto N., Matsuishi T. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820:619–624. doi: 10.1016/j.bbagen.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Allard J.C., Tilak S., Carter A.P. CT and MR of MELAS syndrome. AJNR Am J Neuroradiol. 1988;9:1234–1238. [PMC free article] [PubMed] [Google Scholar]
- 17.Kim I.O., Kim J.H., Kim W.S., Hwang Y.S., Yeon K.M., Han M.C. Mitochondrial myopathy-encephalopathy-lactic acidosis-and strokelike episodes (MELAS) syndrome: CT and MR findings in seven children. AJR Am J Roentgenol. 1996;166:641–645. doi: 10.2214/ajr.166.3.8623642. [DOI] [PubMed] [Google Scholar]
- 18.Pauli W., Zarzycki A., Krzyształowski A., Walecka A. CT and MRI imaging of the brain in MELAS syndrome. Pol J Radiol. 2013;78:61–65. doi: 10.12659/PJR.884010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito H., Mori K., Kagami S. Neuroimaging of stroke-like episodes in MELAS. Brain Dev. 2011;33:283–288. doi: 10.1016/j.braindev.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Kim J.H., Lim M.K., Jeon T.Y., Rha J.H., Rha J.H., Eo H. Diffusion and perfusion characteristics of MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode) in thirteen patients. Korean J Radiol. 2011;12:15–24. doi: 10.3348/kjr.2011.12.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosen L., Phillips S., Enzmann D. Magnetic resonance imaging in MELAS syndrome. Neuroradiology. 1990;32:168–171. doi: 10.1007/BF00588572. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida T., Ouchi A., Miura D., Shimoji K., Kinjo K., Sueyoshi T. MELAS and reversible vasoconstriction of the major cerebral arteries. Intern Med. 2013;52:1389–1392. doi: 10.2169/internalmedicine.52.0188. [DOI] [PubMed] [Google Scholar]
- 23.Castillo M., Kwock L., Green C. MELAS syndrome: imaging and proton MR spectroscopic findings. AJNR Am J Neuroradiol. 1995;16:233–239. [PMC free article] [PubMed] [Google Scholar]
- 24.Tzoulis C., Bindoff L.A. Serial diffusion imaging in a case of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Stroke. 2009;40:e15–e17. doi: 10.1161/STROKEAHA.108.523118. [DOI] [PubMed] [Google Scholar]
- 25.Ross B.D., Jacobson S., Villamil F., Korula J., Kreis R., Ernst T. Subclinical hepatic encephalopathy: proton MR spectroscopic abnormalities. Radiology. 1994;193:457–463. doi: 10.1148/radiology.193.2.7972763. [DOI] [PubMed] [Google Scholar]
- 26.Melhem E.R., Loes D.J., Georgiades C.S., Raymond G.V., Moser H.W. X-linked adrenoleukodystrophy: the role of contrast-enhanced MR imaging in predicting disease progression. AJNR Am J Neuroradiol. 2000;21:839–844. [PMC free article] [PubMed] [Google Scholar]
- 27.Tedeschi G., Bonavita S., Barton N.W., Betolino A., Frank J.A., Patronas N.J. Proton magnetic resonance spectroscopic imaging in the clinical evaluation of patients with Niemann-Pick type C disease. J Neurol Neurosurg Psychiatr. 1998;65:72–79. doi: 10.1136/jnnp.65.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bianchi M.C., Tosetti M., Battini R., Manca M.L., Mancuso M., Cioni G. Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic relevance. AJNR Am J Neuroradiol. 2003;24:1958–1966. [PMC free article] [PubMed] [Google Scholar]
- 29.Pavlakis S.G., Kingsley P.B., Kaplan G.P., Stacpoole P.W., O'Shea M., Lustbader D. Magnetic resonance spectroscopy: use in monitoring MELAS treatment. Arch Neurol. 1998;55:849–852. doi: 10.1001/archneur.55.6.849. [DOI] [PubMed] [Google Scholar]