

Abstract
The mortality rate for Stevens-Johnson syndrome (SJS) is estimated to be ∼12% and for toxic epidermal necrolysis (TEN) it is around 30%. It continues to be a severe life-threatening drug reaction. We present a 60-year-old Caucasian man with a medical history significant for breast cancer status post mastectomy and chemotherapy with docetaxel and cyclophosphamide who presented with severe mucositis and a progressing skin rash consistent with SJS. He was started on high-dose corticosteroids and IVIG but continued to have worsening mucosal ulcerations and severe bleeding from the oral, conjunctival and genital mucosa. He underwent several rounds of plasmapheresis and additional high-dose steroids with mild improvement in the mucocutaneous manifestations. He subsequently developed respiratory failure, which required mechanical ventilation, as well as disseminated intravascular coagulation, diffuse alveolar haemorrhage, with Pneumocystis jirovecii pneumonia which led to his demise on hospital day 15.
Background
Steven-Johnson Syndrome (SJS) is most commonly an idiosyncratic mucocutaneous syndrome along a continuum of bullous diseases that include toxic epidermal necrolysis (TEN) which are differentiated based on the percent of total body surface area involved.1 Although there is no widely accepted consensus defining diagnostic criteria, SJS is characterised by keratinocyte death (acantholysis) which ultimately results in subepidermal separation and the formation of bullae on <10% of the total skin surface along with mucosal involvement.2 SJS and TEN are considered rare, but oftentimes fatal adverse drug reactions occur anywhere between 5 and 56 days after initial drug exposure; however, the average time period is within 14 days of drug exposure.3 4 Unfortunately, there is no identifiable exposure or a known drug trigger in up to 25% of cases with SJS. However, in the absence of a known trigger, a history implicating drugs that can cause SJS and with an appropriate clinical presentation, the diagnosis of SJS can be made. The treatment for SJS remains the same regardless of the implicated triggering medication.
Case presentation
A 60-year-old Caucasian male inmate with medical history notable for breast cancer and hepatitis C-induced liver cirrhosis presented with a chief symptom of a progressively worsening rash, oral ulcerations and odynophagia. He had partial right-sided mastectomy in November of 2015 prior to completing three cycles of chemotherapy with cyclophosphamide and docetaxel at an outside hospital. Unfortunately, he suffered localised severe mucositis without cutaneous involvement after his first two sessions of chemotherapy that improved within 1–2 weeks. It is unclear if chemotherapy doses were reduced, as the outside medical records were unobtainable. Amost 3 weeks prior to admission, after his third cycle of chemotherapy, his oral mucositis rapidly progressed leading to the development of odynophagia.
On initial evaluation his vital signs were significant for a blood pressure of 112/64 mm Hg, heart rate of 109 bpm, temperature of 37.2°C (99°F) and blood oxygen saturation level of 90% on room air. Physical examination demonstrated numerous ulcerations of the oropharynx, erosions of the upper back, chest and neck, and bleeding from the oral, conjunctival and genital mucous membranes. His lungs were clear to auscultation and his cardiac examination was unremarkable except for tachycardia. He was initially admitted to general medicine where an immediate consult to dermatology was placed. The day after admission he developed hypotension, hypoxaemia and metabolic acidemia requiring transfer to the intensive care unit (ICU). In the ICU it was noted that he had increased bleeding from his eyes (figure 1), his mouth (figure 2), genital mucosa (figure 3) as well as a progressing rash on his upper back (figure 4). He was then diagnosed with disseminated intravascular coagulation (DIC). Coagulation profile showed prolonged prothrombin time of 38 s, activated partial prothrombin time of 46.8 s and international normalised ratio of 4. Other blood workup was significant for fibrinogen level of 70 mg/dL and D-dimer of 10.76 µg/mL. His platelet counts markedly dropped to 4×103/µL, with his haemoglobin and haematocrit remaining around 7.0 gm/dL and 22%, respectively, despite ongoing blood products transfusion of packed red blood cells, cryoprecipitate and fresh frozen plasma.
Figure 1.
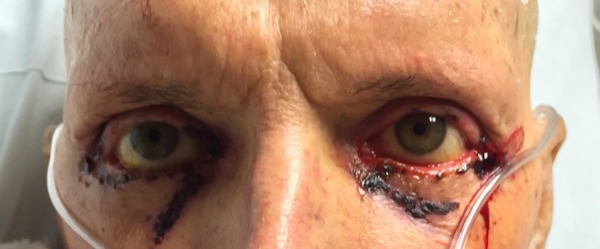
Patient had profound conjunctival erythema with bleeding lateral and medial canthal ulcerations as well as bleeding ulcerations along the bilateral lower lid margins.
Figure 2.
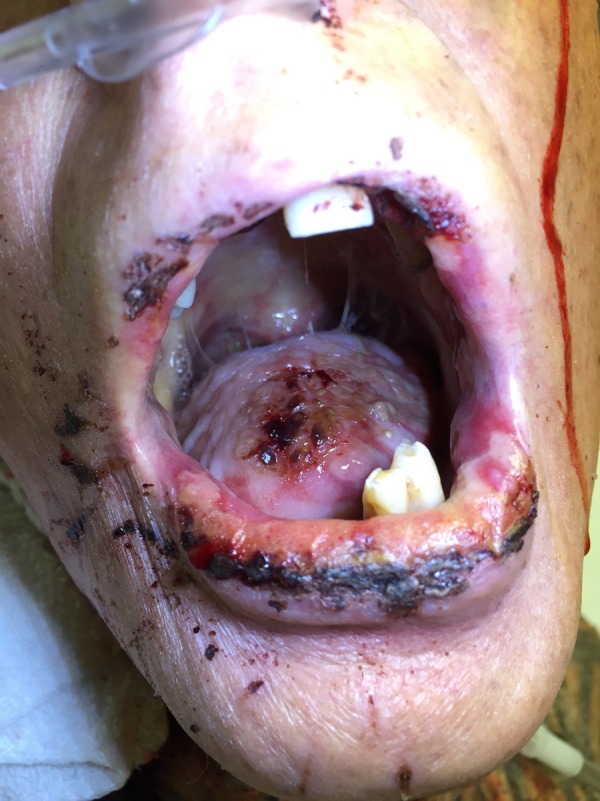
Polymorphous oral lesions consisting of annular and lichenoid ulcerations with haem crust and mild purulent exudate involving the tongue, buccal mucosa, lip mucosa, palate and vermillion lip.
Figure 3.
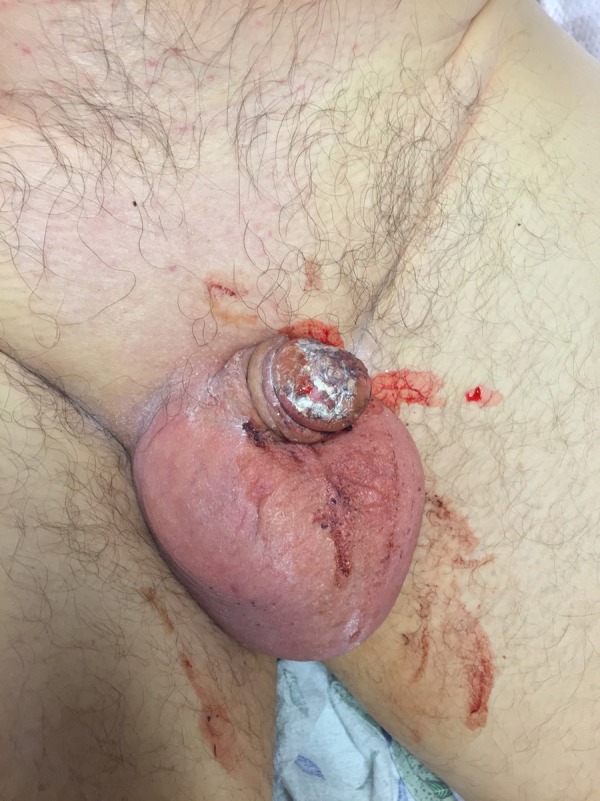
Gross blood from the urethra due to severe genital mucositis.
Figure 4.
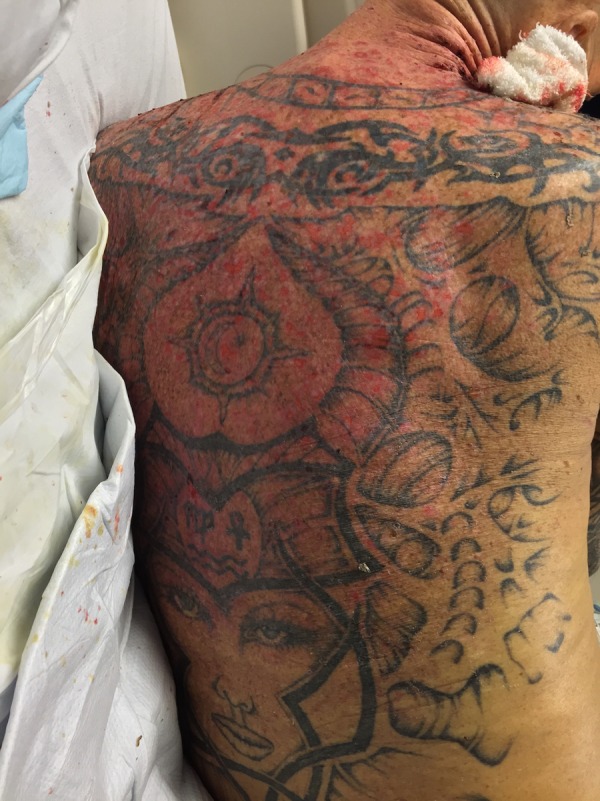
Rash along upper back, which consisted of diffuse erosions and background erythema with a positive Nikolsky sign.
On hospital day 4, he began to demonstrate signs of multiorgan failure with his alanine and aspartate transaminase levels peaked at 12 625 IU/L and 5841 IU/L, respectively, with an alkaline phosphatase of 423 IU/L. His hospital course was further complicated by worsening respiratory failure with haemoptysis and an evidence of pulmonary congestion seen on chest radiograph. A classic ‘crazy paving’ pattern was seen on CT scan (figure 5). A bronchoscopy was performed with a bronchoalveolar lavage (BAL), which had a sequential bloody return of the three subsequent aliquots, consistent with diffuse alveolar haemorrhage. BAL cytology was consistent with Pneumocystis jirovecii. On hospital day 4 and 5 the patient had a worsening renal failure with creatinine of 4 mg/dL and blood urea nitrogen of 94 mg/dL, fluid overload and hypotension, which required norepinephrine and continuous renal replacement therapy.
Figure 5.
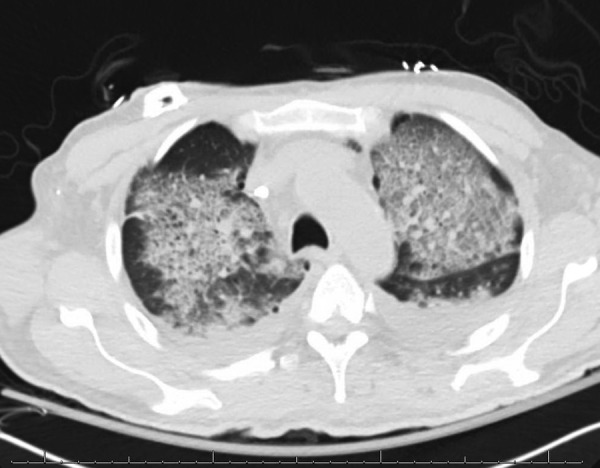
CT scan of the chest demonstrating the ‘crazy paving pattern’ which can be seen in diffuse alveolar haemorrhage and alveolar proteinosis.
Investigations
Dermatology was consulted on admission given the extensive involvement of his mucous membranes and skin. The patient exhibited polymorphous oral ulcerations, which were annular and lichenoid in appearance and erosions with extensive haem crust and purulent exudate involving the buccal mucosa, tongue, mucosal lip, palate and vermillion lip. Blood was also seen draining from bilateral medial conjunctiva and urethra. No anal involvement was seen. The upper back, upper chest and neck displayed shallow erosions with a positive Nikolsky sign. Two 4 mm punch biopsies were performed; one for H&E stains and the other for direct immunofluorescence (DIF). Serum was also drawn for indirect immunofluorescence (IIF) as well as ELISA examining for antibodies to envoplakin, desmoglein 1 and 3.
An extensive infectious disease workup was positive only for Mycoplasma pneumoniae IgG. Herpes simplex virus serology was negative. Additional investigations resulted in a negative cytomegalovirus serology, as well as a negative cryoglobulin, HIV and hepatitis B surface antigen and core antigen.
Biopsy results
H&E exhibited an interface dermatitis with ulceration with elements of acantholysis suggesting a differential diagnosis that included erythema multiforme, SJS and paraneoplastic pemphigus (PNP).
Direct immunofluorescence: Studies were negative, showing no immune complex deposition making SJS more likely.
Indirect immunofluorescence: Negative.
ELISA for envoplakin, desmoglein 1 and 3: Negative.
Differential diagnosis
PNP: The severity of oral mucosal ulceration extending to the vermillion lip with bleeding in addition to erosions on the neck, chest and back made this diagnosis high on the differential, although patient's oncological history was pertinent for breast cancer which is rarely associated with PNP.
SJS: Recent repeated exposure to docetaxel and cyclophosphamide; drugs that are known to cause SJS, with severe recurrent mucositis, rash and a new erosions with positive Nikolsky sign prompted concern for SJS.5 6
Erythema multiforme (EM) major: The combination of oral mucosal ulcerations and history of immunosuppression made Mycoplasma-induced erythema multiform major a possibility, although most cases of EM are in paediatric population.
Pemphigus vulgaris (PV): Owing to the skin and mucosal involvement; PV was a possibility, however, because of the severity of ocular and oral involvement, a paraneoplastic process was favoured.
Treatment
Our patient was initially started on broad-spectrum empiric antibiotics with meropenem, vancomycin and metronidazole. Intravenous immunoglobulin (IVIG) therapy was initiated with a goal of 400 mg/kg/day as well as high-dose methylprednisolone (500 mg daily for total of 5 days). He was only able to tolerate ∼1 day of IVIG therapy due to the development of an anuric renal failure. Additionally, he was started on Bactrim for the P. jirovecii on his BAL sample as well as voriconazole and acyclovir empirically due to his immunocompromised state. Steroid doses were decreased to 50 mg of hydrocortisone after his 5-day course of pulse dose steroids. He continued to receive transfusions of fresh frozen plasma, platelets, cryoprecipitate and packed red blood cells for coagulopathy and bleeding. Since IVIG initially was suspected in aiding the progression of renal failure, he underwent a total of three cycles of plasma exchange without resolution of his bleeding diathesis or his cutaneous manifestations. Since he had an anuric renal failure and being refractory to plasma exchange, the patient was placed on renal replacement therapy (RRT) and was given another 4 days of IVIG with the dose of ∼400 mg/kg/day, which led to a moderate improvement in his mucocutaneous bleeding and ulcerations.
Unfortunately, due to his need for numerous transfusions and intravenous medications he developed gross anasarca despite maximum fluid removal via RRT. On hospital day 15 the decision was made to pursue comfort care measures (withdrawal of care) and he unfortunately died shortly thereafter.
Outcome and follow-up
On hospital day 15 the decision was made to pursue comfort care measures (withdrawal of care) and he died shortly thereafter. No follow-up.
Discussion
The diagnosis of SJS is often based on clinical evidence suggestive of a recent exposure to an agent that has been known to cause SJS as well as histological evidence to support such a diagnosis, while ruling out other possible blistering disorders such as pemphigus and pemphigoid.7 Biopsy results often demonstrate more acute necrosis and ulcerations in TEN than SJS, but both will generally lack the immune complex deposition on immunofluorescence as seen in pemphigus or other autoimmune bullous diseases. Mucosal ulcerations, severe skin desquamation in combination with an offending drug and histological findings of acute epidermal necrosis, acantholysis and interface dermatitis without immune complex deposition are key to the diagnosis of SJS.
EM, which was another initial diagnostic possibility, is commonly associated with HSV infection, but mucosal involvement is more frequently associated with infectious aetiology such as M. pneumoniae.8 Although the PCR for HSV was negative, our patient had a positive M. pneumoniae IgG, but with a negative IgM, which makes the diagnosis of EM less likely. Histologically SJS and EM are difficult to distinguish as they both display acute epidermal acantholysis and dyskeratosis with interface dermatitis. In our patient, the diagnosis of SJS was based on the combination of recent exposure to drugs that are known to cause SJS, cyclophosphamide and docetaxel, with mucositis, rash and histopathological findings consistent with SJS rather than EM.
This patient was admitted to our hospital with a diagnosis of non-resolving chemotherapy-induced mucositis, but normal leucocyte and neutrophil counts 3 weeks after his last cycle made this extremely unlikely. Owing to the necrotic nature of his oral ulcerations, which extended to his vermillion lip in the setting of malignancy, PNP was highest on the differential next to SJS secondary to docetaxel and cyclophosphamide. PNP generally is associated with lymphoproliferative disease such as non-Hodgkin's lymphoma, chronic lymphocytic leukaemia, and multicentric Castleman disease.9 However, it is extremely uncommon to be noted in the presence of solid tumours, in which our patient had a history of breast cancer. Initial H&E results showed a suprabasilar process including pemphigus, SJS and EM in the differential. Further DIF, IIF and ELISA studies ruled out the possibility of a pemphigus disease process. It remains unclear, which of the chemotherapy agents induced SJS in this case, given they were both administered to our patient, simultaneously.
The typical treatment for SJS as well as TEN is the immediate withdrawal of the offending drug agent if still being administered.10 Additional key elements in successful treatment are supportive in nature; revolving around electrolyte replacement, fluids and prevention of infection.11 12 Although still somewhat controversial, the use of corticosteroids especially in higher doses for very short courses have been shown to be beneficial in some studies.13 Likewise, similar evidence is suggestive of a long-term benefit with the administration of IVIG; however, some small trials have shown some efficacy if given early in the course.14 The combination of IVIG and high-dose corticosteroids in small studies had been shown to offer some survival benefit.15 16 Owing to the initial severity of our patient's clinical condition on admission to the ICU, these may have offered little initial survival benefit.
With the development of acute renal failure secondary to IVIG and the refractory nature of his disease after the initial dose of steroids, he underwent three sessions of plasmapheresis. A proposed theory for the benefit of plasmapheresis, in our case, was either to eliminate the presumed drug component or inflammatory mediators that were refractory to the aforementioned treatment.17 There are few studies performed to demonstrate large-scale effectiveness of plasmapheresis; however, smaller trials have shown some benefit. In our patient it is not clear whether it was the IVIG, corticosteroids, plasmapheresis or a combination of all three that led to the improvement in his the mucosal and cutaneous manifestations of the SJS. Despite all efforts, the combination of immunosuppression, high-dose steroids, progressive DIC, organ failure and Pneumocystis pneumonia proved too difficult to overcome leading to our patient's death.
Learning points.
Steven-Johnson syndrome (SJS) and paraneoplastic pemphigus share a similar clinical appearance. Histopathological and immunofluorescence correlation are needed to distinguish the two processes.
Multiple therapies exist for the treatment of SJS; however, none have been well studied in large multicentred trials. Given the frequency of this disorder, a randomised controlled trial may be of significant benefit.
Supportive care and withdrawal of the offending agent are the primary modalities in initial treatment of SJS.
Combination therapy, including high-dose steroids, plasmapheresis and immunoglobulin G may be beneficial in treating the cutaneous manifestations in severe cases of SJS.
Footnotes
Contributors: BJ: wrote the case discussion and learning points; SG: Wrote the case hospital course and investigations; JC: discussed dermatology findings and differential diagnosis; SC: case management and supervised the writing of this case.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Khalili B, Barzin SL. Pathogenesis and recent therapeutic trends in Stevens-Johnson syndrome and toxic epidermal necrolysis. Ann Allergy Asthma Immunol 2006;97:272–81. [DOI] [PubMed] [Google Scholar]
- 2.Letko E, Papaliodis DN, Papaliodis GN et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol 2005;94:419–36. [DOI] [PubMed] [Google Scholar]
- 3.Miliszewski MA, Kirchhof MG, Sikora S et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: an analysis of triggers and implications for improving prevention. Am J Med 2016;129:1221–5. [DOI] [PubMed] [Google Scholar]
- 4.Roujeau JC. Immune mechanisms in drug allergy. Allergol Int 2006;55:27 10.2332/allergolint.55.27 [DOI] [PubMed] [Google Scholar]
- 5.Sawada Y. Docetaxel-induced Steven-Johnson syndrome with regenerating epidermis composed of atypical keratinocytes. JEADV, 2009:1327–49. [DOI] [PubMed] [Google Scholar]
- 6.Assier-Bonnet H, Aractingi S, Cadranel J et al. Steven-Johnson syndrome induced by cyclophosphamide—a report of two cases. Br J Dermatol 1996;135:864–6. [DOI] [PubMed] [Google Scholar]
- 7.Valeyrie-Allanore L, Roujeau JC. Epidermal necrolysis (Stevens-Johnson syndrome and toxic epidermal necrolysis). In: Goldsmith LA, Katz SI, Gilchrest BA et al., eds. Fitzpatrick's dermatology in general medicine. 8th edn New York: McGraw-Hill, 2012. [Google Scholar]
- 8.Assier H, Bastuji-Garin S, Revuz J et al. Erythema multiforme with mucous membrane involvement and Stevens-Johnson syndrome are clinically different disorders with distinct causes. Arch Dermatol 1995;131:539. [PubMed] [Google Scholar]
- 9.Anhalt GJ, Kim SC, Stanley JR et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990;323:1729 10.1056/NEJM199012203232503 [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Doval I, LeCleach L, Bocquet H et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch Dermatol 2000;136:323. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol 2013;69:18–7.e1.. 10.1016/j.jaad.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 12.Roujeau JC, Chosidow O, Saiag P et al. Toxic epidermal necrolysis (Lyell syndrome). J Am Acad Dermatol 1990;23:1039. [DOI] [PubMed] [Google Scholar]
- 13.Corrick F, Anand G. Question 2: would systemic steroids be useful in the management of Stevens-Johnson syndrome? Arch Dis Child 2013;98:828 10.1136/archdischild-2013-304909 [DOI] [PubMed] [Google Scholar]
- 14.Aihara M, Kano Y, Fujita H et al. Efficacy of additional IV immunoglobulin to steroid therapy in Stevens-Johnson syndrome and toxic epidermal necrolysis. J Dermatol 2015;42:768–77. 10.1111/1346-8138.12925 [DOI] [PubMed] [Google Scholar]
- 15.Prins C, Vittorio C, Padilla RS et al. Effect of high-dose intravenous immunoglobulin therapy in Stevens-Johnson syndrome: a retrospective, multicenter study. Dermatology (Basel) 2003;207:96 doi:70957 [DOI] [PubMed] [Google Scholar]
- 16.Schneck J, Fagot JP, Sekula P et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol 2008;58:33 10.1016/j.jaad.2007.08.039 [DOI] [PubMed] [Google Scholar]
- 17.Chaidemenos GC, Chrysomallis F, Sombolos K et al. Plasmapheresis in toxic epidermal necrolysis. Int J Dermatol 1997;36:218. [DOI] [PubMed] [Google Scholar]