

Abstract
The opioid system is widely known to modulate the brain reward system and thus affect the behavior of humans and other animals, including feeding. We hypothesized that the hypothalamic opioid system might also control energy metabolism in peripheral tissues. Mice lacking the kappa opioid receptor (κOR) and adenoviral vectors overexpressing or silencing κOR were stereotaxically delivered in the lateral hypothalamic area (LHA) of rats. Vagal denervation was performed to assess its effect on liver metabolism. Endoplasmic reticulum (ER) stress was inhibited by pharmacological (tauroursodeoxycholic acid) and genetic (overexpression of the chaperone glucose‐regulated protein 78 kDa) approaches. The peripheral effects on lipid metabolism were assessed by histological techniques and western blot. We show that in the LHA κOR directly controls hepatic lipid metabolism through the parasympathetic nervous system, independent of changes in food intake and body weight. κOR colocalizes with melanin concentrating hormone receptor 1 (MCH‐R1) in the LHA, and genetic disruption of κOR reduced melanin concentrating hormone–induced liver steatosis. The functional relevance of these findings was given by the fact that silencing of κOR in the LHA attenuated both methionine choline–deficient, diet‐induced and choline‐deficient, high‐fat diet–induced ER stress, inflammation, steatohepatitis, and fibrosis, whereas overexpression of κOR in this area promoted liver steatosis. Overexpression of glucose‐regulated protein 78 kDa in the liver abolished hypothalamic κOR‐induced steatosis by reducing hepatic ER stress. Conclusions: This study reveals a novel hypothalamic–parasympathetic circuit modulating hepatic function through inflammation and ER stress independent of changes in food intake or body weight; these findings might have implications for the clinical use of opioid receptor antagonists. (Hepatology 2016;64:1086‐1104)
Abbreviations
- AAV
adeno‐associated virus
- ALT
alanine transaminase
- AST
aspartate aminotransferase
- CD‐HFD
choline‐deficient HFD
- CHOP
CCAAT/enhancer binding protein homologous protein
- DMV
dorsomedial vagus
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- GRP78
glucose‐regulated protein 78 kDa
- HFD
high‐fat diet
- ICV
intracerebroventricular
- IL6
interleukin‐6
- JNK
c‐Jun N‐terminal kinase
- KO
knockout
- κOR
kappa opioid receptor
- LHA
lateral hypothalamic area
- LPL
lipoprotein lipase
- MCD
methionine/choline‐deficient
- MCH
melanin concentrating hormone
- MCH‐R
MCH receptor
- NASH
nonalcoholic steatohepatitis
- peIF2α
phosphorylated eukaryotic initiation factor 2 alpha
- pIRE1α
phosphorylated inositol‐requiring protein‐1 alpha
- pPERK
phosphorylated protein kinase R–like ER kinase
- shRNA
short hairpin RNA
- TG
triglyceride
- TNFα
tumor necrosis factor alpha
- TUDCA
tauroursodeoxycholic acid
- UPR
unfolded protein response
- WT
wild type
- XBP1S
X‐box binding protein 1 isoform S.
Anatomical studies have shown that the sympathetic and parasympathetic connections to the liver originate from different brain regions within the hypothalamus and the brainstem.1, 2, 3 Within the hypothalamus, the paraventricular, arcuate, and suprachiasmatic nuclei as well as the lateral hypothalamic area (LHA) project and modulate liver metabolism.4, 5
Extensive research has suggested that the central opioid system is involved in the regulation of energy homeostasis. Agonists and antagonists of the three main classes of opioid receptors (mu, kappa, and delta) increase and decrease food consumption, respectively, when injected into various brain areas.6 Studies in mice specifically lacking each of the opioid receptors have demonstrated their resistance to diet‐induced obesity7, 8, 9 and, importantly, suggested that opioid receptors may regulate energy metabolism in peripheral tissues. For instance, mice lacking mu opioid receptor showed increased expression of key mitochondrial enzymes involved in fatty acid oxidation within skeletal muscle.9 Mice lacking delta opioid receptor showed activation in brown adipose tissue thermogenesis.8 Finally, mice lacking kappa opioid receptor (κOR) had reduced hepatic triglyceride (TG) levels and increased lipid oxidation in fat and liver.7
Evidence has demonstrated an anatomical interaction between melanin concentrating hormone (MCH), a neuropeptide specifically located in the LHA, and the opioid system.10 Functional data have supported these findings and shown that blockade of the kappa opioid receptor (κOR) blunted the orexigenic action of MCH.11 In addition to its role in feeding and energy homeostasis,12, 13, 14 MCH‐deficient mice chronically exposed to a high‐fat diet (HFD) are protected from hepatosteatosis,15 and central pharmacological blockade of the MCH receptor (MCH‐R) alleviated steatohepatitis independently from obesity.16 Consistently, MCH stimulates lipid absorption and deposition in the liver through the parasympathetic nervous system, independent of its orexigenic actions.17
The colocalization of MCH and κOR in the LHA, the important metabolic actions of the κOR system, and the recent approval of the combination of bupropion and naltrexone (an opioid receptor antagonist) by the Food and Drug Administration for the treatment of obesity18 led us to investigate the potential role of κOR as a mediator of hepatic lipid metabolism. Here, we show that genetic down‐regulation of κOR in the LHA attenuates both diet‐induced and MCH‐induced liver disease. Consistently, overexpression of κOR in the LHA is sufficient to induce liver steatosis. Notably, these effects were feeding‐independent and mediated by the parasympathetic nervous system. At the hepatic level, hypothalamic κOR stimulates endoplasmic reticulum (ER) stress, with pharmacological or genetic inhibition of hepatic ER stress blunting the effects of the MCH–κOR pathway in the liver.
Materials and Methods
All the experimental procedures involved in this study were reviewed and approved by the Ethics Committee of the University of Santiago de Compostela, in accordance with the European Union's normative for the use of experimental animals. Male Sprague‐Dawley rats (8‐10 weeks old, 250‐300 g), male C57BL/6 mice (8 weeks old), and κ‐OR mutant mice were used (see the Supporting Information).
CHRONIC INTRACEREBROVENTRICULAR MCH AND PF04455242 INFUSION
Chronic intracerebroventricular (ICV) MCH infusion in both rats and mice and infusion of the κOR antagonist PF04455242 in rats were conducted as described17 (see the Supporting Information).
ADENOVIRAL TAIL VEIN INJECTION IN MICE
Either dominant positive glucose‐regulated protein 78 kDa (GRP78) adenovirus or green fluorescent protein (GFP) control was administered in the tail vein of C57BL/6 mice as described19 (see the Supporting Information).
STEREOTAXIC MICROINJECTION OF ADENOVIRAL AND LENTIVIRAL EXPRESSION VECTORS
Rats and mice were placed in a stereotaxic frame, and lentiviral and adenoviral vectors (short hairpin RNA [shRNA] MCH‐R, shRNA κOR, adenoviral κOR) were injected (see the Supporting Information).
SURGICAL VAGOTOMY IN RATS
The surgical procedure was performed as described17, 20 (see the Supporting Information).
IMMUNOHISTOCHEMISTRY AND VISUALIZATION OF GFP
Brains were processed and immunohistochemistry assays performed to visualize dorsomedial vagus (DMV) nucleus protein levels of c‐FOS (Santa Cruz Biotechnology, Santa Cruz, CA). Cellular counting was performed in the brain using Image‐J software with four rats and five pictures per rat. Livers were processed and immunohistochemistry assays performed to visualize protein levels of GRP78 (Cell Signaling, Danvers, MA) and GFP (Abcam, Cambridge, UK). Immunohistochemistry was conducted as described.17
DETECTION OF κORs IN MCH1‐EXPRESSING CELLS BY IMMUNOHISTOCHEMICAL DOUBLE LABELING
Sections of the lateral hypothalamus of transgenic mice Mchr1-cre/tdTomato were received from Eleftheria Maratos‐Flier (Division of Endocrinology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA). Double immunohistochemistry was processed as described in the Supporting Information.
TISSUE TG CONTENT IN LIVER
TG liver content was assessed using a colorimetric assay (see the Supporting Information).
WESTERN BLOT ANALYSIS
Western blot was performed as described21, 22, 23 (see the Supporting Information).
QUANTITATIVE REAL‐TIME POLYMERASE CHAIN REACTION
Real‐time polymerase chain reaction (Taq‐Man; Applied Biosystems) was performed as described24 using specific sets of primers and probes (Supporting Table S1).
LEVELS OF SERUM METABOLITES
Serum activities of alanine transaminase (ALT) and aspartate transaminase (AST) were measured using the ALT and AST Reagent Kit (Biosystems Reagents) with a Benchmark Plus Microplate Spectrophotometer.
STATISTICS
Results are given as mean ± standard error of the mean. Statistical analysis was performed as follows. For normal distributions, parametric tests were used; for two population comparisons, an unpaired t test was performed and for multiple comparison test, a one‐way analysis of variance (ANOVA) followed by Tukey post hoc test was performed. For nonparametric distributions, Mann‐Whitney test was used for two comparison test and Kruskal‐Wallis followed by Dunn test for multiple comparison population. P < 0.05 was considered statistically significant. Data analysis was performed using GraphPad Prism Software Version 5.0a (GraphPad, San Diego, CA).
Results
MCH‐INDUCED LIPID DEPOSITION IS MEDIATED BY HEPATIC ER STRESS
Central infusion of MCH increased food intake in ad libitum–fed rats, and a second control group of ICV MCH–infused animals was pair‐fed to match the intake of saline‐infused controls (Supporting Fig. S1). Weight gain and the amount of TGs in the liver of ICV MCH ad libitum and MCH pair‐fed rats were significantly higher than those of controls independently of food intake (Supporting Fig. S1), as shown.17 Because it is known that central MCH administration modulates liver metabolism by increasing lipid accumulation and lipid uptake17 and that unfolded protein response (UPR) is activated in several murine models of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis (NASH),25, 26 we assessed the protein levels of UPR sensors in the liver of 1 week ICV MCH–infused rats (10 µg/day). We found that phosphorylated inositol‐requiring protein‐1 alpha (pIRE1α), phosphorylated eukaryotic initiation factor 2 alpha (peIF2α), and the apoptotic marker caspase 3 were significantly elevated after chronic central infusion of MCH (Supporting Fig. S1B) independent of feeding as rats treated with ICV MCH and restricted to the same amount of food (pair‐fed) as the vehicle showed similar results to the MCH‐treated group fed ad libitum.
In order to determine the relevance of UPR metabolism as a mediator of the hepatic actions of central MCH, we inhibited ER stress using pharmacological and virogenetic tools. In a first approach, tauroursodeoxycholic acid (TUDCA), a chemical chaperone known to inhibit ER stress,27, 28 was peripherally administered in combination with ICV MCH infused for 1 week (10 µg/day). The daily dose of TUDCA (250 mg/kg, intraperitoneally) was selected because it did not affect either body weight or food intake (Supporting Fig. S2A,B). Central MCH infusion increased food intake and weight gain in rats cotreated with TUDCA (data not shown), but ICV MCH stimulation failed to increase TG content and oil red O staining in the liver when TUDCA was also coadministered (Supporting Fig. S2C,D). Consistently, the increased protein levels of lipoprotein lipase (LPL), phosphorylated c‐Jun N‐terminal kinase 1 (pJNK1), ER stress, and apoptotic markers in the liver of MCH‐treated rats were also blocked after TUDCA administration (Supporting Fig. S2E).
In a second approach, we tested the relevance of UPR on the hepatic actions of central MCH in mice fed a chow diet in order to determine if this mechanism is maintained in different species. For this, we targeted GRP78, a chaperone that facilitates the proper protein folding acting upstream of the UPR.29, 30 Thus, an adenovirus encoding GRP78 wild‐type (GRP78 WT) together with GFP or control adenovirus expressing GFP alone19 was injected in the tail vein of mice together with ICV MCH for 1 week (2.5 µg/day) as described.17 Infection efficiency in the liver was corroborated by immunohistochemistry of either GFP or GRP78 over prefixed liver slides and by western blot (Fig. 1A). Whereas overexpression of GRP78 in the liver did not modify the effects of MCH on food intake or body weight gain (Fig. 1B,C), it abolished the MCH‐induced liver steatosis (Fig. 1D,E). At the protein level, overexpression of GRP78 also blocked the increased hepatic expression of LPL, pJNK1, ER stress, and apoptotic markers of ICV MCH‐treated rats (Fig. 1F).
Figure 1.
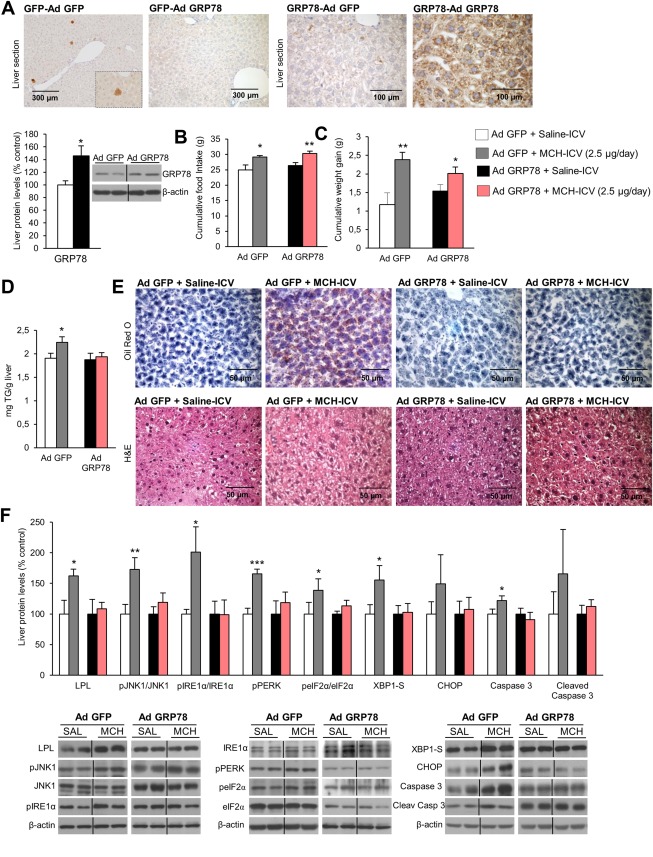
GRP78 overexpression in the liver protects against central MCH‐induced lipid accumulation and ER stress in liver of mice fed a chow diet. Immunostaining of liver fixed section against GFP (left panels) or GRP78 (right panels) on fixed liver sections from mice after tail vein administration of adenoviral vectors that overexpressed either GFP or GRP78, respectively (A). Effect of a 7‐day ICV MCH infusion (2.5 µg/day) combined with hepatic overexpression of GFP or GRP78 on food intake (B), body weight (C), and liver TG content (D). Representative photomicrographs of oil red O and hematoxylin and eosin staining of liver sections (E). Effect of a 7‐day ICV MCH infusion (2.5 µg/day) combined with hepatic overexpression of GFP or GRP78 on liver protein levels of LPL, pJNK, JNK, pIRE1α, IRE1α, phosphorylated protein kinase R–like ER kinase (pPERK), peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 (F). Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Separated photos indicate that gels were run independently. Values are mean ± standard error of the mean of seven or eight animals per group. * P < 0.05, ** P < 0.01, *** P < 0.001 versus controls. Abbreviations: Ad, adenovirus; H&E, hematoxylin and eosin.
GENETIC INHIBITION OF MCH‐R IN THE LHA ATTENUATES METHIONINE/CHOLINE‐DEFICIENT DIET–INDUCED STEATOHEPATITIS THROUGH THE VAGUS NERVE
Genetic activation of MCH‐Rs or infusion of MCH specifically in the LHA, but not in other hypothalamic areas such as the arcuate or the ventromedial nuclei, modulates hepatic lipid metabolism.17 Therefore, we next investigated if inhibition of MCH‐R in this brain area would be sufficient to improve the hepatic condition in rats fed a methionine/choline‐deficient (MCD) diet, which causes steatohepatitis. We stereotaxically delivered a lentiviral virus encoding a shRNA against MCH‐R in the LHA in rats fed an MCD diet. Infection efficiency in the LHA was assessed by expression of GFP and decreased levels of MCH‐R in the LHA (Fig. 2A). Although inhibition of MCH‐R in the LHA did not affect food intake or body weight (Fig. 2B), it attenuated MCD diet–induced hepatic lipid content (Fig. 2C). In agreement with these results indicating reduced hepatic damage, circulating levels of AST and ALT were diminished when MCH‐R was down‐regulated in the LHA of rats fed the MCD diet (Fig. 2D). The lipid depots in the oil red O staining sections as well as the size of the lipid droplets on hematoxylin–eosin staining sections showed a strong macrovesicular steatosis in liver sections of MCD‐fed rats; however, MCD‐fed rats treated with the shRNA MCH‐R in the LHA showed a marked reduction of the macrovesicular lipid size (Fig. 2E). Consistent with that, trichrome Masson staining (to determine liver fibrosis) showed increased collagen deposition around the central vein of the portal areas in the liver of MCD diet–fed rats (Fig. 2E, lower panel, black arrows) but not in rats treated with shRNA MCH‐R in the LHA also fed the MCD diet (Fig. 2E). At the biochemical level, down‐regulation of MCH‐R in the LHA inhibited the hepatic protein levels of LPL, pJNK1, ER stress, and apoptotic markers (Fig. 2F).
Figure 2.
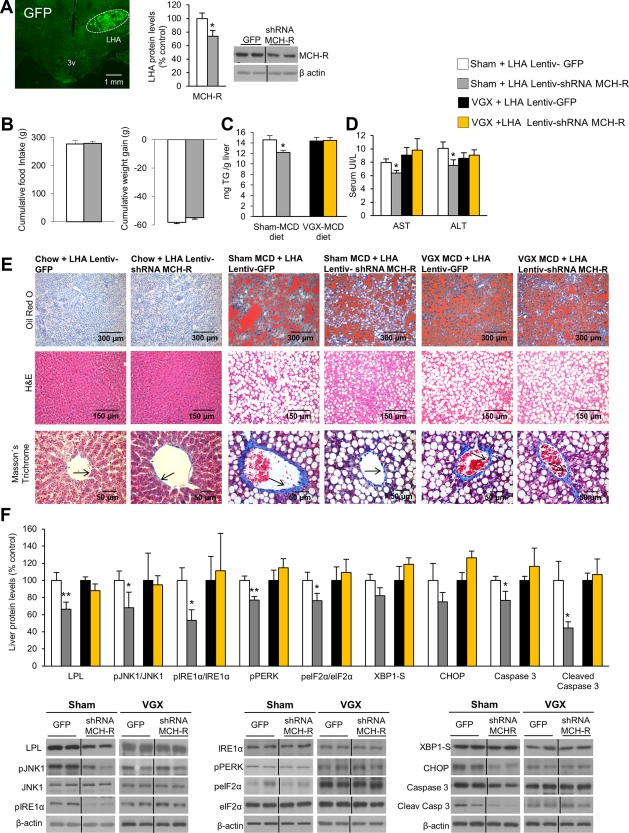
Genetic down‐regulation of MCH‐R in the LHA ameliorates MCD diet–induced NASH and ER stress through the vagus nerve. Representative photomicrographs of brain section showing the injection of a lentivirus that encodes GFP precisely placed in the LHA (×1.25 magnification) and MCH‐R protein levels in the LHA 3 weeks after lentiviral injection encoding either GFP or shRNA MCH‐R (A). Food intake and body weight (B); TG liver content (C); serum levels of AST and ALT (D); representative photomicrograph of liver sections with oil red O, hematoxylin and eosin, and Masson's trichrome staining (E); and liver protein levels of LPL, pJNK, JNK, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 (F) in rats fed the MCD diet infected with a lentivirus encoding GFP or shRNA MCH‐R in the LHA in combination with either sham (controls) or vagotomy. Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Separated photos indicate that gels were run independently. Values are mean ± standard error of the mean of seven or eight animals per group. * P < 0.05, ** P < 0.01, *** P < 0.001 versus controls. Abbreviations: H&E, hematoxylin and eosin; 3v, third ventricle; VGX, vagotomy.
We next hypothesized that the effect of MCH‐R on hepatic lipid metabolism involved vagal innervation. In order to test this, we used rats undergoing vagotomy, with sections of both the dorsal and ventral branches of the vagus nerve dissected, fed the MCD diet 3 weeks after the stereotaxic injection of lentiviral vectors inhibiting MCH‐R in the LHA. The effectiveness of vagotomy was corroborated by assessing the expected morphological changes from stomach enlargement due to increased content related to reduced peristalsis (Supporting Fig. S3). Our findings demonstrate that vagotomy blocked the hepatic effects of shRNA MCH‐R injected into the LHA, as shown by the lack of action on hepatic TG levels (Fig. 2C), serum AST and ALT (Fig. 2D), oil red O staining, fibrosis (Fig. 2E), and protein levels in the liver (Fig. 2F).
GENETIC INHIBITION OF MCH‐R IN THE LHA ATTENUATES CHOLINE‐DEFICIENT HFD–INDUCED LIVER INJURY
Because MCD is known to cause multiple hepatic effects such as oxidative stress31 impaired hepatic action of adiponectin,32 increased fatty acid uptake and reduced very low‐density lipoprotein secretion,33 and a defect in c‐Met,34 we wanted to use another model that induces liver damage in a manner more consistent with dietary observations in humans. Therefore, we next genetically inhibited MCH‐R in the LHA of rats fed a diet combining choline deficiency with an HFD (CD‐HFD).
The inhibition of MCH‐R in the LHA of rats fed the CD‐HFD did not affect food intake or body weight (Fig. 3A) but decreased circulating levels of ALT (Fig. 3B). Consistently, the staining of collagen was also diminished when MCH‐R was down‐regulated in the LHA of rats fed the CD‐HFD, without differences in oil red O staining or the amount of hepatic TGs (Fig. 3C; Supporting Fig. S4). The down‐regulation of MCH‐R in the LHA significantly inhibited the hepatic protein levels of pJNK1/JNK1, X‐box binding protein 1 isoform S (XBP1S), and cleaved caspase 3 and the expression of genes involved in inflammation such as tumor necrosis factor alpha (TNFα), interleukin‐6 (IL6), and CD36 (Fig. 3D).
Figure 3.
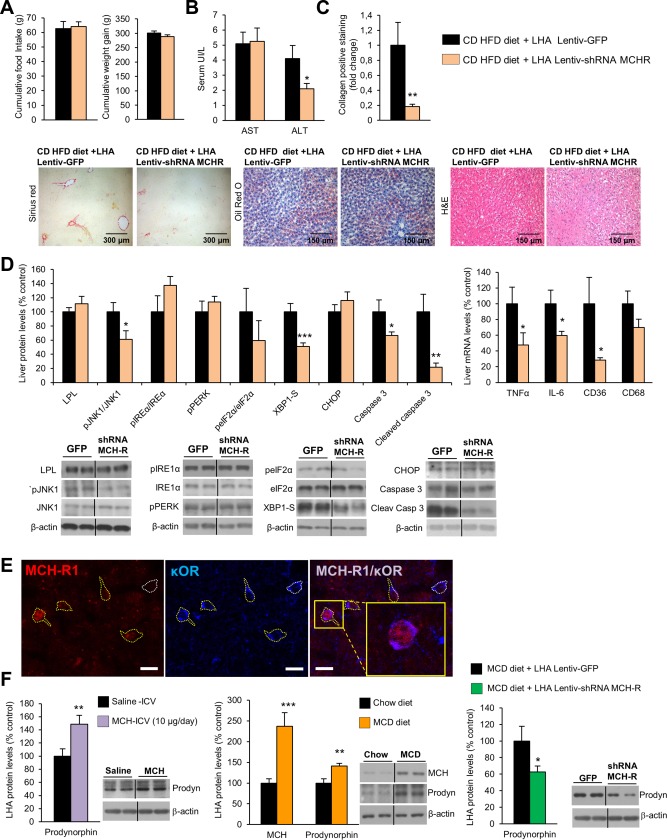
Genetic down‐regulation of MCH‐R in the LHA ameliorates CD‐HFD–induced liver damage and ER stress, MCHR1 co‐localizes with κOR, and MCH regulates prodynorphyn levels in the LHA. Food intake and body weight (A); serum levels of AST and ALT (B); collagen‐positive staining and representative photomicrograph of liver sections with sirius red, oil red O, and hematoxylin and eosin staining (C); liver protein levels of LPL, pJNK1, JNK1, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 and hepatic gene expression of TNFα, IL6, CD36 and CD68 (D) in rats fed a CD‐HFD infected with a lentivirus encoding GFP or shRNA MCH‐R in the LHA. Single (left and middle panels) and dual‐channel (right panel) photomicrographs of a representative brain section showing immunostaining for tdTomato (red) and κOR (blue) in the LHA of mice exhibiting tdTomato in MCHR1‐CRE expressing cells. The majority of tdTomato‐positive cells are also immunoreactive for κOR (delineated by yellow dotted line). White dotted line shows a κOR‐positive cell negative for tdTomato. To demonstrate a double‐labeled cell, the enframed area in the right panel is further magnified. Scale bar = 10 µm (E). Prodynorphyn protein levels in the LHA of rats infused with ICV MCH during 1 week, MCH and prodynorphyn protein levels in the LHA of rats fed the MCD diet, and prodynorphyn protein levels in the LHA of rats fed a MCD diet with inhibition of MCH‐R in the LHA (F). Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Separated photos indicate that gels were run independently. Values are mean ± standard error of the mean of 7‐10 animals per group. * P < 0.05, *** P < 0.001 versus controls. Abbreviation: H&E, hematoxylin and eosin.
MCH‐R1 COLOCALIZES WITH κOR IN THE LHA, AND MCH INDUCES PRODYNORPHIN EXPRESSION
An anatomical and functional association between MCH and the κ opioid system has been demonstrated.11, 35, 36 However, it is unknown if κOR modulates the peripheral actions of MCH or whether hypothalamic κOR is able to control peripheral metabolism. Using the reporter protein tdTomato, MCH‐R1‐CRE expressing cells were identified in the lateral hypothalamus of transgenic mice, with MCH‐R1 expression not found in astrocytes (Supporting Fig. S4B); and we tested whether they express κOR or not by immunohistochemical double labeling (Fig. 3E). Confocal microscopic analysis of the sections revealed that the majority of MCH‐R1 expressing cells (Fig. 3E, panel left) are positive for this opioid receptor subtype (Fig. 3E, panels middle and right). Because prodynorphin is the natural ligand of κOR, we assessed if its expression was modulated by MCH. Chronic central infusion of MCH significantly increased prodynorphin in the LHA (Fig. 3F). In the LHA of rats fed the MCD we found significantly higher levels of both MCH and prodynorphin (Fig. 3F). When inhibiting MCH‐R by injecting shRNA lentivirus into the LHA of rats fed the MCD diet, prodynorphin protein levels significantly decreased (Fig. 3F).
GLOBAL κOR KNOCKOUT MICE ARE RESISTANT TO MCH‐INDUCED LIPID DEPOSITION AND ER STRESS IN LIVER
Because the results described above indicate that MCH stimulates prodynorphin and down‐regulation of MCH‐R inhibits prodynorphin in the LHA, we next assessed if central MCH was able to modulate hepatic lipid metabolism when the κOR system is disrupted. To test this hypothesis, we centrally injected MCH for 1 week in WT and κOR‐deficient mice (2.5 µg/day). MCH stimulated food intake and body weight gain in both WT and κOR knockout (KO) mice (Fig. 4A). However, whereas the TG content in the liver was significantly increased in WT mice treated with ICV MCH, there were no changes in the liver TG content of κOR‐KO mice (Fig. 4B). Of note, we previously described that κOR‐KO mice fed an HFD had lower TG content in the liver when compared to their littermates,7 but in the current study we used κOR‐KO mice fed a chow diet and did not detect differences in hepatic TG content. Accordingly, protein levels of key enzymes modulating the metabolism of fatty acids and ER stress were significantly increased in WT mice treated with ICV MCH but remained unchanged in the liver of κOR‐KO mice infused with MCH (Fig. 4C).
Figure 4.
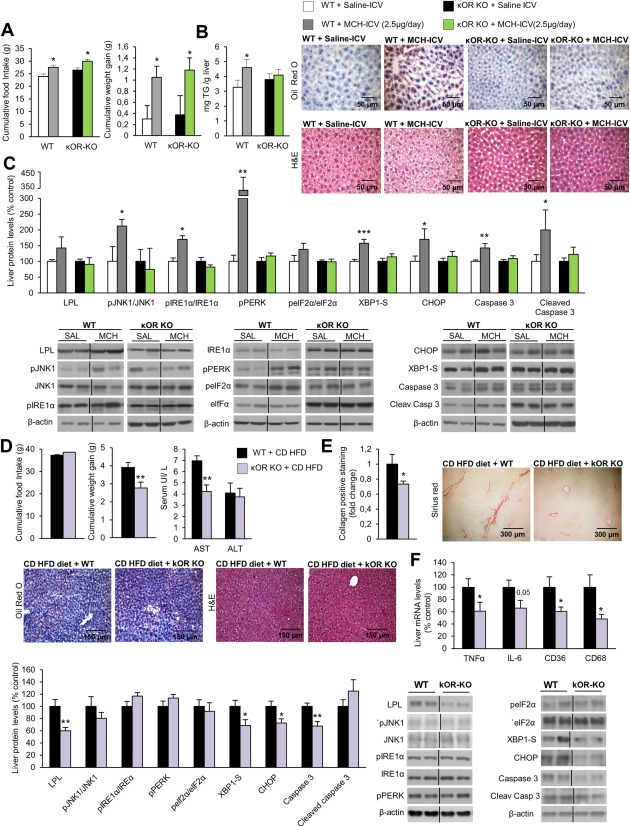
κOR‐KO mice are resistant to MCH‐induced and CD‐HFD–induced liver damage. Effect of a 7‐day ICV MCH infusion (2.5 µg/day) in either WT or κOR‐KO mice fed a chow diet on food intake and body weight (A); TG liver content and oil red O and hematoxylin and eosin liver section staining represented in the microphotographs (B); and liver protein levels of LPL, pJNK, JNK, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 (C). Effect of CD‐HFD (2 weeks) in WT and κOR‐KO mice on food intake and body weight, serum levels of AST, and ALT (D); collagen‐positive staining and representative photomicrograph of liver sections with sirius red, oil red O, and hematoxylin and eosin staining (E); liver protein levels of LPL, pJNK1, JNK1, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 and hepatic gene expression of TNFα, IL6, CD36, and CD68 (F). Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Separated photos indicate that gels were run independently. Values are mean ± standard error of the mean of 7‐10 animals per group. * P < 0.05, ** P < 0.01, *** P < 0.001 versus controls. Abbreviations: H&E, hematoxylin and eosin; SAL, saline.
To further explore if κOR‐KO mice were also resistant to diet‐induced liver damage, we next challenged WT and κOR‐KO mice to a CD‐HFD for 2 weeks. In agreement with a previous report, κOR‐KO mice fed the CD‐HFD gained less weight than WT mice7 (Fig. 4D). In addition, κOR‐KO mice had decreased circulating levels of AST (Fig. 4D). The staining of collagen in liver sections was also diminished in κOR‐KO mice fed the CD‐HFD, with no differences in oil red O staining or in the amount of hepatic TGs (Fig. 4E; Supporting Fig. S5). κOR‐KO mice fed the CD‐HFD showed a significant reduction in the hepatic protein levels of LPL, XBP1s, CCAAT/enhancer binding protein homologous protein (CHOP), and caspase 3 and in the expression of proinflammatory genes such as TNFα, CD36, and CD68 (Fig. 4F).
GENETIC DOWN‐REGULATION OF κOR IN THE LHA PROTECTS AGAINST MCH‐INDUCED LIPID STORAGE AND ER STRESS IN LIVER
An adeno‐asociated virus (AAV) encoding an shRNA against κOR was stereotaxically delivered in the LHA of rats. The efficiency of the stereotaxic injections in the LHA was corroborated by GFP and κOR protein levels in this hypothalamic area (Fig. 5A). Three weeks after the injection with the AAV, osmotic pumps and ICV cannulae were implanted to infuse MCH for 1 week. Food intake and body weight change were increased in the MCH‐infused rats and in those with κOR down‐regulation (Fig. 5B). The DMV nucleus, located in the hindbrain, is a critical place controlling parasympathetic vagal efferent fibers to the gastrointestinal tract37 (Supporting Fig. S6A). ICV MCH increased C‐FOS immunoreactivity in the DMV nucleus of control rats; however, ICV MCH failed to increase C‐FOS when the κOR was down‐regulated in the LHA (Fig. 5C). At the peripheral level, the increase of liver TG levels induced by central MCH was blocked when κOR was silenced in the LHA (Fig. 5D,E). At the protein level, down‐regulation of κOR in the LHA protects against MCH‐induced lipid uptake and ER stress in the liver (Fig. 5F). Importantly, down‐regulation of κOR in the LHA did not blunt the effects of central MCH on white adipose tissue (Supporting Fig. S6B).
Figure 5.
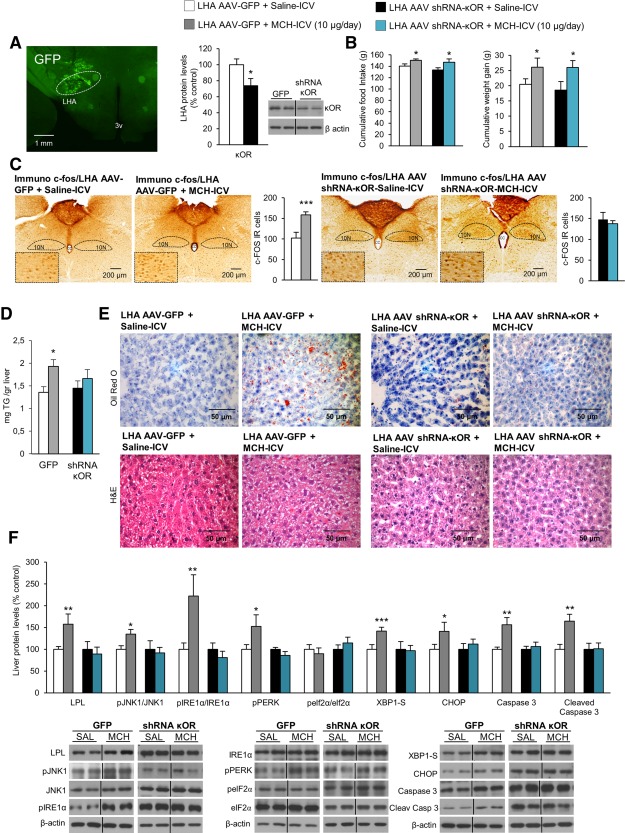
Inhibition of κOR in the LHA protects against MCH‐induced liver steatosis in rats fed a chow diet. Representative photomicrograph of brain section showing the injection of an AAV that encodes GFP precisely placed in the LHA (×1.25 magnification) and κOR protein levels in the LHA 3 weeks after the AAV injection that encodes either GFP or shRNA κOR (A). Food intake and body weight (B); c‐FOS immunoreactive cells in the DMV nucleus (C); liver TG content (D); oil red O and hematoxylin and eosin staining liver sections (E); and liver protein levels of LPL, pJNK, JNK, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 (F) of rats after a 7‐day ICV MCH infusion (10 µg/day) in combination with AAV‐GFP or AAV‐shRNA κOR in the LHA. Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Separated photos indicate that gels were run independently. Values are mean ± standard error of the mean of seven or eight animals per group. * P < 0.05, ** P < 0.01, *** P < 0.001 versus controls. Abbreviations: CC, central canal; H&E, hematoxylin and eosin; IR, immunoreactive; 10N, vagus nerve; SAL, saline; 3v, third ventricle.
CENTRAL PHARMACOLOGICAL ANTAGONISM AND GENETIC INHIBITION OF κOR IN THE LHA AMELIORATE MCD DIET–INDUCED NASH
After demonstration that the down‐regulation of κOR in the LHA protects against MCH‐induced liver steatosis, we next sought to investigate its effect in mice fed the MCD diet. First, we performed a dose–response assay of a specific κOR antagonist, PF04455242,38 and found that a single ICV injection of this compound at a dose of 3.4 nmol was able to decrease body weight without changing food intake in rats fed a chow diet (Supporting Fig. S7). Thus, we centrally infused PF04455242 (3.4 nmol/day) for 1 week in rats fed the MCD diet. The chronic treatment did not modify food intake or body weight (Fig. 6A) but decreased circulating levels of AST (Fig. 6B) and reduced hepatic levels of TG (Fig. 6C). Given that central pharmacological blockade of κOR was sufficient to ameliorate MCD‐induced liver steatosis, we next aimed to knock down κOR specifically in the LHA. After 3 weeks of the AAV shRNA‐κOR delivery in the LHA, rats were fed the MCD diet for 3 weeks. The efficiency of the stereotaxic injections in the LHA was corroborated by GFP (Fig. 6D). The inhibition of κOR in the LHA did not modify food intake or body weight (Fig. 6D) but significantly reduced TG liver content and reduced collagen deposition around the central vein liver lobules (Fig. 6E) as well as messenger RNA levels of the main inflammatory markers (IL6, CD36, CD68) in the liver of rats fed the MCD diet (Fig. 6F). Consistent with these findings, protein levels of LPL, pJNK1, and ER stress markers were also decreased in the liver of rats subjected to κOR down‐regulation in the LHA (Fig. 6F).
Figure 6.
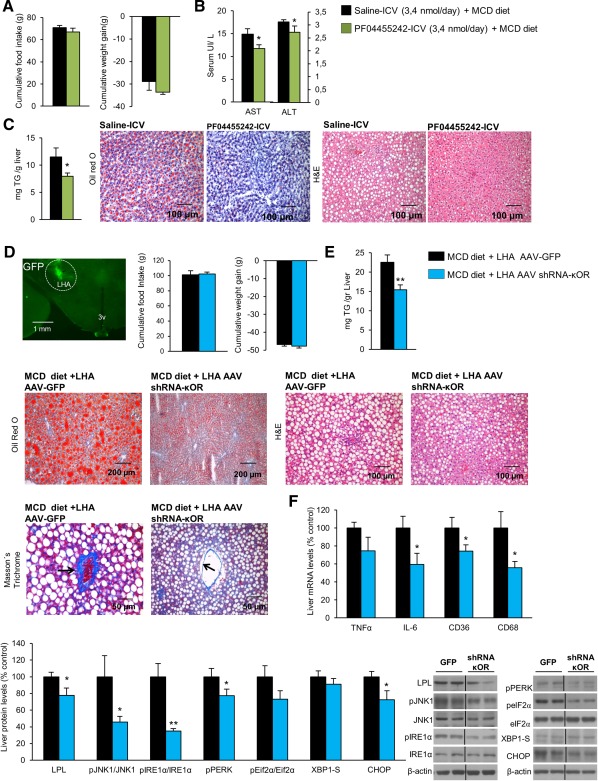
Central pharmacological antagonism and down‐regulation of κOR in the LHA alleviates MCD diet–induced NASH. Effect of a 7‐day PF04455242 ICV infusion (3.4 nmol/day) in rats fed the MCD diet on food intake and body weight (A), serum levels of AST and ALT (B), and TG liver content and oil red O and hematoxylin and eosin liver section staining represented in the microphotographs (C). Representative photomicrograph of brain section showing the injection of a lentivirus that encodes GFP precisely placed in the LHA (A) (×1.25 magnification). Food intake and body weight (D); TG liver content and representative photomicrograph of liver sections with oil red O, hematoxylin and eosin, and Massońs trichrome staining (E); liver messenger RNA levels of the inflammation markers TNFα, IL6, CD36, and CD68 (E); and liver protein levels of LPL, pJNK, JNK, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, and CHOP (F) of rats fed the MCD diet for 3 weeks after κOR inhibition in the LHA using an AAV vector. Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Values are mean ± standard error of the mean of seven or eight animals per group. * P < 0.05, ** P < 0.01, *** P < 0.001 versus controls. Abbreviations: H&E, hematoxylin and eosin; 3v, third ventricle.
GENETIC INHIBITION OF κOR IN THE LHA AMELIORATES CD‐HFD–INDUCED LIVER INJURY
Using a similar approach to that described in Fig. 3, we inhibited κOR in the LHA of rats fed the CD‐HFD. No changes were detected in either food intake or body weight (Fig. 7A), but circulating levels of ALT were decreased (Fig. 7B). Consistently, the staining of collagen was also diminished when κOR was down‐regulated in the LHA of rats fed the CD‐HFD, without differences in oil red O staining or the amount of TG in the liver (Fig. 7C; Supporting Fig. S8). At the biochemical level, down‐regulation of κOR in the LHA significantly inhibited the hepatic protein levels of LPL, pJNK1/JNK1, pIRE1α/IRE1α, XBP1‐S, CHOP, and cleaved caspase 3 (Fig. 7D) as well as the expression of genes involved in inflammation such as TNFα, IL6, and CD36 (Fig. 7E).
Figure 7.
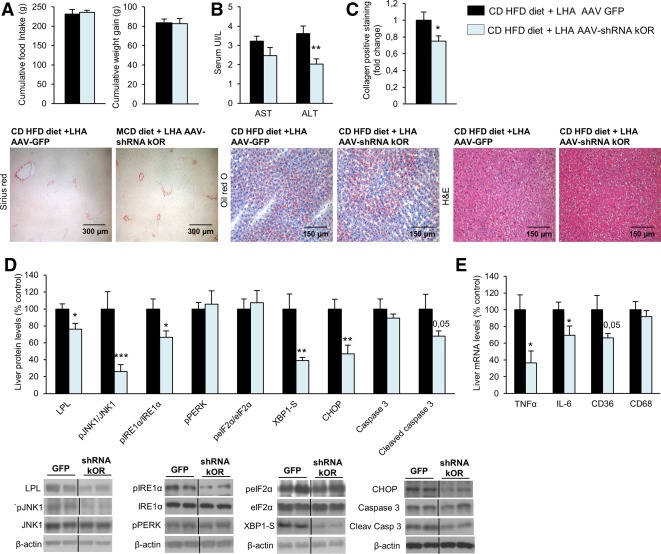
Genetic down‐regulation of κOR in the LHA ameliorates CD‐HFD–induced liver damage and ER stress. Food intake and body weight (A); serum levels of AST and ALT (B); collagen‐positive staining and representative photomicrograph of liver sections with sirius red, oil red O, and hematoxylin and eosin staining (C); liver protein levels of LPL, pJNK1, JNK1, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 (D); and hepatic gene expression of TNFα, IL6, CD36, and CD68 (E) in rats fed the CD‐HFD infected with a lentivirus encoding GFP or shRNA κOR in the LHA. Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Separated photos indicate that gels were run independently. Values are mean ± standard error of the mean of 8‐10 animals per group. * P < 0.05, *** P < 0.001 versus controls. Abbreviation: H&E, hematoxylin and eosin.
GENETIC OVEREXPRESSION OF κOR IN THE LHA TRIGGERS LIVER ADIPOSITY THROUGH STIMULATION OF ER STRESS
We next assessed if the overexpression of κOR in this hypothalamic area was able to induce lipid accumulation in the liver. The efficiency of the stereotaxic injections in the LHA was corroborated by GFP and increased κOR protein levels in the LHA (Fig. 8A). Our results show that administration of an adenoviral vector overexpressing κOR into the LHA did not change food intake or body weight (Fig. 8B) but increased hepatic TG content (Fig. 8C) in rats fed the chow diet. The overexpression of κOR in the LHA also increased protein levels of LPL, pJNK1, and ER stress markers (Fig. 8D).
Figure 8.
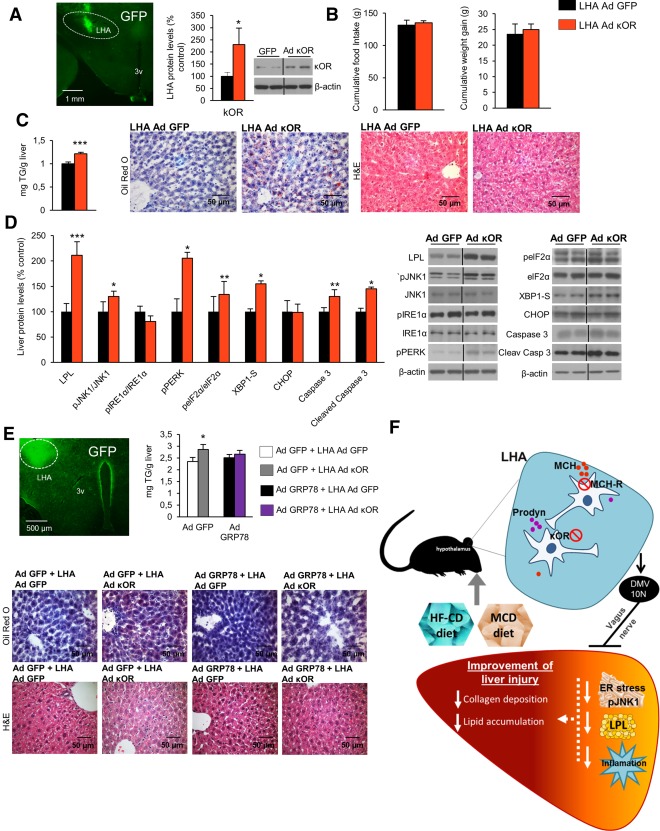
κOR overexpression in the LHA induces liver steatosis, and attenuation of hepatic ER stress blunts its effect. Representative photomicrograph of brain section showing the injection of adenoviral vector encoding GFP precisely placed in the LHA (×1.25 magnification) and κOR protein levels in the LHA of rats 1 week after injection of an adenoviral vector overexpressing κOR in the LHA of rats fed a chow diet (A). Effect of a 7‐day adenoviral overexpression of κOR in the LHA on food intake and body weight (B), liver TG content and oil red O and hematoxylin and eosin staining liver sections (C), and liver protein content of LPL, pJNK, JNK, pIRE1α, IRE1α, pPERK, peIF2α, eIF2α, XBP1S, CHOP, caspase 3, and cleaved caspase 3 (D). Representative photomicrograph of brain section showing the injection of adenoviral vector encoding GFP precisely placed in the LHA (×1.25 magnification), hepatic TG content, and representative photomicrographs of oil red O and hematoxylin and eosin staining of liver sections (×40 magnification) (E) in mice after tail vein administration of adenoviral vectors that overexpressed either GFP or GRP78 in the liver combined with overexpression of κOR in the LHA. Working model of the hypothalamic role of κOR in the control of liver metabolism (F). Protein β‐actin levels were used to normalize protein levels. Dividing lines indicate splicings within the same gel. Values are mean ± standard error of the mean of seven or eight animals per group. * P < 0.05, ** P < 0.01, *** P < 0.001 versus controls. Abbreviations: Ad, adenovirus; H&E, hematoxylin and eosin; 10N, vagus nerve; 3v, third ventricle.
Finally, in order to investigate the relevance of ER stress as a mediator of the hepatic actions of hypothalamic κOR, we overexpressed κOR in the LHA of mice where the chaperone GRP78 was also overexpressed in the liver, and no differences between food intake or body weight were detected (Supporting Fig. S9). The efficiency of the stereotaxic injections in the LHA was corroborated by GFP (Fig. 8E). The overexpression of κOR in the LHA augmented the hepatic lipid content, whereas the overexpression of GRP78 in the liver blunted the effects of hypothalamic κOR (Fig. 8E). Overall, these results indicate that hypothalamic κOR mediates both diet‐induced and MCH‐induced liver damage through inflammation and ER stress (Fig. 8F).
Discussion
In this study we report for first time that hypothalamic κOR directly controls hepatic lipid metabolism, independent of weight and feeding changes. Specifically, we show that genetic down‐regulation of κOR in the LHA ameliorates diet‐induced and MCH‐induced liver steatosis, whereas its overexpression induces hepatic lipid accumulation. This occurs through effects on hepatic inflammation and ER stress as genetic overexpression of GRP78 blunts hypothalamic κOR‐induced hepatic lipid storage (Fig. 8F). Interestingly, those actions were independent of feeding behavior.
The opioid system is mainly recognized as a key player in neural reward processes leading to addictive behavior.6 Numerous studies have established that both systemic and brain administration of κOR antagonists reduces food intake and body weight in rodent models, whereas κOR agonists cause opposite effects.6 However, the potential role of κOR in the control of liver metabolism is largely unknown. κOR‐deficient mice were protected against the development of diet‐induced obesity and liver steatosis,7 but given the lower adiposity found in κOR null mice fed an HFD, this effect has been proposed as secondary. κOR and dynorphin (its endogenous ligand) are widely expressed throughout the central nervous system, including the hypothalamus, where both preprodynorphin and κOR are located in the LHA.39 Our present findings demonstrate that the down‐regulation of κOR in the LHA was sufficient to protect liver damage induced by the two different dietary interventions, the MCD diet and the CD‐HFD. Importantly, these effects are independent of changes in body weight and feeding. The protection against MCD diet–induced and CD‐HFD–induced liver damage was correlated with reduced inflammation and ER stress. In this sense, it is important to highlight that the effect of κOR knockdown in the LHA was not exactly the same in rats fed the MCD diet or the CD‐HFD. Whereas the inhibition of MCH‐R and κOR decreased the amount of TGs in the liver of rats fed the MCD diet, it failed to show differences in hepatic TGs after knockdown of MCH‐R and κOR in the LHA of rats fed the CD‐HFD. This difference is likely due to the direct input of lipids from the HFD to the liver and the different molecular mechanisms by which each type of diet causes liver injury. In any case, the integrative nature of the data is given by the fact that inhibition of κOR in the LHA is able to protect from liver damage using different mice models.
Consistent with loss‐of‐function experiments, the overexpression of κOR in the LHA (in both rats and mice) increased hepatic lipid storage along with stimulation of lipid uptake and ER stress. To determine the physiological relevance of hepatic ER stress as a modulator of the central actions of κOR, we performed a selective disruption of hepatic ER stress by genetically overexpressing GRP78, the most abundant ER‐resident chaperone that can prevent accumulation of unfolded/missfolded proteins within the ER.19, 29, 30 We found that overexpression of κOR in the LHA was unable to alter liver metabolism in rats concurrently overexpressing GPR78. Thus, these results demonstrate that ER stress is essential for the hepatic actions of hypothalamic κOR.
Pharmacologic or genetic manipulation of MCH causes important alterations in feeding behavior, body weight, glucose metabolism, and lipid deposition in the liver.12, 13, 14, 17, 40 Our previous findings indicated that activation of the MCH system in the LHA stimulates TG synthesis and uptake through JNK1.17 Herein, we report that brain MCH controls liver metabolism through hepatic ER stress. Numerous studies have highlighted the relevance of ER stress in the development of hepatic steatosis.28, 41, 42, 43, 44 ER stress is a consequence of the accumulation of unfolded proteins on the ER lumen produced by biological, physiological, or pathological stimuli.45, 46 ER stress causes JNK1 activation through IRE1α,28, 42 and, as mentioned above, MCH induced LPL expression in a JNK1‐dependent manner.17 Therefore, the results indicating that pharmacological or genetic blockade of ER stress blunts the hepatic action of MCH are in concordance with our previous molecular findings.
Consistent with studies indicating that pharmacological blockade of MCH‐R47 or absence of MCH15, 48 ameliorates hepatic steatosis, the down‐regulation of MCH‐R in the LHA also reduced MCD diet‐induced and CD‐HFD–induced liver damage. Also in agreement with our previous report indicating that the MCH system regulates liver metabolism by its action within the LHA and through the vagus nerve,17 we found here that LHA MCH‐R requires an intact vagus nerve to improve MCD diet–induced steatohepatitis and fibrosis. Although the LHA is the hypothalamic area responsible for the hepatic actions of MCH, the neuronal pathways occurring within this area remain completely unknown. It has been shown that MCH interacts with the opioid system to modulate food intake.11 Among the three different opioid receptors, κOR exhibits significant colocalization with MCH and with MCH‐R10, 36 in the LHA. Indeed, in our study we confirmed that MCHR1 and κOR colocalize in the LHA, suggesting a direct interaction between these two systems. Furthermore the expression of prodynorphin in the LHA was stimulated by MCH and reduced after silencing MCH‐R1 in the LHA, indicating that MCH is upstream of the prodynorphin/κOR system. We next explored the functionality of this interaction and found that chronic infusion of MCH in the lateral ventricle failed to promote lipid deposition in the liver of κOR‐deficient mice, as well as in animals in which κOR was genetically down‐regulated in the LHA. Accordingly, we also found that stimulation of MCH‐induced ER stress in the liver was blunted after disruption of the κOR. The mechanism by which κOR blunts the effect of MCH at the central level involves the inability of MCH to activate the vagus nerve. This is demonstrated by the fact that inhibition of κOR in the LHA dampened MCH‐induced C‐FOS levels in the DMV, a brainstem area that is related to the parasympathetic vagal functions in the gastrointestinal tract.37 The knowledge regarding the connection between parasympathetic signaling and ER stress is extremely limited, and the results seem to be dependent on the type and status of the cell as well as the type of muscarinic receptors.49, 50 The interaction between the parasympathetic system and ER stress in the liver will require further investigation. Overall, these results demonstrate that MCH modulates the prodynorphin/κOR system in the LHA to control hepatic lipid metabolism. These data indicate that the interaction between MCH and the prodynorphin/κOR system occurs at a preautonomic level and controls the parasympathetic branch of the autonomic nervous system.
In summary, this study reveals a novel hypothalamic circuit, where κOR specifically located within the LHA directly controls hepatic lipid storage and mediates both diet‐induced and MCH‐induced liver damage independent of changes in food intake and body weight. To exert its actions, this neuronal circuit modulates hepatic inflammation and ER stress through the vagal parasympathetic nervous system. Overall, these findings suggest that in addition to its potential to treat obesity18 the opioid system might be a new pharmaceutical target to treat liver steatosis (Fig. 8F).
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28716/suppinfo.
Supporting Information
Acknowledgment
We thank Dr. Eleftheria Maratos‐Flier from Harvard University for kindly providing mice exhibiting tdTomato in MCHR1‐CRE expressing cells.
Potential conflict of interest: Nothing to report.
Supported by Ministerio de Economia y Competitividad (BFU2014‐5587, to C.D.; SAF2015‐71026‐R, to M.L.; BFU2015‐70664‐R, to M.L.), Xunta de Galicia (2015‐CP079; EM 2012/039 and 2015‐CP080, to R.N.), the National Development Agency (BONUS HU 08/2‐2011‐0006, to Z.L.), the National Science Foundation of Hungary (OTKA K101326, K100722), Centro de Investigación Biomédica en Red (CIBER) de Fisiopatología de la Obesidad y Nutrición (CIBERobn, an initiative of the Instituto de Salud Carlos III of Spain which is supported by FEDER funds), the European Community's Seventh Framework Programme (ERC StG‐2011‐OBESITY53‐281408, to R.N.), and Instituto de Salud Carlos III/SERGAS (“Sara Borrell” CD14/00091 and CD14/00077, to O.A.‐M, C.C).
REFERENCES
- 1. Puschel GP. Control of hepatocyte metabolism by sympathetic and parasympathetic hepatic nerves. Anat Rec A Discov Mol Cell Evol Biol 2004;280:854–867. [DOI] [PubMed] [Google Scholar]
- 2. la Fleur SE, Kalsbeek A, Wortel J, Buijs RM. Polysynaptic neural pathways between the hypothalamus, including the suprachiasmatic nucleus, and the liver. Brain Res 2000;871:50–56. [DOI] [PubMed] [Google Scholar]
- 3. Bruinstroop E, Fliers E, Kalsbeek A. Hypothalamic control of hepatic lipid metabolism via the autonomic nervous system. Best Pract Res Clin Endocrinol Metab 2014;28:673–684. [DOI] [PubMed] [Google Scholar]
- 4. Bruinstroop E, la Fleur SE, Ackermans MT, Foppen E, Wortel J, Kooijman S, et al. The autonomic nervous system regulates postprandial hepatic lipid metabolism. Am J Physiol Endocrinol Metab 2013;304:E1089–E1096. [DOI] [PubMed] [Google Scholar]
- 5. Stanley S, Pinto S, Segal J, Pérez CA, Viale A, DeFalco J, et al. Identification of neuronal subpopulations that project from hypothalamus to both liver and adipose tissue polysynaptically. Proc Natl Acad Sci 2010;107:7024–7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bodnar RJ. Endogenous opioids and feeding behavior: a 30‐year historical perspective. Peptides 2004;25:697–725. [DOI] [PubMed] [Google Scholar]
- 7. Czyzyk TA, Nogueiras R, Lockwood JF, McKinzie JH, Coskun T, Pintar JE, et al. κ‐Opioid receptors control the metabolic response to a high‐energy diet in mice. FASEB J 2010;24:1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Czyzyk TA, Romero‐Pico A, Pintar J, McKinzie JH, Tschop MH, Statnick MA, et al. Mice lacking delta‐opioid receptors resist the development of diet‐induced obesity. FASEB J 2012;26:3483–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tabarin A, Diz‐Chaves Y, Carmona Mdel C, Catargi B, Zorrilla EP, Roberts AJ, et al. Resistance to diet‐induced obesity in mu‐opioid receptor‐deficient mice: evidence for a “thrifty gene.” Diabetes 2005;54:3510–3516. [DOI] [PubMed] [Google Scholar]
- 10. Parks GS, Wang L, Wang Z, Civelli O. Identification of neuropeptide receptors expressed by melanin‐concentrating hormone neurons. J Comp Neurol 2014;522:3817–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lopez CA, Guesdon B, Baraboi ED, Roffarello BM, Hetu M, Richard D. Involvement of the opioid system in the orexigenic and hedonic effects of melanin‐concentrating hormone. Am J Physiol Regul Integr Comp Physiol 2011;301:R1105–R1111. [DOI] [PubMed] [Google Scholar]
- 12. Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, et al. Melanin‐concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 2001;107:379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos‐Flier E. Mice lacking melanin‐concentrating hormone are hypophagic and lean. Nature 1998;396:670–674. [DOI] [PubMed] [Google Scholar]
- 14. Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, et al. A role for melanin‐concentrating hormone in the central regulation of feeding behaviour. Nature 1996;380:243–247. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y, Ziogas DC, Biddinger S, Kokkotou E. You deserve what you eat: lessons learned from the study of the melanin‐concentrating hormone (MCH)–deficient mice. Gut 2010;59:1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito M, Gomori A, Suzuki J, Tsujioka S, Sasaki M, Matsuda M, et al. Antagonism of central melanin‐concentrating hormone 1 receptor alleviates steatohepatitis in mice. J Endocrinol 2008;198:309–315. [DOI] [PubMed] [Google Scholar]
- 17. Imbernon M, Beiroa D, Vazquez MJ, Morgan DA, Veyrat‐Durebex C, Porteiro B, et al. Central melanin‐concentrating hormone influences liver and adipose metabolism via specific hypothalamic nuclei and efferent autonomic/JNK1 pathways. Gastroenterology 2013;144:636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yanovski SZ, Yanovski JA. Naltrexone extended‐release plus bupropion extended‐release for treatment of obesity. JAMA 2015;313:1213–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Contreras C, Gonzalez‐Garcia I, Martinez‐Sanchez N, Seoane‐Collazo P, Jacas J, Morgan DA, et al. Central ceramide‐induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep 2014;9:366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seoane LM, Al‐Massadi O, Caminos JE, Tovar SA, Dieguez C, Casanueva FF. Sensory stimuli directly acting at the central nervous system regulate gastric ghrelin secretion. an ex vivo organ culture study. Endocrinology 2007;148:3998–4006. [DOI] [PubMed] [Google Scholar]
- 21. Nogueiras R, Wiedmer P, Perez‐Tilve D, Veyrat‐Durebex C, Keogh JM, Sutton GM, et al. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest 2007;117:3475–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopez M, Lage R, Saha AK, Perez‐Tilve D, Vazquez MJ, Varela L, et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 2008;7:389–399. [DOI] [PubMed] [Google Scholar]
- 23. Lopez M, Varela L, Vazquez MJ, Rodriguez‐Cuenca S, Gonzalez CR, Velagapudi VR, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med 2010;16:1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nogueiras R, Perez‐Tilve D, Veyrat‐Durebex C, Morgan DA, Varela L, Haynes WG, et al. Direct control of peripheral lipid deposition by CNS GLP‐1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet‐induced obesity. J Neurosci 2009;29:5916–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mosbah IB, Alfany‐Fernandez I, Martel C, Zaouali MA, Bintanel‐Morcillo M, Rimola A, et al. Endoplasmic reticulum stress inhibition protects steatotic and non‐steatotic livers in partial hepatectomy under ischemia‐reperfusion. Cell Death Dis 2010;1:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 2011;54:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress–induced caspase‐12 activation. Hepatology 2002;36:592–601. [DOI] [PubMed] [Google Scholar]
- 28. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006;313:1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev 2006;86:1133–1149. [DOI] [PubMed] [Google Scholar]
- 30. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- 31. George J, Pera N, Phung N, Leclercq I, Yun Hou J, Farrell G. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol 2003;39:756–764. [DOI] [PubMed] [Google Scholar]
- 32. Larter CZ, Yeh MM, Williams J, Bell‐Anderson KS, Farrell GC. MCD‐induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J Hepatol 2008;49:407–416. [DOI] [PubMed] [Google Scholar]
- 33. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline–deficient diet. J Lipid Res 2008;49:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kroy DC, Schumacher F, Ramadori P, Hatting M, Bergheim I, Gassler N, et al. Hepatocyte specific deletion of c‐Met leads to the development of severe non‐alcoholic steatohepatitis in mice. J Hepatol 2014;61:883–890. [DOI] [PubMed] [Google Scholar]
- 35. Chang GQ, Karatayev O, Ahsan R, Gaysinskaya V, Marwil Z, Leibowitz SF. Dietary fat stimulates endogenous enkephalin and dynorphin in the paraventricular nucleus: role of circulating triglycerides. Am J Physiol Endocrinol Metab 2007;292:E561–E570. [DOI] [PubMed] [Google Scholar]
- 36. Chee MJS, Pissios P, Maratos‐Flier E. Neurochemical characterization of neurons expressing melanin‐concentrating hormone receptor 1 in the mouse hypothalamus. J Comp Neurol 2013;521:2208–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. ter Horst GJ, Luiten PGM, Kuipers F. Descending pathways from hypothalamus to dorsal motor vagus and ambiguus nuclei in the rat. J Auton Nerv Syst 1984;11:59–75. [DOI] [PubMed] [Google Scholar]
- 38. Grimwood S, Lu Y, Schmidt AW, Vanase‐Frawley MA, Sawant‐Basak A, Miller E, et al. Pharmacological characterization of 2‐methyl‐N‐((2'‐(pyrrolidin‐1‐ylsulfonyl)biphenyl‐4‐yl)methyl)propan‐1‐amine (PF‐04455242), a high‐affinity antagonist selective for kappa‐opioid receptors. J Pharmacol Exp Ther 2011;339:555–566. [DOI] [PubMed] [Google Scholar]
- 39. Arvidsson U, Riedl M, Chakrabarti S, Vulchanova L, Lee JH, Nakano AH, et al. The kappa‐opioid receptor is primarily postsynaptic: combined immunohistochemical localization of the receptor and endogenous opioids. Proc Natl Acad Sci USA 1995;92:5062–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Domingos AI, Sordillo A, Dietrich MO, Liu ZW, Tellez LA, Vaynshteyn J, et al. Hypothalamic melanin concentrating hormone neurons communicate the nutrient value of sugar. Elife 2013;2:e01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rutkowski DT, Wu J, Back S‐H, Callaghan MU, Ferris SP, Iqbal J, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress–mediated suppression of transcriptional master regulators. Dev Cell 2008;15:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 2011;54:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, et al. The unfolded protein response transducer IRE1α prevents ER stress–induced hepatic steatosis. EMBO J 2011;30:1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid‐induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest 2008;118:316–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinez de Morentin PB, Lopez M. “Mens sana in corpore sano”: exercise and hypothalamic ER stress. PLoS Biol 2010;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999;13:1211–1233. [DOI] [PubMed] [Google Scholar]
- 47. Gomori A, Ishihara A, Ito M, Matsushita H, Mashiko S, Iwaasa H, et al. Blockade of MCH1 receptor signalling ameliorates obesity and related hepatic steatosis in ovariectomized mice. Br J Pharmacol 2007;151:900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alon T, Friedman JM. Late‐onset leanness in mice with targeted ablation of melanin concentrating hormone neurons. J Neurosci 2006;26:389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wylie PG, Challiss RA, Blank JL. Regulation of extracellular signal–regulated kinase and c‐Jun N‐terminal kinase by G‐protein‐linked muscarinic acetylcholine receptors. Biochem J 1999;338(Pt. 3):619–628. [PMC free article] [PubMed] [Google Scholar]
- 50. Bi X, He X, Xu M, Zhao M, Yu X, Lu X, et al. Acetylcholine ameliorates endoplasmic reticulum stress in endothelial cells after hypoxia/reoxygenation via M3 AChR‐AMPK signaling. Cell Cycle 2015;14:2461–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28716/suppinfo.
Supporting Information