

Summary
Excitation-transcription coupling, linking stimulation at the cell surface to changes in nuclear gene expression, is conserved throughout eukaryotes. How closely related coexpressed transcription factors are differentially activated remains unclear. Here, we show that two Ca2+-dependent transcription factor isoforms, NFAT1 and NFAT4, require distinct sub-cellular InsP3 and Ca2+ signals for physiologically sustained activation. NFAT1 is stimulated by sub-plasmalemmal Ca2+ microdomains, whereas NFAT4 additionally requires Ca2+ mobilization from the inner nuclear envelope by nuclear InsP3 receptors. NFAT1 is rephosphorylated (deactivated) more slowly than NFAT4 in both cytoplasm and nucleus, enabling a more prolonged activation phase. Oscillations in cytoplasmic Ca2+, long considered the physiological form of Ca2+ signaling, play no role in activating either NFAT protein. Instead, effective sustained physiological activation of NFAT4 is tightly linked to oscillations in nuclear Ca2+. Our results show how gene expression can be controlled by coincident yet geographically distinct Ca2+ signals, generated by a freely diffusible InsP3 message.
Graphical Abstract
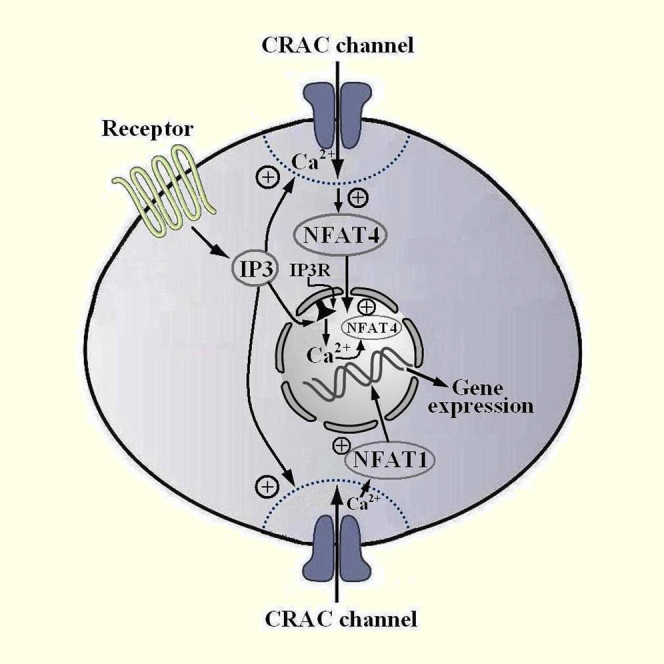
Highlights
-
•
NFAT1 is activated by local Ca2+ entry, whereas NFAT4 also needs nuclear Ca2+
-
•
Nuclear Ca2+ increases via InsP3 receptors located on the inner nuclear membrane
-
•
NFAT1 and NFAT4 show very different rephosphorylation (deactivation) kinetics
-
•
Slow deactivation of NFAT1 affords a form of short-term memory to gene expression
NFAT1 and NFAT4 are widely coexpressed transcription factors. NFAT1 is activated by Ca2+ microdomains near CRAC channels, whereas NFAT4 additionally requires nuclear Ca2+. Kar et al. now show that nuclear Ca2+ is raised by InsP3 receptors on the inner nuclear membrane. The dependence of NFAT isoforms on distinct sub-cellular Ca2+ signals regulates gene expression in cell- and stimulus-specific contexts.
Introduction
Proteins of the NFAT family of transcription factors regulate expression of a multitude of genes that are essential for vertebrate development and function (Müller and Rao, 2010). The family consists of five members, of which four (NFAT1–NFAT4) are activated by intracellular Ca2+ signals. In resting cells, NFAT proteins are extensively phosphorylated and thereby trapped within the cytoplasm. Upon stimulation of cell-surface receptors that increase cytoplasmic Ca2+, the proteins are dephosphorylated by Ca2+-calmodulin-activated calcineurin, the target for immunosuppressants cyclosporine A and tacrolimus. Dephosphorylation of cytoplasmic NFAT exposes a lysine-rich nuclear localization sequence, enabling the transcription factor to migrate into the nucleus, in combination with the protein importin β (Rao et al., 1997). Nuclear export occurs after rephosphorylation by nuclear resident protein kinases, which unmasks a nuclear export sequence (Gwack et al., 2006), enabling CRM1 and Ran-GTP nuclear shuttle proteins to transport NFAT across the nuclear envelope via the nuclear pore complex (Kehlenbach et al., 1998).
Once within the nucleus, NFATs regulate gene transcription, both alone and in combination with other transcription factors such as the AP-1 complex and FOXP3 (Bettelli et al., 2005, Macián et al., 2001). Whether NFAT proteins cooperate with AP-1 in particular has important functional consequences. Studies with an engineered NFAT1 protein that was unable to interact with AP-1 led to anergy and exhaustion in CD4+ and CD8+ T cells (Macián et al., 2002, Martinez et al., 2015). By contrast, NFAT/AP-1 cooperation resulted in a productive immune response. In addition to this form of protein-protein crosstalk, we have recently found that NFAT1 and NFAT4, two homologs that are often coexpressed in cells and within the same spatial domain, are regulated by different patterns of cytoplasmic Ca2+ signal (Kar and Parekh, 2015). Ca2+ microdomains near open store-operated Ca2+ release-activated Ca2+ (CRAC) channels activate NFAT1 without the need for a rise in either bulk cytoplasmic or nuclear Ca2+ (Kar et al., 2011, Kar and Parekh, 2015). On the other hand, NFAT4 requires both CRAC channel-generated Ca2+ microdomains and a nuclear Ca2+ rise for sustained nuclear residency following stimulation with thapsigargin (Kar and Parekh, 2015), a widely used non-physiological agent that inhibits SERCA pumps and leads to store depletion and subsequent opening of CRAC channels. The nuclear Ca2+ increase that occurs upon stimulation with thapsigargin is a two-step process: Ca2+ first enters the cytoplasm through plasmalemmal CRAC channels and then diffuses into the nucleus, likely through nuclear pores. In this study, we asked what was the source of nuclear Ca2+ following stimulation with a physiological trigger. Unexpectedly, we find that the nuclear Ca2+ needed to activate NFAT4 is mobilized from the nuclear membrane itself. Our results reveal that two geographically distinct Ca2+ signals, one at the cell periphery and the other constrained within the nucleoplasm, are necessary and sufficient to drive NFAT4 activation and gene expression independent of the interspersed cytoplasmic Ca2+ signal.
Results
Physiological Stimulation Activates NFAT4 Independent of Bulk Cytoplasmic Ca2+
Activation of cell-surface G protein-coupled cysteinyl leukotriene type I (cysLT1) receptors with the pro-inflammatory molecule leukotriene C4 (LTC4) evokes repetitive baseline cytoplasmic Ca2+ oscillations that are similar to those widely observed in other cells types and in response to different phospholipase C-coupled agonists (Thomas et al., 1996). In principal, these receptors can raise nuclear Ca2+ and thus maintain activation of NFAT4 in one of two ways: by an increase in bulk cytoplasmic Ca2+, arising mainly from Ca2+ release through inositol 1,4,5-trisphosphate (InsP3) receptors embedded in the endoplasmic reticulum (with a contribution from Ca2+ entry through CRAC channels), which then invades the nucleus (Model 1; Figure 1A), or by Ca2+ release directly into the nucleoplasm from nuclear InsP3 receptor channels (Model 2; Figure 1A).
Figure 1.

Buffering Cytoplasmic Ca2+ Does Not Impair Agonist-Evoked NFAT4 Nuclear Migration
(A) Cartoons summarize the two possible models for increasing nuclear Ca2+ following cysLT1 receptor activation (see text for details).
(B) Images show NFAT4-GFP nuclear migration for the different conditions indicated.
(C) Graphs summarize aggregate data from between 7 and 9 cells for each condition. Colors of graphs correspond to labels in (B). Time denotes time after stimulation.
(D) Cytoplasmic Ca2+ signals, measured with C-GCaMP3, following thapsigargin stimulation are shown for a control cell and one expressing PV-NES.
(E) Histogram compares rate of rise of the cytoplasmic Ca2+ signal following Ca2+ readdition to thapsigargin-treated cells, as in (D). Bars are averages of 19 (control) and 11 (PV-NES) cells.
(F) Cytoplasmic Ca2+ oscillations to 160 nM LTC4 are shown for the conditions indicated. Untagged PV constructs were used here.
(G) Number of oscillations per 200 s bin after stimulation are compared. Each point is the mean of 10–21 cells.
(H) Peak amplitude of each oscillation is compared between the different conditions. Peak amplitude represents (F/F0) − 1.
(I–M) As in (D)–(H), but now nuclear Ca2+ was measured instead, using N-GCaMP3. In (J), control is mean from 22 cells and PV-NES from 19 cells. In (L), each point is the mean of between 11 and 26 cells. For most graphs, error bars are contained within the symbols. For the graphs and histograms, data are represented as mean ± SEM. See also Figures S1 and S2.
Model 1 predicts that an increase in cytoplasmic Ca2+ buffering should prevent NFAT4 activation by cysLT1 receptor stimulation because this manipulation would reduce the rate and extent of Ca2+ diffusion from the cytoplasm into the nucleus. By contrast, the expectation from Model 2 is that NFAT4 activation will be unaffected by changes in cytoplasmic Ca2+ buffering capacity. To test these predictions, we overexpressed the cytoplasmic Ca2+ buffer parvalbumin that had been genetically manipulated to express only in the cytoplasm through addition of a nuclear export sequence (PV-NES) (Kar and Parekh, 2015). Although stimulation with thapsigargin led to robust migration of NFAT4-GFP from the cytoplasm into the nucleus in control cells, expression of PV-NES largely suppressed NFAT4 migration (Figures 1B and 1C) (Kar and Parekh, 2015). PV-NES therefore buffers cytoplasmic Ca2+ sufficiently well to prevent a rise in bulk cytoplasmic Ca2+ following CRAC channel opening from invading the nucleus to maintain active NFAT4. Activation of cysLT1 receptors with LTC4 also resulted in robust nuclear accumulation of NFAT4 (Figures 1B and 1C). However, this was unaffected by the presence of PV-NES (Figures 1B and 1C). Ca2+ microdomains near open CRAC channels remained essential for NFAT4 activation by LTC4 in the presence of PV-NES because channel block with Synta66 prevented nuclear NFAT4 migration (Figures 1B and 1C).
To confirm that PV-NES indeed reduced stimulus-evoked Ca2+ signals, we measured bulk cytoplasmic Ca2+ with the fluorescent Ca2+ indicator dye Fura-2. The Ca2+ response was significantly decreased by the presence of PV-NES following stimulation with thapsigargin (Figure S1). The intracellular Ca2+ signal measured with Fura-2 includes contributions from the cytoplasm and the nucleus. To measure Ca2+ in each compartment, we used the genetically encoded fluorescent indicator protein GCaMP3, engineered to express either a nuclear export sequence (GCaMP3-NES) or nuclear localization sequence (GCaMP3-NLS), to report Ca2+ in the cytoplasm or nucleus, respectively. In control cells expressing GCaMP3-NES, thapsigargin activated a robust Ca2+ signal due to Ca2+ influx, but the response was reduced by ∼60% following expression of PV-NES (Figures 1D and 1E). Similarly, cytoplasmic Ca2+ oscillations to LTC4 ran down more quickly (Figures 1F and 1G) and were smaller in size (Figures 1F and 1H) in the presence of PV-NES. The nuclear Ca2+ rise induced by thapsigargin (measured with GCaMP3-NLS) was also substantially reduced by PV-NES (Figures 1I and 1J), as expected since this increase is secondary to the cytoplasmic Ca2+ rise.
LTC4 evoked numerous nuclear Ca2+ oscillations in control cells, and these were largely unaffected by the presence of PV-NES (Figures 1K–1M), as was the case for NFAT4 migration (Figures 1B and 1C). When parvalbumin was targeted to the nucleus instead (PV-NLS), NFAT4 migration in response to LTC4 challenge was suppressed (Figure S2) (Kar and Parekh, 2015), as were the nuclear Ca2+ oscillations (Figures 1K–1M). The cytoplasmic Ca2+ oscillations evoked by LTC4 were unaffected by the presence of PV-NLS (Figures 1F–1H).
Equivalent results were obtained when we overexpressed a different cytoplasmic Ca2+-binding protein, calbindin D28K-GFP: NFAT4-cherry failed to accumulate in the nucleus after stimulation with thapsigargin (middle set of images in Figure 2A; Figure 2B). By contrast, activation of cysLT1 receptors triggered robust migration of NFAT4-cherry into the nucleus in cells expressing calbindin D28K-GFP, which was prevented by Synta66 (Figures 2A and 2B).
Figure 2.

The Cytoplasmic Ca2+ Binding Protein Calbindin D28K Prevents Nuclear Migration of NFAT4, but Not NFAT1
(A) Images compare NFAT1- or NFAT4-cherry movement in cells expressing calbindin D28K-GFP and were taken at different times after stimulation, as indicated. NFAT constructs used are shown on the right of each row of images, where N1 and N4 denote NFAT1 and NFAT4. Stimuli are also shown on the right.
(B) Aggregate data from experiments as in (A) are summarized. Each point is the mean of between 8 and 13 individual cells. The N1-Thap curve has been offset upward by 0.03, to increase clarity.
(C) In cells expressing PV-NES (untagged) or calbindin D28K-GFP (indicated on the right of each image), stimulation with LTC4 after thapsigargin (1 μM) failed to show any NFAT4 movement. LTC4 was applied 20 min after pre-treatment with thapsigargin. Upper images show rest and then 20 and 40 min after thapsigargin stimulation (control recording from the same cell preparation). In the control and PV-NES experiments, NFAT4-GFP was expressed. In coexpression with calbindin D28K-GFP, NFAT4-cherry was used instead.
(D) Aggregate data are compared for the conditions shown. Each point is the mean of between 9 and 15 cells. For the graphs, data are represented as mean ± SEM.
NFAT1 activation, which is driven only by local Ca2+ entry through CRAC channels, was unaffected by the presence of PV-NES (Kar and Parekh, 2015) or calbindin D28K-GFP (upper set of images in Figure 2A; Figure 2B), regardless of whether the stimulus was thapsigargin or LTC4.
Collectively, these data show that cysLT1 receptor stimulation activates NFAT4 even when a rise in bulk cytoplasmic Ca2+ has been impaired. The findings are therefore incompatible with the various scenarios embodied in Model 1 (Figure 1), where nuclear Ca2+ is totally contingent upon a cytoplasmic Ca2+ rise. Instead, our data are consistent with Model 2, where the nuclear Ca2+ increase required for maintained NFAT4 activation is independent of cytoplasmic Ca2+. Model 2 posits that nuclear Ca2+ is increased by activation of nuclear InsP3 receptors and is therefore causally attendant to a rise in nucleoplasmic InsP3. The nuclear envelope is a functional Ca2+ store due to the presence of SERCA pumps on the outer membrane and InsP3 receptors on both the inner and outer membranes (Alonso and García-Sancho, 2011, Gerasimenko et al., 1996), with InsP3 releasing stored Ca2+ directly into the nucleoplasm (Echevarría et al., 2003, Humbert et al., 1996, Zima et al., 2007). Although LTC4 triggered NFAT4 accumulation in the nucleus in PV-NES- and calbindin-expressing cells, it failed to do so if cells were pre-exposed to thapsigargin (Figures 2C and 2D). This supports Model 2 in that sustained activation of NFAT4 by receptor stimulation requires a functional nuclear Ca2+ store.
Buffering Nuclear InsP3 Prevents the Nuclear Ca2+ Rise to Agonist and Subsequent Sustained NFAT4 Activation
We designed experiments to test Model 2 more directly. A key prediction is that buffering nuclear InsP3 should impair InsP3 from activating InsP3 receptors in the inner nuclear membrane, preventing the rise in nuclear Ca2+ and thus sustained nuclear NFAT4 activation. To address this, we dampened InsP3 increases in either the cytoplasm or the nucleus by exploiting genetically encoded InsP3 buffers. We expressed a construct containing the InsP3 binding site of the InsP3 receptor tagged with RFP and engineered to express either a cytoplasmic or nuclear localization sequence, which we refer to as IP3 buffer-NES-RFP and IP3 buffer-NLS-RFP, respectively. Expression of IP3 buffer-NES-RFP was localized to the cytoplasm (Figures 3A and 3B), whereas IP3 buffer-NLS-RFP was restricted to the nucleus (Figures 3A and 3B). Coexpression of both buffers resulted in a similar distribution between nucleus and cytoplasm. Stimulation with thapsigargin, which evokes Ca2+ signals independent of InsP3, resulted in robust nuclear accumulation of NFAT1-GFP or NFAT4-GFP despite the presence of IP3 buffer-NES-RFP, IP3 buffer-NLS-RFP, or both (Figure 3C; aggregate data are summarized in Figure 3K). Therefore these buffers per se do not interfere with NFAT migration.
Figure 3.

Buffering Nuclear InsP3 Prevents Nuclear Accumulation of NFAT4
(A) Confocal images compare sub-cellular distribution of RFP-tagged InsP3 buffer containing either a nuclear export sequence (IP3-NES; upper panel), InsP3 buffer containing a nuclear localization sequence (IP3-NLS, middle panel), or both (lower panel). DAPI was used to stain the nucleus.
(B) Fluorescence intensities of the InsP3 buffers are compared between the cytoplasmic and nuclear compartments, denoted C and N, respectively. Each bar is the mean of between 9 and 15 cells.
(C) Migration of NFAT1-GFP (denoted N1) and NFAT4-GFP (N4) into the nucleus is compared following stimulation with thapsigargin for 40 min in the presence of the different IP3 buffers (RFP-tagged), indicated on the left.
(D) Cytoplasmic Ca2+ oscillations are compared between cells expressing RFP-tagged IP3-NLS, IP3-NES, or both.
(E) The number of oscillations to LTC4 produced each 200 s bin are compared for the different conditions. Each point is the mean of between 8 and 11 cells.
(F) The amplitude of each Ca2+ oscillation is compared for the conditions shown. Data for (E) and (F) were extracted from experiments as in (D).
(G) Nuclear Ca2+ oscillations are compared for the different conditions indicated.
(H) The number of nuclear Ca2+ oscillations per 200 s bin is shown for each condition. Each point is the mean of between 10 and 26 cells.
(I) The peak amplitude of each nuclear Ca2+ oscillation is compared. Data for (H) and (I) were extracted from experiments as in (G). In (G)–(I), control denotes mock-transfected cells.
(J) Images compare movement of NFAT1-GFP or NFAT4-GFP for the conditions shown. IP3 buffers were tagged with RFP. LTC4 was applied at 160 nM for 40 min and ionomycin (ionom) was 2 μM.
(K) Histogram summarizes data for the various conditions shown. Rest denotes the unstimulated state. N1 and N4 indicate NFAT1-GFP and NFAT4-GFP. Open bars above filled histograms denote the rescue of NFAT movement following stimulation with ionomycin for 20 min. Each bar is the mean from three independent experiments.
In (B), (E), (F), (H), (I), and (K), data are represented as mean ± SEM. See also Figure S3.
After expression of InsP3 buffer-NES-RFP, cytoplasmic (Figures 3D–3F) and nuclear (Figures 3G–3I) Ca2+ signals to cysLT1 receptor activation were significantly reduced. Activation of either NFAT1 or NFAT4 was also suppressed by InsP3 buffer-NES-RFP (Figures 3J and 3K), as expected since cytoplasmic InsP3 is required to deplete the endoplasmic reticulum Ca2+ store for CRAC channel activation following cysLT1 receptor stimulation. Movement could be rescued by a dose of ionomycin that raises bulk cytoplasmic Ca2+ to high levels independent of store-operated Ca2+ influx (Figures 3J and 3K; Kar and Parekh 2015). Markedly different results were obtained when InsP3 buffer-NLS-RFP was expressed instead. Cytoplasmic Ca2+ oscillations to LTC4 were now largely unaffected (Figures 3D–3F), whereas the nuclear Ca2+ signals were suppressed (Figures 3G–3I). Despite cytoplasmic Ca2+ signals being normal in the presence of IP3 buffer-NLS, activation of cysLT1 receptors now failed to stimulate NFAT4 accumulation in the nucleus (Figures 3J and 3K). Movement could subsequently be rescued by ionomycin. Simultaneous measurements of cytoplasmic and nuclear Ca2+ in the same cells confirmed that Ca2+ oscillated in both compartments after stimulation with LTC4, but only the nuclear Ca2+ oscillations were abolished by InsP3 buffer-NLS (Figure S3). Oscillations in cytoplasmic and nuclear Ca2+ in both compartments were suppressed by coexpression of InsP3 buffer-NES and InsP3 buffer-NLS (Figure S3). Migration of NFAT1 into the nucleus in response to LTC4 was unaffected by InsP3 buffer-NLS-RFP (Figures 3J and 3K), consistent with it requiring only local Ca2+ entry through CRAC channels for activation. Nuclear migration of either NFAT1 or NFAT4 was suppressed by coexpression of both InsP3 buffers (Figures 3J and 3K). In aggregate, these data suggest that the nuclear Ca2+ rise required for NFAT4 residency within the nucleus is accomplished through Ca2+ release by nuclear InsP3. In support of this, immunostaining revealed that type I InsP3 receptors localized to the nucleus as well as the cytoplasm (Figure 4A). InsP3 receptors were also detected in both nuclear and cytoplasmic extract (Figure 4B).
Figure 4.

Nuclear Ca2+ Oscillations Are Generated by Nuclear InsP3 Receptors and Are Effective in Maintaining NFAT4 Activity
(A) Immunostaining reveals the presence of type I InsP3 receptors in the cytoplasm and nucleus. No staining is seen when secondary antibody (Sec. AB) alone is used. Secondary antibody was conjugated with Alexa 488 for visualization.
(B) Western blots of cytoplasmic and nuclear extract show the presence of type I InsP3 receptors in both compartments. Similar results were obtained in two independent experiments.
(C) Immunogold labeling reveals the presence of type I InsP3 receptors in the inner nuclear membrane. Left-hand panel denotes a control micrograph and the right-hand one after immunogold labeling of type I InsP3 receptors. Red arrows denote InsP3R on the inner nuclear membrane, black arrow shows receptor on the outer nuclear membrane, and gray ones denote receptor in the endoplasmic reticulum.
(D) Comparison of nuclear Ca2+ signals to LTC4 in a control cell and in one in which the cytoplasm and nucleoplasm had been loaded with EGTA via the form EGTA-AM.
(E) Loading the cell with EGTA reduces NFAT4-cherry nuclear migration in response to LTC4. Migration can be rescued by stimulating cells with ionomycin (2 μM; 20 min).
(F) Aggregate data from several experiments as in (E) are summarized. Open bar above EGTA-AM denotes the extent of rescue by ionomycin. Data are represented as mean ± SEM.
(G) Graph plots NFAT4-cherry nuclear accumulation against the integrated nuclear Ca2+ signal in control cells and cells loaded with EGTA. Each point depicts NFAT4 movement and integrated nuclear Ca2+ from the same cell. The y axis represents peak NFAT nuclear/cytoplasmic ratio after stimulation with LTC4 minus the NFAT nuclear/cytoplasmic ratio before stimulation.
(H) Nuclear Ca2+ oscillations evoked by LTC4 run down quickly when Ca2+ entry through CRAC channels is blocked with 10 μM BTP2.
(I) Estimated nuclear Ca2+ concentration for an oscillatory response in a control cell is compared with a plateau-type response in an EGTA-loaded cell, following stimulation with 160 nM LTC4. To calibrate the nuclear Ca2+ signal in intact cells, following expression of NGCaMP3, Rmin and Rmax were obtained by stimulating cells with 5 μM ionomycin either in Ca2+-free external solution (with fluorescence measured ∼15 min later) or 10 mM Ca2+ external solution, respectively. The KD for nuclear GCaMP3 was taken as 660 nM from the literature. The estimated Ca2+ concentrations are, at best, only an approximation (see text). Arrow denotes LTC4 application.
(J) The graph plots the predicted build-up of nuclear Ca2+-calmodulin-calcineurin following stimulation with LTC4 in a control cell (red trace) and in one loaded with EGTA (blue trace).
(K) Contour plot depicts active nuclear calcineurin as a function of nuclear Ca2+ concentration and total nuclear calcineurin available.
See also Figure S4, Table S1, and description of the model in Supplemental information.
In order for InsP3 buffer-NLS to suppress nuclear Ca2+ oscillations, leaving cytoplasmic ones intact, InsP3 receptors must be embedded within the inner nuclear membrane facing the nucleoplasm. To assess this, we used electron microscopy with immunogold labeling of type I InsP3 receptors, an approach that provides the spatial resolution to resolve the specific nuclear location of proteins. Whereas no detectable immunogold labeling was seen in control cells not exposed to InsP3 receptor antibody (Figure 4C, left-hand panel, labeled immunogold−), the inner nuclear membrane revealed particles when InsP3 antibody was present (Figure 4C, right-hand panel, labeled immunogold+).
In some studies, the nuclear Ca2+ rise is secondary to the cytoplasmic Ca2+ increase, which rapidly propagates into the nucleus (Lin et al., 1994, Lipp et al., 1997). On the other hand, nuclear Ca2+ can increase independently of cytoplasmic Ca2+ (Echevarría et al., 2003, Humbert et al., 1996, Zima et al., 2007). In our experiments with cysLT1 receptor activation, cytoplasmic Ca2+ oscillations did not invade the nucleoplasm to the extent needed to maintain NFAT4 residency because InsP3 buffer-NLS-RFP greatly reduced the nuclear Ca2+ signals (Figures 3G–3I) and suppressed transcription factor accumulation (Figure 3K), despite cytoplasmic Ca2+ signaling remaining intact (Figures 3D–3F; Figure S3). Furthermore, buffering cytoplasmic Ca2+ with parvalbumin-NES abolished cytoplasmic calcium oscillations to LTC4 (Figures 1F–1H), yet nuclear Ca2+ signaling (Figures 1K–1M) and NFAT4 activation (Figure 1C) were unaffected. We also considered the possibility that InsP3 released Ca2+ into the cytoplasm from receptors located in the outer nuclear membrane and Ca2+ then diffused into the nucleus through nuclear pores. Because PV-NES had no inhibitory effect on nuclear Ca2+ signals evoked by agonist, this possibility is unlikely.
How does cysLT1 receptor activation increase nuclear Ca2+ independently of cytoplasmic Ca2+? Our finding that InsP3 buffer-NES-RFP suppressed the nuclear Ca2+ rise to LTC4 demonstrates that InsP3 is first produced in the cytoplasm and then diffuses into the nucleus, rather than local generation of InsP3 within the nucleus itself. The lifetime of InsP3 in the cytoplasm is sufficiently long for it to diffuse many times across a cell with dimensions similar to that of a HEK cell before it is broken down (Allbritton et al., 1992).
For the Same Integrated Nuclear Ca2+ Signal, Nuclear Ca2+ Oscillations Are More Effective in Maintaining NFAT4 Activation than a Bulk Nuclear Ca2+ Rise
We asked whether nuclear Ca2+ oscillations conferred a signaling advantage to sustained NFAT4 activation compared with a non-oscillatory Ca2+ increase that raised nucleoplasmic Ca2+ to a similar extent over the same stimulation period (integrated Ca2+ signal). Simultaneous measurements of nuclear Ca2+ using GCaMP3-NLS and nuclear accumulation of NFAT4-cherry showed that oscillatory nuclear Ca2+ signals induced by LTC4 (Figure 4D) evoked robust nuclear accumulation of NFAT4 (Figures 4E and 4F) for only a small integrated nuclear Ca2+ signal (Figure 4G; labeled control). In cells loaded with the slow Ca2+ chelator EGTA, which increases the buffering capacity of the nucleoplasm (Figure S3 in Kar and Parekh, 2015), nuclear Ca2+ oscillations to LTC4 were replaced by a steady increase in nuclear Ca2+ (Figure 4D). Integration of these signals showed similar or greater total nuclear Ca2+ over the same time period compared with oscillatory responses (Figure 4G), yet NFAT4 accumulation was substantially less (Figures 4E–4G).
The Nuclear Ca2+ Store Is Replenished by Store-Operated Ca2+ Entry
Ca2+ entry through CRAC channels is required to refill the endoplasmic reticulum with Ca2+. Since the endoplasmic reticulum is contiguous with the outer nuclear membrane, we asked whether Ca2+ entry through CRAC channels was likewise essential for repletion of the nuclear store. Whereas numerous nuclear Ca2+ oscillations were seen in control cells stimulated with LTC4, the Ca2+ signals ran down quickly in the presence of the CRAC channel blocker BTP2 (Figure 4H; Figure S4). Similar results were obtained when cells were stimulated either in the presence of another CRAC channel blocker (Synta66) or in the absence of external Ca2+ (data not shown).
Ca2+ influx contributes little to the size or decay rate of each cytoplasmic Ca2+ oscillation evoked by LTC4 (Di Capite et al., 2009). The finding that the nuclear store is replenished by Ca2+ flux through CRAC channels at the cell surface is therefore a remarkable example of Ca2+ tunneling (Mogami et al., 1997), where local Ca2+ entry can be transported to distant cytoplasmic regions via uptake into the endoplasmic reticulum.
Mathematical Modeling of Nuclear Calcineurin Activation
We simulated active calcineurin build-up in the nucleus (Figures 4I–4K) for different patterns of nuclear Ca2+ signal, using a reduction of the Saucerman and Bers model (Saucerman and Bers, 2008), as suggested by Bazzazi et al. (Bazzazi et al., 2015; see Supplemental Information for further details). To model active calcineurin levels, an estimate of nuclear Ca2+ concentration was needed. We therefore attempted to calibrate the GCaMP3 signal, as described in the legend to Figure 4I. The concentrations shown in Figure 4I are, however, only a rough estimate because of the inherent difficulties in calibrating a fluorescent probe within the nucleoplasm of an intact cell (Bootman et al., 2009), coupled with GCaMP3 being a non-ratiometric probe. Notwithstanding these limitations, the model suggests calcineurin activity oscillates in the nucleus (Figure 4J) with a pattern similar to the nuclear Ca2+ oscillations (Figure 4I). By contrast, nuclear calcineurin activity is very low when a small-amplitude but long-lasting nuclear Ca2+ rise occurs. A contour plot comparing active calcineurin with Ca2+ concentration and total nuclear calcineurin (Figure 4K) revealed that the same level of active calcineurin could be achieved by having either high total nuclear calcineurin combined with low nuclear Ca2+ or low total nuclear calcineurin coupled with high nuclear Ca2+. The contour plot also shows that large nuclear Ca2+ spikes enable robust activation of a small number of nuclear calcineurin molecules, whereas more sustained smaller Ca2+ rises are much less effective. This suggests an advantage to large oscillatory Ca2+ signaling: in an individual cell having a fixed total number of effector molecules like calcineurin, a small number of effectors will be activated strongly by large-amplitude Ca2+ oscillations, compared with weak recruitment of a greater number of effectors in response to smaller-amplitude but more prolonged Ca2+ signals. For an effector that is an enzyme like calcineurin, a small fraction of active molecules should be sufficient to engender a strong downstream response.
NFAT4 Is Rephosphorylated within the Nucleus Much Faster than NFAT1
NFAT4 is exported out of the nucleus ∼8 times faster than NFAT1 (Kar and Parekh, 2015, Yissachar et al., 2013), an effect due partly to differences in the respective SP-3 motifs (Kar and Parekh, 2015). The nuclear export of each isoform is a two-step process involving rephosphorylation within the nucleoplasm followed by exit through the nuclear pores in a complex with shuttle proteins CRM1 and Ran-GTP. The relative contributions of these steps are unknown. We hypothesized that the main difference in NFAT nuclear export rates was due to differences in nuclear rephosphorylation rather than nuclear transport, and this was why nuclear Ca2+ and thus active calcineurin was so important for NFAT4 accumulation. To address this, we exposed cells to leptomycin B, an effective inhibitor of CRM1-dependent export (Fornerod et al., 1997). In the presence of leptomycin B, NFAT4 accumulated within the nucleus following CRAC channel activation at a similar rate to control cells (Figures 5A and 5B). Following nuclear accumulation, we triggered nuclear export by termination of Ca2+ influx combined with inhibition of calcineurin with cyclosporine A (Kar et al., 2011). Compared with control cells, where NFAT4 exited the nucleus with a time constant of ∼10 min (Kar and Parekh, 2015), nuclear export was suppressed for ∼20 min in leptomycin B-treated cells (Figures 5A and 5B). Thereafter, NFAT4 export proceeded gradually, reflecting either delayed leptomycin B-insensitive transport or loss of leptomycin B activity over time. Regardless, the 20 min hiatus in NFAT4 export in leptomycin B enabled us to measure the kinetics of NFAT4 rephosphorylation within the nucleus in the absence of export. Gel shifts from western blots of nuclear extract revealed that NFAT4 was rephosphorylated quickly (Figure 5C), with a half-time of ∼4.5 min (Figure 5D). By contrast, only modest nuclear phosphorylation of NFAT1 occurred within 20 min (Figures 5E and 5F). An NFAT4 construct in which the SP-3 region of NFAT4 had been replaced with that from NFAT1 was rephosphorylated more slowly than NFAT4 but faster than NFAT1 (Figures 5G and 5H; half-time of ∼15 min), supporting the view that the SP-3 region is an important determinant of nuclear NFAT rephosphorylation (Kar and Parekh, 2015). For the 20 min that NFAT-GFP export was suppressed (Figure 5B), no increase in cytoplasmic NFAT-GFP was detectable (Figures S5A–S5C). Hence, over this time frame, leptomycin B fully inhibits NFAT nuclear export.
Figure 5.

NFAT4 Is Rephosphorylated within the Nucleus Considerably More Quickly Than NFAT1
(A) Images compare nuclear export of NFAT4-GFP between a control cell and one exposed to leptomycin B (LMB; 100 nM). Nuclear export was initiated by removal of external Ca2+ in the presence of cyclosporine A.
(B) Aggregate data from several experiments are compared.
(C) Western blot shows gel shift of NFAT4-GFP from nuclear extract after stimulation and then at different times (2–20 min) after initiation of export (see bottom of G for timings).
(D) Graph plots relative gel shift as a function of time up to 20 min after the initiation of nuclear export. A decreased gel shift denotes rephosphorylation.
(E) Gel shift for nuclear NFAT1-GFP under conditions identical to (C).
(F) Graph shows relative gel shift for NFAT1-GFP, as in (D).
(G) Gel shift for the SP3NFAT1-NFAT4-GFP construct.
(H) Graph plots relative gel shift for SP3NFAT1-NFAT4-GFP, as in (D). For the gels in (C), (E), and (G), conditions for each lane are shown only in (G). For the resting condition (indicated as Rest), no detectable NFAT was seen in any of the gels because the gels show nuclear extract and, at rest, there is very little NFAT in the nucleus.
(I) Blot compares kinetics of phosphorylation of serine 165 in NFAT4 from the nuclear fraction, denoted as P-S165.
(J) As in (I), but phosphorylation of serine 177 (P-S177) in NFAT1 was measured.
(K) Graphs plot the kinetics of phosphorylation of serine 165 NFAT4 and SP3N1-N4 or serine 177 in NFAT1.
In (C), (E), (G), (I), and (J), HH3 (histone H3) was used as a nuclear marker and ERK was taken as a cytoplasmic marker. Rest denotes the non-stimulated state. Thap + LMB represents the gel shift seen 40 min after stimulation with thapsigargin in the presence of LMB and external Ca2+, when NFAT had fully accumulated in the nucleus. Nuclear extract was isolated at the different times indicated. In all panels, 100 nM leptomycin B was added 10 min before thapsigargin and then maintained throughout. In the graphs, data are represented as mean ± SEM. See also Figure S5.
In the Ar-5 T cell clone, significant rephosphorylation of NFAT1 occurs within 15 min (Loh et al., 1996), which is considerably faster than the rate we find in HEK293 cells (Figure 5F). In RBL-2H3 cells, NFAT1 was exported out of the nucleus with a rate constant of ∼0.06 min−1 (Yissachar et al., 2013), which is ∼4-fold faster than the rate constant (∼0.014 min−1) for the same isoform in HEK293 cells. Hence, the kinetics of NFAT1 export varies between different cell types, perhaps reflecting the activities of nuclear kinases.
Although gel-shift analysis is widely used to measure NFAT phosphorylation status, shifts can be induced by other post-translational modifications. Additionally, since multiple serines are phosphorylated and then dephosphorylated (Okamura et al., 2000), the shift is a smear rather than a discrete change, which renders quantification somewhat selective. Because we analyzed the shifts for NFAT1 and NFAT4 in the same way, the relative difference in dephosphorylation/rephosphorylation kinetics seems valid. Nevertheless, we utilized an alternative approach to compare the kinetics of NFAT4 rephosphorylation/dephosphorylation with that of NFAT1. Serine 165 on NFAT4 is close to serine 177 on NFAT1; both are phosphorylated at rest and dephosphorylated by calcineurin, and commercial antibodies to these phosphorylated residues are available. We therefore tracked the phosphorylated state of each NFAT isoform using phospho-specific antibodies. At rest, both isoforms were phosphorylated and restricted to the cytoplasmic fraction (Figures S5D and S5E), with no detectable phosphorylated form in the nucleus (Figures 5I and 5J). Stimulation with thapsigargin for 30 min in the presence of leptomycin B led to a marked decrease in phosphorylation of both NFAT isoforms in cytoplasmic extract (Figures S5D and S5E), with no detectable change in the phosphorylated nuclear levels (Figures 5I and 5J). This is because only dephosphorylated NFAT migrates into the nucleus, and we are tracking phosphorylated residues. However, subsequent removal of external Ca2+ coupled with addition of cyclosporine A in the continued presence of leptomycin B led to rapid rephosphorylation of nuclear NFAT4 (Figure 5I) with a half-time of ∼1.1 min (Figure 5K). Serine 177 on NFAT1, by contrast, was phosphorylated considerably more slowly (Figures 5J and 5K), with a half-time of ∼35 min. In the SP3N1-NFAT4 chimera, serine 165 was rephosphorylated more slowly than NFAT4 but faster than NFAT1 (Figure 5K and Figures S5F and S5G; half-time of ∼6.4 min), consistent with the gel-shift data in Figure 5H.
It is important to note that these phosphorylation studies detect only one phosphorylated residue in each NFAT isoform and do not represent the kinetics of total rephosphorylation. It is possible that serine 165 is the first residue in NFAT4 to be rephosphorylated, whereas serine 177 in NFAT1 is among the last, which could explain the different kinetics. The fact that serine 165 in the SP3N1-NFAT4 chimera was phosphorylated ∼6-fold more slowly than in NFAT4, which is broadly similar to the 3.5-fold slower kinetics seen in the gel-shift assay, suggests NFAT4 is indeed phosphorylated more quickly than NFAT1 within the nucleus.
Differences in Cytoplasmic Rephosphorylation Rates of NFAT Isoforms
Taking advantage of leptomcyin B, our new data in Figure 5 have enabled us to quantify the kinetics of NFAT rephosphorylation within the nucleus (k6 reaction in Figure 6A). We therefore designed experiments to measure NFAT rephosphorylation kinetics in the cytoplasm (k2 reaction in Figure 6A). Acetate inhibits NFAT migration into the nucleus by preventing association of the NFAT-importin β complex with tubulin α (Ishiguro et al., 2011). NFAT dephosphorylation proceeded normally, but the dephosphorylated protein was trapped within the cytoplasm (Ishiguro et al., 2011). We reasoned that this approach might enable us to measure the kinetics of NFAT rephosphorylation within the cytoplasm, independent of any nuclear flux. We first confirmed that acetate inhibited translocation of NFAT1 and NFAT4 into the nucleus (Figures 6B and 6C). We then activated CRAC channels with thapsigargin in the presence of acetate to drive NFAT dephosphorylation. After 20 min, a time sufficient for NFAT dephosphorylation (Kar et al., 2011), we rapidly inhibited calcineurin activity by terminating Ca2+ influx in the presence of cyclosporine A. This enabled the measure of NFAT rephosphorylation within the cytoplasm (k2). NFAT4 was rephosphorylated quickly, with a half-time of ∼4 min (Figures 6D and 6E). By contrast, NFAT1 was rephosphorylated considerably more slowly, with only ∼40% recovery by 20 min (Figures 6D and 6E).
Figure 6.

Cytoplasmic Rephosphorylation of NFAT Isoforms
(A) The various steps regulating NFAT dynamics are shown.
(B) Treatment with acetate (5 mM) inhibits movement of NFAT1-GFP or NFAT4-GFP to the nucleus in response to thapsigargin stimulation.
(C) Aggregate data are compared. Each bar represents mean data from three independent experiments. Acetate was applied 10 min before stimulation and then maintained throughout.
(D) NFAT isoform rephosphorylation was measured in cytoplasmic extract at different times after termination of calcineurin activity.
(E) Graphs summarize averaged data from two independent experiments, as in (D).
(F) Blots compare the kinetics of reposhosphorylation of Ser165 on NFAT4 and Ser177 on NFAT1 in cytoplasmic extract.
(G) The graph summarizes data from two independent gels, as in (F). Zero intensity at time 0 corresponds to full dephosphorylation of NFAT after thapsigargin stimulation (F).
(H) Images compare NFAT1-GFP and NFAT4-GFP nuclear accumulation in experiments using a two-pulse protocol. In the single-pulse protocol, LTC4 was applied for 2 min in Ca2+-containing external solution (pulse 1) and then washed for 38 min in Ca2+-free solution (when the image was taken). In the paired-pulse protocol, after the first pulse, cells were washed for 8 min in Ca2+-free solution and then a second identical LTC4 pulse was applied for 2 min in external Ca2+. Cells were then exposed to Ca2+-free solution for a further 28 min before images were taken. Aggregate data from several independent experiments are shown in the histogram. Each bar is the mean of between 7 and 15 cells.
In (C), (E), (G), and (H), data are represented as mean ± SEM. See also Figure S6.
Similar results were seen with the phospho-serine antibodies against NFAT1 and NFAT4. NFAT4 was rephosphorylated significantly more quickly than NFAT1 (Figures 6F and 6G), consistent with the gel-shift data (Figure 6E).
NFAT1 but Not NFAT4 Nuclear Accumulation Shows Paired-Pulse Facilitation
A consequence of the faster k2 reaction for NFAT4 than NFAT1 is that NFAT4 will be rephosphorylated in the cytoplasm considerably more quickly following deactivation of calcineurin. NFAT proteins are extensively phosphorylated by protein kinases (Okamura et al., 2000). A slower k2 for NFAT1 means that, following a brief pulse of Ca2+ that is insufficient for complete NFAT1 dephosphorylation, the rephosphorylation process would proceed slowly. Hence, a second identical Ca2+ pulse a few minutes later should result in further net dephosphorylation and thus complete activation of NFAT1, imparting a form of paired-pulse facilitation to NFAT1 nuclear migration. Our previous experiments demonstrated robust paired-pulse facilitation to NFAT1 (Kar et al., 2012), although the underlying molecular basis was unclear. On the other hand, the faster rate of rephosphorylation of NFAT4 would result in more extensive rephosphorylation during the interpulse interval, and this would not result in additional dephosphorylation when a second identical Ca2+ pulse is applied, preventing paired-pulse facilitation. To test this, we measured NFAT4 nuclear dynamics following brief single or paired pulses of LTC4. Activation of cysLT1 receptors to a single 2 min pulse failed to induce any detectable NFAT1 or NFAT4 nuclear migration over a subsequent 38 min period (Kar et al., 2012; Figure 6H). However, two identical LTC4 pulses, applied 8 min apart, resulted in prominent NFAT1 but not NFAT4 nuclear accumulation 28 min later (38 min after the first pulse; Figure 6H). Similar paired-pulse facilitation was obtained when two identical Ca2+ pulses were applied to thapsigargin-treated cells instead (Figure S6).
Cytokine Expression Is Differentially Regulated by Nuclear Ca2+ in a Cell-Specific Context
An important physiological ramification of our findings is that expression of the same gene induced by the same agonist should be differentially regulated by nuclear Ca2+ in different cell types, depending on which NFAT protein dominates. 16-HBE bronchial airway epithelial cells express significantly more NFAT4 than NFAT1 (Samanta et al., 2014), whereas the converse is the case in RBL-1 mast cells (data not shown). We therefore stimulated 16-HBE and RBL-1 mast cells with LTC4 at a concentration that evoked oscillatory Ca2+ signals and measured expression of the interleukin-5 (IL-5) gene, which contains the GGAA core nucleotides for an NFAT binding site within the promoter (De Boer et al., 1999), in the absence or presence of nuclear InsP3 buffer. In 16-HBE cells, LTC4 stimulation led to a robust increase in transcription of IL-5 (Figure 7A) as well as in protein expression (Figure 7C). Pre-treatment with the calcineurin inhibitor cyclosporine A suppressed these responses (Figures 7A and 7C), demonstrating transcription was through the calcineurin-NFAT pathway. Expression of nuclear InsP3 buffer prevented LTC4-induced IL-5 transcription and subsequent protein expression (Figures 7A and 7C), consistent with a major role for nuclear Ca2+ signaling. By contrast, thapsigargin-evoked expression of IL-5 was unaffected by nuclear InsP3 buffer but remained sensitive to cyclosporine A (Figures 7B and 7D). Therefore InsP3 nuclear buffer does not interfere with the transcriptional process itself.
Figure 7.

Cytokine Expression Is Regulated By nuclear Ca2+ in a Cell-Specific Context
Data in (A)–(D) are from 16-HBE bronchial epithelial cells.
(A) RT-PCR shows IL-5 expression is increased by LTC4 and this is prevented by cyclosporine A or after expression of the IP3-NLS buffer. Data have been normalized to rest levels (% control).
(B) As in (A), but now thapsigargin is the stimulus.
(C) Western blot compares IL-5 protein expression following stimulation with LTC4. IL-5 expression has been normalized to ERK levels.
(D) As in (C), but the stimulus is thapsigargin. Data in (E)–(H) are from RBL-1 mast cells.
(E) LTC4 increases IL-5 transcription, and this is unaffected by IP3-NLS but suppressed by cyclosporine A.
(F) As in (E), but thapsigargin is the stimulus.
(G) Western blot compares IL-5 protein expression following stimulation with LTC4.
(H) As in (F), but thapsigargin is the stimulus.
In all panels, histograms summarize data from three independent experiments. In all histograms, data are represented as mean ± SEM.
In RBL-1 cells, where NFAT1 dominates, LTC4 also increased expression of IL-5 (Figures 7E and 7G). These responses were unaffected by nuclear InsP3 buffer but were suppressed by cyclosporine A (Figures 7E and 7G). Thapsigargin-induced IL-5 expression was also unaffected by the presence of nuclear InsP3 buffer (Figures 7F and 7H).
Although NFAT and c-fos often interact synergistically to regulate gene expression, block of the non-receptor tyrosine kinase Syk, a maneuver that abolishes cysLT1 receptor activation of c-fos expression (Ng et al., 2012), had no effect on IL-5 gene expression to LTC4 in RBL-1 cells (3.73-fold ± 0.10-fold increase in IL-5 mRNA relative to basal following LTC4 stimulation in control cells and 3.69-fold ± 0.13-fold increase in the presence of Syk inhibitor, p > 0.1; corresponding changes in protein expression were 6.14 ± 0.42 and 6.29 ± 0.57; p > 0.1). Collectively, the data indicate that NFAT-driven gene expression in different cell types can be differentially regulated by nuclear Ca2+. Moreover, the same gene (IL-5) can be differentially controlled by nuclear Ca2+, depending on the NFAT isoform present.
Conclusion
Stimulation of cysLT1 receptors increases cytoplasmic InsP3, which lowers the endoplasmic reticulum Ca2+ content sufficiently for CRAC channels in the plasma membrane to open, a step necessary and sufficient for NFAT1 activation (Kar et al., 2011, Kar and Parekh, 2015). The closely related NFAT4 protein additionally requires a nuclear Ca2+ rise (Kar and Parekh, 2015). Our new data demonstrate that cytoplasmic InsP3 diffuses into the nucleus to activate InsP3 receptors spanning the inner nuclear membrane, which then increase nucleoplasmic Ca2+, enabling prolonged NFAT4 activation. NFAT4 activation is therefore dependent on two spatially segregated Ca2+ signals: one at the plasma membrane and the other in the nucleus several micrometers away. The different requirements for nuclear Ca2+ and thus nuclear Ca2+-calcineurin reflect the different rates of rephosphorylation of NFAT1 and NFAT4 in the nucleus, with rephosphorylation of the latter proceeding several-fold more quickly. The slower rephosphorylation rate of NFAT1 suggests this family member would be considerably better suited for imparting short-term memory to gene expression (Kar et al., 2012). Similar differences in cytoplasmic rephosphorylation rates are also important, playing a major role in the development of paired-pulse facilitation for NFAT1 only.
Remarkably, the spatially restricted Ca2+ signals activate NFAT4 without a role for bulk oscillatory Ca2+ signals in the relatively vast expanse of the enveloping cytoplasm. A corollary of this is that cytoplasmic Ca2+ oscillations, long considered the universal currency of physiological Ca2+ signaling, are not essential for the form of receptor-driven excitation-transcription coupling we describe. The oscillatory cytoplasmic Ca2+ signal here may be a consequence of the need to deplete the Ca2+ stores sufficiently for CRAC channels to activate (Bird et al., 2009, Brandman et al., 2007, Parekh et al., 1997), coupled with other roles such as the enhancement of mitochondrial ATP production that is needed to support energy-dependent Ca2+ responses (Hajnóczky et al., 1995).
Our data also show that expression of the same gene in response to the same agonist is controlled differently, depending on which NFAT protein dominates. Whereas NFAT1 activation is governed only by Ca2+ microdomains near CRAC channels, a stronger stimulus is needed for NFAT4 nuclear accumulation, because InsP3 has to overcome breakdown by cytoplasmic enzymes to enter the nucleus and release nuclear Ca2+. The dependence of different NFAT proteins on different stimulus intensities (Kar and Parekh, 2015) will both increase the bandwidth of excitation-transcription coupling and recruit different gene expression profiles. Finally, our results demonstrate how a ubiquitous and globally diffusible second messenger like InsP3 can nevertheless be used to activate different transcription factor isoforms through generation of coordinated yet spatially distinct sub-cellular Ca2+ signals.
Experimental Procedures
Cells
HEK293 (Kar et al., 2011), RBL-1 (Kar et al., 2012), and 16-HBE (Samanta et al., 2014) cells were cultured as described. HEK293 cells were used in Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6 and RBL-1 and 16-HBE cells in Figure 7.
cDNA Constructs and Transfection
HEK293 and 16-HBE cells were transfected using lipofectamine, as described (Kar et al., 2011, Samanta et al., 2014). RBL-1 cells were transfected using the Amaxa system (Kar et al., 2012). cDNA for NFAT4-GFP, parvalbumin constructs, and cytoplasmic GCaMP3 were from Addgene (deposited by Dr. Anjana Rao, Dr. Anton Bennett, and Dr. Jin Zhang, respectively). Nuclear and cytoplasmic InsP3 buffer were gifts from Dr. Maria de Fatima Leite, Federal University of Minas Gerais, Brazil. NFAT1-GFP was a gift from Dr. Jennings Worley. Alterations to these constructs were done by Mutagenex. All plasmids were used at 1 μg, and experiments commenced 24–48 hr after transfection.
NFAT Migration
Tagged NFAT protein levels in the cytoplasm and nucleus were measured using the IMAGO charge-coupled device camera-based system from TILL Photonics, with a × 100 oil immersion objective (numerical aperture 1.3) (Kar and Parekh, 2015). Regions of interest of identical size were drawn in the cytoplasm and nucleus of each cell, and fluorescence was computed. Nuclear localization was confirmed by costaining with the nuclear dye DAPI. Unless otherwise indicated, we calculated the nuclear/cytoplasmic ratio of tagged NFAT as a function of stimulus time.
Cytoplasmic and Nuclear Ca2+ Measurements
Cytoplasmic and nuclear Ca2+ measurements, using GCaMP3 constructs, were carried out at room temperature by using the IMAGO charge-coupled device camera-based system from TILL Photonics (Kar et al., 2012). Cells were placed in standard external solution composed of 145 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM D-glucose, and 10 mM HEPES at pH 7.4, with NaOH and were excited at 489 nm (typically 100 ms exposure), at 0.5 Hz. Fura-2 experiments (Figure S1) were carried out as described (Kar et al., 2012). Ca2+ signals are plotted either as F/F0, which denotes the increase in fluorescence divided by the resting level for the GCaMP3 reporters, or R, which denotes the 356/380 nm ratio, for Fura-2. Simultaneous measurements of cytoplasmic and nuclear Ca2+ were performed with fluo-4, as described (Kar and Parekh, 2015).
Immunogold Electron Microscopy
Briefly, cells were fixed with 3% paraformaldehyde/0.05% glutaraldehyde in 0.1 M phosphate buffer for 10 min, stained with uranyl acetate (2% w/v in distilled water), dehydrated through increasing concentrations of methanol (70%–100%), and embedded in LR Gold (London Resin Company). Ultrathin sections (50–80 nm) were prepared by use of a Reichert Ultracut S ultramicrotome (Leica), mounted on 200-mesh nickel grids, incubated at room temperature for 2 hr with a monoclonal anti-InsP3 receptor type I antibody (dilution 1:100; Abcam) and for 1 hr with anti-mouse IgG −15 nm gold complex (British Biocell International). All antisera were diluted in 0.1 M phosphate buffer containing 0.1% egg albumin (pH 7.2). As a control the primary antibody was replaced with non-immune sera. After immunolabeling, sections were lightly counterstained with lead citrate and uranyl acetate and examined with a JEOL transmission electron microscope (JEM-1010, JEOL), and images were collected with a Gatan Orius digital camera (Gatan).
Cytoplasmic Staining
HEK293 cells were fixed with paraformaldehyde (4%) at room temperature and permeabilized with PBS/Triton X-100 (0.5%). Cells were then stained overnight with anti-InsP3 receptor type I antibody (Abcam), used at 1:100 dilution. Primary antibody was then visualized with Alexa Fluor 488 goat anti-mouse secondary antibody (1:500)
Western Blot
Total cell lysates (40 μg) were analyzed by SDS-PAGE on a 10% or 12% (IL-5) gel. Membranes were blocked with 5% nonfat dry milk in PBS plus 0.1% Tween 20 (PBST) buffer for 1 hr at room temperature (Kar and Parekh, 2015). For phospho-NFAT blots, we used TBST (Tris buffer saline with Tween 20). Membranes were washed with PBST three times and then incubated with primary antibody for 24 hr at 4°C. Antibodies against ERK (1:5,000, rabbit polyclonal; Santa Cruz Biotechnology), GFP (1:2,000, rabbit polyclonal; Cell Signaling), HH3 (Histone H3; 1:5,000, rabbit polyclonal; Abcam), InsP3 type I receptor (1:1,000, mouse monoclonal; Abcam), anti-phospho Ser177 NFAT1 (Santa Cruz; 1:2,000 dilution), anti-phospho Ser165 NFAT4 (Abcam; 1:2,000), and IL-5 (Santa Cruz; 1:500) were used. Secondary antibodies for rabbit or mouse were used at 1:5,000 or 1:3,000 dilutions. The membranes were then washed with PBST again and incubated with peroxidase-linked anti-rabbit IgG from Santa Cruz Biotechnology or anti-mouse IgG from BD Bioscience for 1 hr at room temperature. After washing with PBST, the bands were detected by an enhanced chemiluminescence (ECL) plus western blotting detection system (Amersham Biosciences). Blots were analyzed by UN-Scan software. Nuclear and cytoplasmic extracts were separated as before (Kar and Parekh, 2015) and assessed by the presence of HH3 in nuclear but not cytoplasmic extract and ERK2 in the cytoplasmic but not nuclear extract.
Gel shifts were quantified in ImageJ. Intensity of the region corresponding to the dephosphorylated state (lower band; obtained after stimulation with thapsigargin in 2 mM external Ca2+ for 20 min; see relevant figure legends) was measured and the region (same dimensions and position) was transposed to adjacent lanes. Data are presented relative to the intensity of the dephosphorylated state. Gel analysis was carried out on blots from at least two independent experiments.
RT-PCR
16-HBE cells transfected with cys LT1 receptors or wild-type RBL-1 cells were stimulated with thapsigargin or LTC4 in DMEM for 20 min, washed five times with DMEM (without thapsigargin or LTC4), and then kept in an incubator at 37°C for 6–8 hr. Cells were then washed with PBS, and total RNA was extracted using an RNeasy Mini Kit (QIAGEN). RNA was quantified spectrophotometrically by absorbance at 260 nm. Total RNA (1 μg) was reverse transcribed using the iScriptTM cDNA Synthesis Kit (Bio-Rad), according to the manufacturer’s instructions. Two-microliter samples (cDNA), from RT-PCR, were subjected to polymerase chain reaction (PCR) with the specific primers and GoTaq Green master mix (Promega). The mRNA levels were normalized to GAPDH for 16-HBE cell and β-actin for RBL-1 cells.
IL-5 (human): Forward-5′-CTGAGGATTCCTGTTCCTGT-3′; Reverse-5′-CAACTTTCTATTATCCACTC-3′
GAPDH (human): Forward-5′-GGTATCGTGGAAGGACTCAT-3′; Reverse-5′-CCACCCTGTTGCTGTAGCCAAATTC-3′
IL-5 (rat): Forward-5′-AGACCGGTCATACATGCACA-3′; Reverse-5′-TCGTCTCATTGCTCGTCAAC-3′
β-actin (rat): Forward- 5′-TTGTAACCAACTGGGACGATATG-3′; Reverse- 5′-GATCTTGATCTTCATGGTGCTAGG-3′
Mathematical Modeling
The details of mathematical modeling are explained in the Supplemental Information.
Statistical Analysis
Data are presented as the mean ± SEM. Statistical significance was determined by using Student’s t test. ∗p < 0.05 and ∗∗p < 0.01.
Author Contributions
P.K. and A.B.P. designed experiments. P.K. carried out the experiments. Electron microscopy was conducted by H.C.C. Analyses of data were performed by P.K. and A.B.P. Discussion of the model and how to identify various rate constants involved P.K., G.R.M., and A.B.P. The modeling was executed by G.R.M. A.B.P. wrote the manuscript.
Acknowledgments
This work was supported by an MRC Programme grant to A.B.P. (grant number LO1047X). G.R.M is supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and Royal Society (grant number 101222/Z/13/Z). We thank Dr. Juris Galvanovskis for help with confocal microscopy measurements of nuclear Ca2+.
Published: November 17, 2016
Footnotes
Supplemental Information includes six figures, one table, supplemental equations, and a zipped data file containing calcineurin code and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.11.011.
Accession Numbers
Original imaging data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/v8sfxkh9pw.1.
Supplemental Information
References
- Allbritton N.L., Meyer T., Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- Alonso M.T., García-Sancho J. Nuclear Ca(2+) signalling. Cell Calcium. 2011;49:280–289. doi: 10.1016/j.ceca.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Bazzazi H., Sang L., Dick I.E., Joshi-Mukherjee R., Yang W., Yue D.T. Novel fluorescence resonance energy transfer-based reporter reveals differential calcineurin activation in neonatal and adult cardiomyocytes. J. Physiol. 2015;593:3865–3884. doi: 10.1113/JP270510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E., Dastrange M., Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. USA. 2005;102:5138–5143. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird G.S., Hwang S.Y., Smyth J.T., Fukushima M., Boyles R.R., Putney J.W.J., Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 2009;19:1724–1729. doi: 10.1016/j.cub.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman M.D., Fearnley C., Smyrnias I., MacDonald F., Roderick H.L. An update on nuclear calcium signalling. J. Cell Sci. 2009;122:2337–2350. doi: 10.1242/jcs.028100. [DOI] [PubMed] [Google Scholar]
- Brandman O., Liou J., Park W.S., Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer M.L., Mordvinov V.A., Thomas M.A., Sanderson C.J. Role of nuclear factor of activated T cells (NFAT) in the expression of interleukin-5 and other cytokines involved in the regulation of hemopoetic cells. Int. J. Biochem. Cell Biol. 1999;31:1221–1236. doi: 10.1016/s1357-2725(99)00069-2. [DOI] [PubMed] [Google Scholar]
- Di Capite J., Ng S.-W., Parekh A.B. Decoding of cytoplasmic Ca(2+) oscillations through the spatial signature drives gene expression. Curr. Biol. 2009;19:853–858. doi: 10.1016/j.cub.2009.03.063. [DOI] [PubMed] [Google Scholar]
- Echevarría W., Leite M.F., Guerra M.T., Zipfel W.R., Nathanson M.H. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 2003;5:440–446. doi: 10.1038/ncb980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornerod M., Ohno M., Yoshida M., Mattaj I.W. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Gerasimenko O.V., Gerasimenko J.V., Tepikin A.V., Petersen O.H. Calcium transport pathways in the nucleus. Pflugers Arch. 1996;432:1–6. doi: 10.1007/s004240050098. [DOI] [PubMed] [Google Scholar]
- Gwack Y., Sharma S., Nardone J., Tanasa B., Iuga A., Srikanth S., Okamura H., Bolton D., Feske S., Hogan P.G., Rao A. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–650. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G., Robb-Gaspers L.D., Seitz M.B., Thomas A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Humbert J.P., Matter N., Artault J.C., Köppler P., Malviya A.N. Inositol 1,4,5-trisphosphate receptor is located to the inner nuclear membrane vindicating regulation of nuclear calcium signaling by inositol 1,4,5-trisphosphate. Discrete distribution of inositol phosphate receptors to inner and outer nuclear membranes. J. Biol. Chem. 1996;271:478–485. doi: 10.1074/jbc.271.1.478. [DOI] [PubMed] [Google Scholar]
- Ishiguro K., Ando T., Maeda O., Watanabe O., Goto H. Cutting edge: tubulin α functions as an adaptor in NFAT-importin β interaction. J. Immunol. 2011;186:2710–2713. doi: 10.4049/jimmunol.1003322. [DOI] [PubMed] [Google Scholar]
- Kar P., Parekh A.B. Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol. Cell. 2015;58:232–243. doi: 10.1016/j.molcel.2015.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P., Nelson C., Parekh A.B. Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J. Biol. Chem. 2011;286:14795–14803. doi: 10.1074/jbc.M111.220582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P., Nelson C., Parekh A.B. CRAC channels drive digital activation and provide analog control and synergy to Ca(2+)-dependent gene regulation. Curr. Biol. 2012;22:242–247. doi: 10.1016/j.cub.2011.12.025. [DOI] [PubMed] [Google Scholar]
- Kehlenbach R.H., Dickmanns A., Gerace L. Nucleocytoplasmic shuttling factors including Ran and CRM1 mediate nuclear export of NFAT In vitro. J. Cell Biol. 1998;141:863–874. doi: 10.1083/jcb.141.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Hajnóczky G., Thomas A.P. Propagation of cytosolic calcium waves into the nuclei of hepatocytes. Cell Calcium. 1994;16:247–258. doi: 10.1016/0143-4160(94)90088-4. [DOI] [PubMed] [Google Scholar]
- Lipp P., Thomas D., Berridge M.J., Bootman M.D. Nuclear calcium signalling by individual cytoplasmic calcium puffs. EMBO J. 1997;16:7166–7173. doi: 10.1093/emboj/16.23.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh C., Shaw K.T.-Y., Carew J., Viola J.P.B., Luo C., Perrino B.A., Rao A. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J. Biol. Chem. 1996;271:10884–10891. doi: 10.1074/jbc.271.18.10884. [DOI] [PubMed] [Google Scholar]
- Macián F., López-Rodríguez C., Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- Macián F., García-Cózar F., Im S.H., Horton H.F., Byrne M.C., Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- Martinez G.J., Pereira R.M., Äijö T., Kim E.Y., Marangoni F., Pipkin M.E., Togher S., Heissmeyer V., Zhang Y.C., Crotty S. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity. 2015;42:265–278. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H., Nakano K., Tepikin A.V., Petersen O.H. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- Müller M.R., Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 2010;10:645–656. doi: 10.1038/nri2818. [DOI] [PubMed] [Google Scholar]
- Ng S.W., Bakowski D., Nelson C., Mehta R., Almeyda R., Bates G., Parekh A.B. Cysteinyl leukotriene type I receptor desensitization sustains Ca2+-dependent gene expression. Nature. 2012;482:111–115. doi: 10.1038/nature10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura H., Aramburu J., García-Rodríguez C., Viola J.P., Raghavan A., Tahiliani M., Zhang X., Qin J., Hogan P.G., Rao A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Parekh A.B., Fleig A., Penner R. The store-operated calcium current I(CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Rao A., Luo C., Hogan P.G. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Samanta K., Bakowski D., Parekh A.B. Key role for store-operated Ca2+ channels in activating gene expression in human airway bronchial epithelial cells. PLoS ONE. 2014;9:e105586. doi: 10.1371/journal.pone.0105586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman J.J., Bers D.M. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys. J. 2008;95:4597–4612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A.P., Bird G.S., Hajnóczky G., Robb-Gaspers L.D., Putney J.W.J., Jr. Spatial and temporal aspects of cellular calcium signaling. FASEB J. 1996;10:1505–1517. [PubMed] [Google Scholar]
- Yissachar N., Sharar Fischler T., Cohen A.A., Reich-Zeliger S., Russ D., Shifrut E., Porat Z., Friedman N. Dynamic response diversity of NFAT isoforms in individual living cells. Mol. Cell. 2013;49:322–330. doi: 10.1016/j.molcel.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Zima A.V., Bare D.J., Mignery G.A., Blatter L.A. IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J. Physiol. 2007;584:601–611. doi: 10.1113/jphysiol.2007.140731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.