

Abstract
The lung is composed of airways and lung parenchyma, and the extracellular matrix (ECM) contains the main building blocks of both components. The ECM provides physical support and stability to the lung, and as such it has in the past been regarded as an inert structure. More recent research has provided novel insights revealing that the ECM is also a bioactive environment that orchestrates the cellular responses in its environs. Changes in the ECM in the airway or parenchymal tissues are now recognized in the pathological profiles of many respiratory diseases, including asthma, chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF). Only recently have we begun to investigate whether these ECM changes result from the disease process, or whether they constitute a driving factor that orchestrates the pathological outcomes. This review summarizes our current knowledge of the alterations in the ECM in asthma, COPD, and IPF, and the contributions of these alterations to the pathologies. Emerging data suggest that alterations in the composition, folding or rigidity of ECM proteins may alter the functional responses of cells within their environs, and in so doing change the pathological outcomes. These characteristics highlight potential avenues for targeting lung pathologies in the future. This may ultimately contribute to a better understanding of chronic lung diseases, and novel approaches for finding therapeutic solutions. © 2016 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: extracellular matrix, lung, collagen, myofibroblast, asthma, chronic obstructive pulmonary disease, niche, fibrosis
Introduction
The extracellular matrix (ECM) provides structural support and stability to the lung, and as such it has previously been regarded as a non‐stimulatory tissue. Recent research has provided novel insights indicating that the ECM is also a bioactive milieu that sets the scene for cellular responses in its environs.
In this review, we summarize the pathological alterations and the consequent functional changes that occur in three major lung diseases: asthma, chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF). In the near future, targeting the ECM might be an alternative for the treatment of chronic lung diseases.
Several chronic inflammatory diseases of the lungs develop with an altered composition of the ECM. Differing initial insults, or endogenous responses in different regions of the lungs, result in distinct compositions in the remodelled ECM in each disease, causing diverse functional outcomes. Several studies have addressed the composition of the ECM in chronic lung diseases such as asthma, COPD, and IPF, by studying biopsy samples, resected tissues or autopsy samples taken from the lungs at one time point (Figure 1). These cross‐sectional studies have presented sometimes contradictory results, as different methods of analysis and quantification were used, in diseases in different stages or in different anatomical locations. Nevertheless, they provide a basis for understanding ECM composition in human chronic pulmonary diseases.
Figure 1.
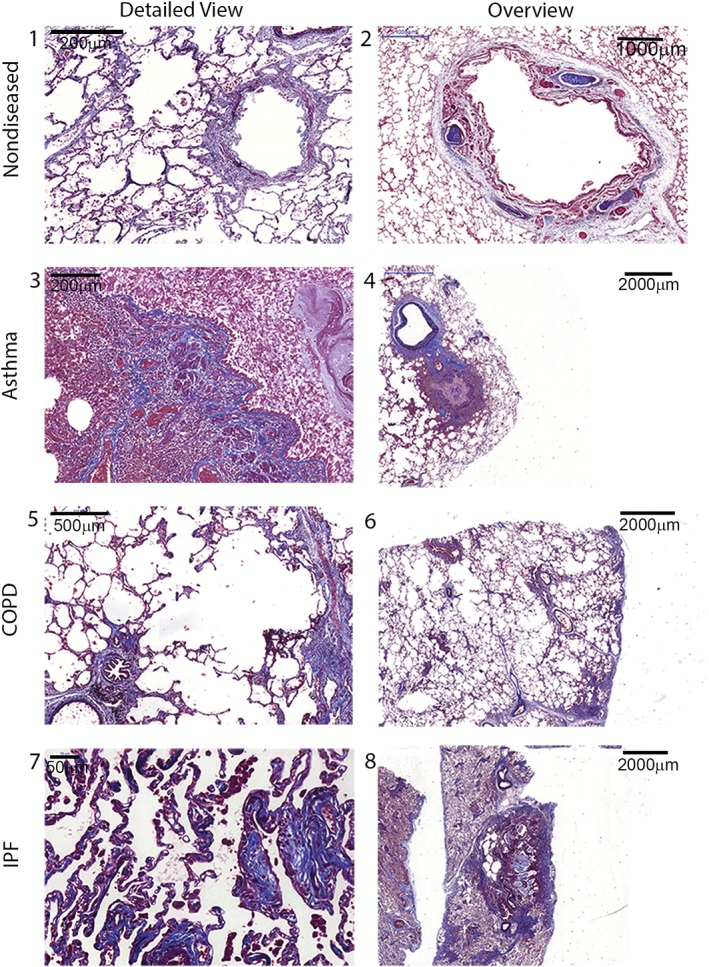
Histochemical staining of lung tissue sections. (1, 2) Large cartilaginous airway, small airway and surrounding alveolar parenchyma of a normal lung. Note the normal thickness of the mucosa, and the absence of inflammation. The alveoli have a normal size and architecture, and no fibrosis. (3, 4) Large airway and lung parenchyma of a patient with asthma. There is a thickened airway due to inflammation, and some matrix deposition, mainly at the submucosal level. The parenchyma shows an area of hyperinflation, but no fibrosis. (5, 6) Small airway and lung parenchyma in a patient with COPD. There is parenchymal destruction (emphysema) and some peribronchiolar fibrosis. (7, 8) Small airways and the lung parenchyma of a patient with IPF. There is architectural disorganization of the parenchyma around a dense fibrotic area, with cystic areas involving part of the parenchyma. The detail shows fibroblastic foci with areas of collagen deposition along alveolar walls. Verhoeff–Masson staining. Collagens are in blue, muscle is in red, and EFs are in black.
The ECM in diseases of the lungs
Asthma
Thickening of the airway basement membrane (BM) was the initial structural alteration described in asthma 1. It consists of thickening of the subepithelial lamina reticularis that lies beneath the true basal lamina of the bronchial epithelium, involving accumulation of collagens I, III and IV and fibronectin 2. In non‐asthmatics, the thickness of the lamina reticularis is approximately 5–6 µm, whereas it is consistently thickened in biopsies of asthmatics, with mean values of ∼9 µm 3. BM thickening develops early in the disease course, and persists after remission 4. However, in both adults and children, there is a lack of association between BM thickness and disease duration or severity 5. The ultrastructure of the collagen fibres in the thickened BM in asthma consists of an accumulation of (thinner) reticulin fibres rather than the (thicker) interstitial fibres, suggesting that the accumulation of collagen in asthma is different from that in interstitial lung diseases 6.
An altered composition of major ECM components has been described in all lung compartments in asthma [central airways, airway smooth muscle (ASM), distal parenchyma, and vessels], and the results vary according to asthma severity, control of disease, age, and use of corticosteroids 7. The main effector in asthma is the ASM, which secretes ECM components and, in turn, is functionally responsive to the ECM's composition, with the ECM deposited by asthma‐derived ASM enhancing growth and cytokine output as compared with that in non‐asthmatics 8, 9. There is no relative increase in the ECM in relation to the increased ASM mass in cases of fatal asthma 10, but there are altered contents of fibronectin, elastin fibres (EFs), matrix metalloproteinase (MMP)‐9, and MMP‐12 11. Patients with moderate asthma have more proteoglycans (PGs) within the ASM than those with severe asthma, suggesting that PG deposition may be protective 12. In mild asthmatics, despite no differences in ECM gene expression between asthmatics and controls, there are structure–function relationships between the fractional composition of the ECM within the ASM layer and indices of bronchoconstriction and bronchodilation in asthmatics 13. In fatal asthma, more pronounced changes in the small airways are observed in relation to PG, glycoprotein and collagen contents than in the central airways 14, 15. These changes, which are associated with rupture of alveolar attachments and a decreased elastin content, imply important involvement of the small airways in asthma pathophysiology, but further research is needed to understand the role of these ECM changes 16.
Lack of control and asthma severity are associated with changes in ECM composition, in both the central and the distal airway compartments. In severe asthma, there is increased deposition of collagen I and III and an increased ASM mass in bronchial biopsies 17, 18. In uncontrolled asthma, important differences have been reported in PG composition in both central airways and the alveolar parenchyma, such as increased percentage areas of collagen, versican and decorin as compared with patients with controlled asthma 19.
Access to airway tissue with functional correlates in asthma is restricted to endobronchial biopsies in the research setting, making structure–function studies related to the ECM difficult to conduct. The use of bronchial thermoplasty to treat severe asthma reduces ASM mass in biopsies 3 months after the procedure 20, but there are still no data regarding which changes in the ECM occur in the treated mucosa. Such information is important for the future development of targeted interventions that do not require this invasive procedure.
Chronic obstructive pulmonary disease
The instillation of elastolytic enzymes, with the subsequent development of emphysema, in the lungs of animals led to the recognition that the destruction of EFs was central in the pathogenesis of COPD 21. The ultrastructure of EFs is abnormal in COPD patients, with finely disrupted fibres, vacuoles and electron‐dense deposits, and signs of abnormal elastogenesis 22, with several studies showing reductions in EFs in the lung parenchyma of individuals with COPD as compared with ‘healthy smokers’, associated with loss of lung function 23, 24. Indeed, in severe COPD, increased gene expression of elastin is not accompanied by an increase in EF content per unit lung volume 25. A large study of lung gene expression showed that several genes related to elastogenesis were upregulated in the lungs of COPD patients, including that for fibulin‐5, a glycoprotein involved in EF assembly 26. The main cell type responsible for ECM production in COPD is thought to be the fibroblast/myofibroblast, but the ASM and epithelial cells also contribute to the ECM pool detected in these gene expression studies.
Interestingly, elastic reduction is not limited to the lung parenchyma, but is also observed in the small and large airways of COPD patients 24. This suggests that the loss of EFs in COPD is involved in airway obstruction not only by causing loss of airway–parenchyma coupling and hence airway collapse upon forced expiration, with subsequent air trapping, but also by altering the structural support of the airways.
Reports on the collagen content in COPD lungs are conflicting, with increased total collagen having been found in lungs of patients with emphysema 27. However, expression levels of genes related to collagen production were decreased in the lungs of COPD patients 28. Annoni et al reported that COPD patients have less collagen I in the airways, potentially favouring airway collapsibility 24. Similarly, Annoni et al 24 observed a decreased versican content in the lung parenchyma in moderate COPD, whereas Merrilees et al described an increased content in the same lung area, although they used different quantification methods 29. Van Straaten et al described a decrease in the contents of the PGs decorin and biglycan in patients with severe emphysema 30. The contents of other ECM components, such as tenascin and fibronectin, are increased in COPD patients 24.
At the microscopic level, advances in imaging technology are enabling a greater understanding of the three‐dimensional assembly of the ECM in COPD. Second harmonic generation (SHG), which has been used in the physical sciences for decades, has recently been validated for studying collagen I structural changes in COPD 31, 32 (Figure 2; supplementary material, Movies S1–S6). In addition, the intrinsic fluorophore properties of elastin have been harnessed through the use of two‐photon and multiphoton fluorescence techniques to reveal complex collagen and elastin networks not previously identified by conventional microscopy 31, 32.
Figure 2.
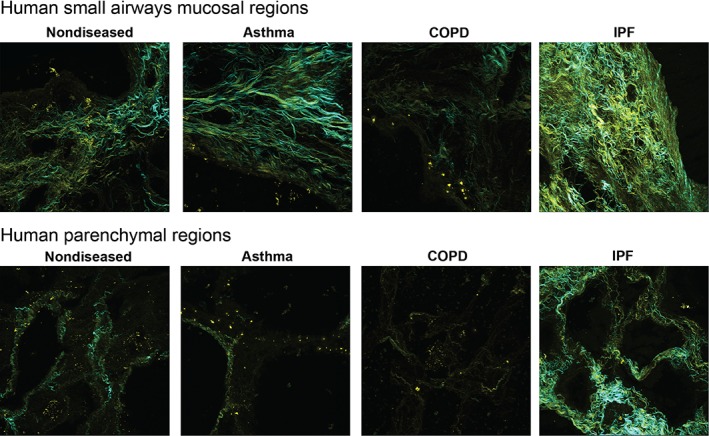
Representative SHG images of regions from the small airways mucosa or lung parenchyma from healthy donors or patients with asthma, COPD, or IPF. Yellow: backward immature/disorganized collagen. Cyan: mature/organized collagen.
Characterizing the functional implications of the alterations in ECM proteins in COPD lungs will provide a greater understanding of the mechanisms driving the perpetuation of lung destruction in the parenchymal regions, while fibrotic deposits line the small airways.
Idiopathic pulmonary fibrosis
Massive deposition of heterogeneously distributed ECM components in the alveolar parenchyma is a major characteristic of IPF. Structural alterations are present mainly in the alveolar regions, but also affect the terminal airways. These pathological alterations lead to a restrictive pattern and lower volumes on spirometry, with reduced gas transfer 33.
Fibroblastic foci, which constitute a singular characteristic of IPF, among other interstitial lung diseases, are accumulations of myofibroblasts, adjacent to areas of apoptotic or hyperplastic alveolar epithelial cells. Activated fibroblasts and myofibroblasts are believed to be the major producers of the ECM.
Immunohistochemical studies in the 1980s described depositions of collagens (mainly types I and III) and fibronectin along alveolar septa in chronic fibrotic diseases. Collagen III seems to predominate in early IPF, whereas collagen I dominates in late‐stage disease. (Figure 2; supplementary material, Movies S3‐S6). Authors initially reported that fibrosis occurred in foci near the air–tissue interface, localized outside remnants of a disrupted basal lamina and therefore within airspaces, providing support for the current concept of local, recurrent alveolar epithelial injury being central in the pathogenesis of IPF 34, 35. Roberts et al reported that fibroblast foci stained heavily for versican, to a lesser degree for hyaluronan, and very little for decorin and biglycan. The versican‐rich areas contained little mature collagen, but the myofibroblasts in these areas stained for type I procollagen, suggesting early collagen synthesis 36. The tenascin‐C content was also increased in the fibroblast foci 37. These data reinforce the idea that, in fibroblast foci, active matrix remodelling occurs and that early versican deposition may drive the process.
The content of EFs was also increased in fibrotic areas in IPF patients, and correlated with the degree of collagen deposition in biopsies. In addition, patients with high EF scores had worse outcomes than those with low scores, and EF score was an independent predictor of poor prognosis. EFs are needed to provide physiological elastic recoil within the lungs, but excess amounts of EFs affect the ‘hardness’ or ‘stiffness’ of the lungs, and increase the work of breathing in the early inspiratory phase, owing to enhanced elastic recoil 38.
It remains to be established whether the altered composition of the ECM is particular to IPF or occurs in other chronic fibrotic interstitial lung diseases. Estany et al studied lung gene expression of glycoproteins such as tenascin, fibronectin, and versican, and found no differences between IPF and chronic hypersensitivity pneumonitis 37.
Parker et al recently introduced the interesting concept that the remodelled ECM activates a positive pro‐fibrotic feedback loop 39. They cultivated control and IPF‐derived fibroblasts in de‐cellularized matrix from controls and IPF patients. Surprisingly, IPF‐derived matrix had a greater impact on gene expression than the origin of the cell, with prominent alterations in translational control being seen. Genes coding for ECM proteins in IPF were also targets of miR‐29, which was downregulated in IPF‐derived fibroblasts, suggesting a higher level of ECM protein expression. This study opens avenues for the development of new therapeutic strategies.
Responses of the ECM to corticosteroids
Corticosteroids constitute a mainline therapeutic strategy for managing inflammatory diseases in the lungs. However, the effectiveness of these treatments in preventing or reversing the ECM remodelling described above in asthma, COPD and IPF is controversial. Although collagens do not respond to corticosteroid treatment in severe asthma, a 2‐week course of budesonide increased the content of the PGs versican and biglycan, showing the selective action of corticosteroids on airway structure in asthma 40. Similarly, COPD patients receiving 30 months of inhaled fluticasone had increases in airway collagen III and versican contents as compared with patients receiving placebo 41. Treatment with steroids may hence contribute to stabilization of the airway structure in COPD.
In vitro data on the influence of corticosteroids on ECM production are also conflicting. Corticosteroids induced ECM protein production from ASM cells 42, 43, 44, 45, and the combination of budesonide and fluticasone induced collagen I and fibronectin production 46. Interestingly, Goulet et al reported that, in primary human lung fibroblasts, glucocorticoids increased total ECM and collagen deposition in the presence of serum, but not in the absence of serum 47. In contrast, glucocorticoids reduced the production of ECM proteins from bovine tracheal ASM cells 48, fibroblasts 49, and sputum cells 50.
The efficiency of current therapeutic approaches for addressing ECM changes in lung diseases is an underexplored area. Finding mechanisms for targeting the detrimental ECM changes in the lung has the potential to provide a wealth of information leading to future breakthroughs in the management of lung pathologies.
Cellular sources of ECM proteins
Fibroblasts and myofibroblasts are the main producers of ECM components in the lung, but airway epithelial cells and ASM cells are also important sources. Fibroblasts are key players in regulating the homeostasis of ECM proteins by coordinately secreting MMPs and their inhibitors and integrating signals from the ECM, a topic that has been reviewed elsewhere 51. Integrins are key molecules that mediate cell–cell and cell–ECM interactions, which also influence the ECM balance in the cellular microenvironment. The role of integrins in airway remodelling and their potential as therapeutic targets has recently been reviewed 52. No distinguishing differences in the cellular sources of ECM between asthma, COPD and IPF have been identified to date. We have a limited understanding of the mechanisms governing the switch from healthy to diseased stroma. When tissues are damaged and normal wound‐healing responses become dysregulated, fibrosis occurs. Most chronic fibrotic disorders have in common a persistent agent (often unknown) that sustains the production of growth factors, proteolytic enzymes, angiogenic factors, and fibrogenic cytokines, which stimulate the deposition of ECM that progressively remodels tissues 53 (Figure 3).
Figure 3.
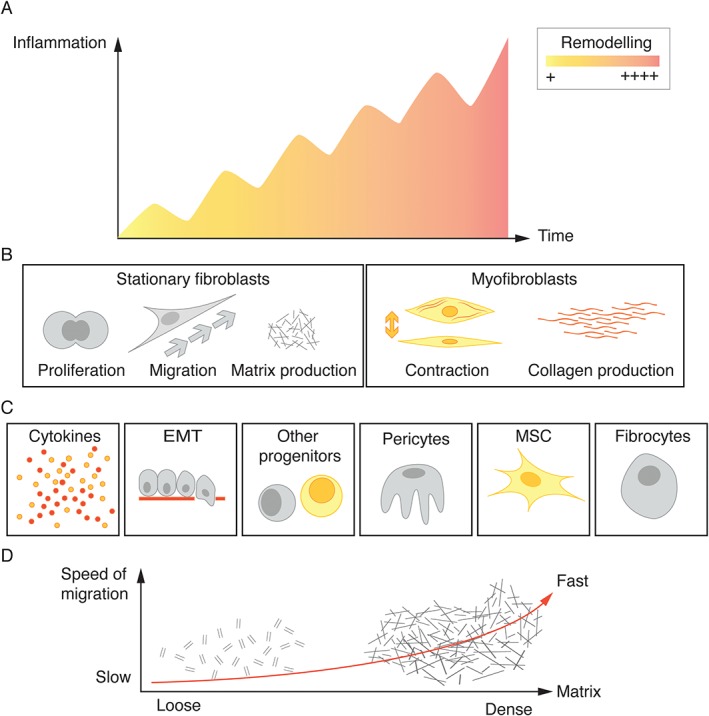
Model of events contributing to fibrosis in lung disease. (A) The common view that persistent inflammatory responses contribute to fibrosis. (B) In response to injury and fibrotic events, fibroblasts migrate towards the site of injury, proliferate, and ultimately differentiate into myofibroblasts. These cells specialize in ECM production. (C) Several mesenchymal progenitor cells in the lung may be involved in fibrotic events and contribute to the fibroblast pool in lung fibrosis, including fibrocytes, MSCs, pericytes, and potentially other progenitors. Epithelial cells may contribute to the fibroblast pool through EMT. (D) Migration is influenced in a complex way by tissue stiffness.
In response to injury, fibroblasts migrate towards the site of injury, and ultimately differentiate into myofibroblasts, which specialize in ECM production. The origin and differentiation potential of fibroblasts are very dynamic, and may result in unique patterns of the ECM in different lung disorders 54. Lung‐resident fibroblasts contribute greatly to the ECM in health, and are able to differentiate into muscle‐like contractile myofibroblasts, adopt a migratory phenotype, or proliferate in response to injury cues 55, 56, 57. Myofibroblasts deposit collagen types I and III during fibrotic events, but also collagen type IV, various PGs, and glycoproteins such as laminins and fibronectin.
Several mesenchymal progenitor cells in the lung have been discovered to be involved in fibrotic events, including fibrocytes, mesenchymal stromal cells (MSCs), and pericytes. One particular distinct perivascular cell type, which expresses ADAM12 and platelet‐derived growth factor receptor‐α, has a specific profibrotic fate. Interestingly, the cellular progeny of ADAM12‐positive cells is restricted to profibrotic cells, and not other mesenchymal derivatives such as adipocytes, suggesting that these cells are distinct from MSCs or pericytes 58. The MSCs in lung are located perivascularly, maintain the capacity to differentiate into bone, adipose and chondrocyte tissue in vitro, and, importantly, are organ‐resident 59, but their role is not clear 60. Fibrocytes are progenitor cells originating from bone marrow, and show both haematopoietic cell surface markers (CD45 and CD34) and mesenchymal markers (prolyl‐4‐hydroxylase and α‐smooth muscle actin), and typically also the chemokine receptor CXCR4 61. Importantly, they home to locations with ongoing tissue remodelling in IPF through the CXCL12–CXCR4 chemokine axis and differentiate into fibroblasts 62. CXCR4 is expressed on 90% of all fibrocytes, making it a potential target in IPF 63. Fibrocytes have also been observed in asthma and bronchiolitis obliterans, suggesting that these cells are important in the perpetuation of lung disease 62, 64, 65. The interaction of fibrocytes and the feedback that they receive from the lung ECM niche in different diseases in which fibrosis is involved is of great importance. Importantly, epithelial cells may contribute to the fibroblast pool in lung disease through epithelial‐to‐mesenchymal transition (EMT), a process whereby epithelial cells gradually lose epithelial markers (loss of E‐cadherin) and acquire a mesenchymal (fibroblast‐like) cell phenotype (gain of N‐cadherin) 66. Several different strategies targeting fibrosis are emerging, and new tools that mimic the in vivo situation to manipulate disease mechanisms are being developed 67, 68, 69. These approaches focusing on this previously underexplored source of ECM‐producing cells deserve greater attention.
The differences in occurrence, levels, location patterns and activity of the described cells, i.e. fibroblasts, myofibroblasts, MSCs, pericytes, and fibrocytes, mirror the state of the disease of the patient, and are not specific for a distinct lung disease such as COPD/asthma or IPF. However, these types of more complex comparison study are clearly warranted in the future, whereby patients with different lung diseases, in the same state of disease, and from whom biopsy sampling is possible, are compared to allow fingerprinting of distinct disease mechanisms and phenotypes, and also new biomarkers.
Shaping of the ECM
The many components of the ECM dictate the need for a highly regulated assembly process to bring all of the pieces together in a structured, organ‐specific manner (reviewed recently in 70). Although our understanding of the roles that many of the components play within the structure is limited, it is clear that the assembly of the ECM provides unique tissue‐specific microenvironments.
Importantly, the ECM is a highly dynamic environment, with some molecules changing rapidly and other components maintaining a steady state. Parts of the ECM are being constantly remodelled and subjected to post‐transcriptional changes 71. Collagen turnover rates in the lung have been estimated to be up to 10–15% per day 72. Interestingly, collagen synthesis rates decrease with age, and the proportion of newly synthesized collagen degraded in a mouse increased from 27% in a young animal (1 month) to 82% in an old animal (15 months) 73. PG turnover has been reported to occur a slower rate than collagen turnover in other organs, ranging from 3 to 70 days 74, 75. In contrast, EF proteins are remarkably long‐lived, with an estimated mean stability duration of 74 years in humans 76, and do not show a correlation with age 77.
The dynamic nature of the ECM is directed by the synthesis rates of the individual components and also by the surfeit of proteases that are released by both resident mesenchymal and inflammatory cells in the lung. MMPs, in particular, have received considerable attention in the endeavour to elucidate mechanisms driving ECM remodelling in the respiratory system 78. Although it is recognized that this is a level of control of the ECM state that can be disrupted during disease, this will not be reviewed extensively here. Interestingly, enhanced ECM degradation has recently been reported in COPD exacerbations, suggesting that there may be a link between the rate of ECM turnover during disease exacerbations and disease progression 79. This is an under‐recognized element of lung pathology that should be a focus for research in the future.
ECM stiffness
The stiffness of the ECM in the lung is directly related to the biomechanical properties of the tissue. The fibrillary ECM proteins (e.g. collagen and fibronectin) are responsible for the tensile strength, whereas the elastic recoil is provided by the elastin molecules 21, 80. Changes in the levels and ratios of these proteins, as seen in asthma, COPD, and IPF, impact on the relative contributions of the proteins to ECM stiffness 81. Normal human lung parenchyma has a stiffness of 0.44–0.75 kPa (depending on whether the alveolar wall, airway wall or airway epithelium is measured), whereas IPF lung tissue has a stiffness of 50 kPa 82, and emphysematous tissue has a reduced stiffness 83. The enhanced collagen deposition in IPF is associated with the enhanced stiffness in this ECM 82. Similarly, the increased fractional area of tenascin and fibronectin in the COPD ECM, which indicates relative elastin destruction, may be linked to the reduced stiffness 84.
The ECM also acts as a reservoir for growth factor deposition, with transforming growth factor (TGF)‐β being well recognized as being secreted in an inactive form that is bound in the ECM by the latent TGF‐β‐binding protein 85. ECM biomechanical forces are important for activation of TGF‐β, and stiffer matrices promote enhanced activation of TGF‐β, suggesting that the stiffer ECM in IPF generates a feedback loop that exacerbates pro‐fibrotic signalling in this microenvironment 86.
The ECM structural assembly is regulated by enzymatic and non‐enzymatic cross‐linking, which also influence the stiffness of the ECM by altering the physical characteristics of the ECM and strengthening protein–protein interactions within the structure. These alterations can change the resistance to degradation by proteolysis, enhance cell–matrix interactions, and lead to the emergence of neoepitopes, as reviewed in 87. As decorin cross‐links collagen fibrils, the decreases in decorin and biglycan contents in smoking‐related emphysema impact on collagen cross‐linking 30, 88. Lysyl oxidases and transglutaminases are the enzyme families responsible for collagen cross‐linking in the ECM. Selective reduction of lysyl oxidase‐like 2 levels was recently investigated as a therapeutic approach for IPF 89 (ClinicalTrials.gov NCT01769196). Although a lack of efficacy led to the early termination of this trial, ECM stiffness is an under‐recognized area that deserves greater attention as a potential therapeutic target in lung diseases.
Cellular behaviours that are directed by the ECM
The ECM is a bioactive entity that has the capacity to regulate many cellular behaviours that impact on lung pathologies (Figure 4).
Figure 4.
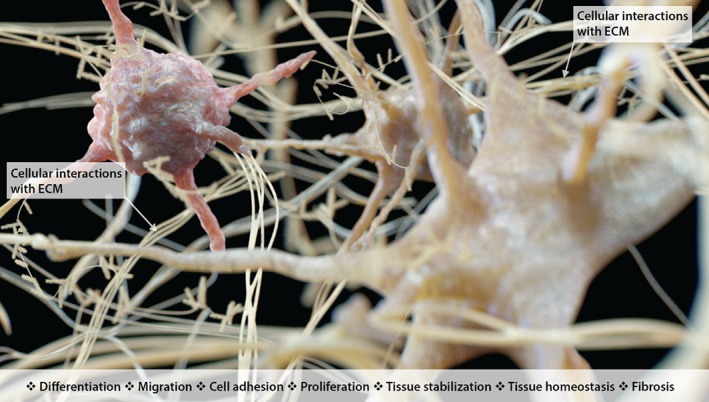
A graphical representation of the complex cellular interactions with the ECM in the lung: potential impact on differentiation, migration, adhesion, proliferation, tissue stabilization, homeostasis, and fibrosis. Cellular interactions with the ECM provide a positive feedback loop to direct activities of the cells, alter tissue homeostasis, and drive fibrosis progression.
Migration
Cell migration is dependent on complex tissue environments and their interactions with cellular biomechanical/chemical properties. Cells migrate in different modes, but the common underlying process involves polarization of the actomyosin‐driven shape change of the cell body 90. Of great importance, migration is influenced in a complex way by the confinement of cells in the tissue, adhesion ligand density, such as integrin binding to collagens, laminins, and fibronectins, and stiffness and topology of the matrix 91, 92. The ECM is therefore highly involved in cell migration, and biglycan and decorin induce morphological and cytoskeletal changes in fibroblasts, resulting in an increase in cell migration. Several intracellular signalling molecules are upregulated in response to decorin and biglycan, including the focal adhesion proteins paxillin and zyxin, and some of the small Rho GTPases such as RhoA, Rac1, and Cdc42 93.
Proof of concept in disease
The mobility of fibroblasts in lung disease is commonly affected and influenced by an altered ECM. Elongated fibroblasts in the broncheoalveolar lavage fluid of mild asthmatics migrated twice as far as fibroblasts originating from bronchial biopsies from the same patients, and these fibroblasts also had increased production of biglycan, versican, and decorin, along with induced expression of RhoA and Rac1. These fibroblasts were not found in healthy controls 57. Interestingly, similar results have been found in systemic sclerosis 94.
Proliferation
The ECM is a strong determinant of cell proliferation. Many ECM proteins, including collagen I, fibronectin, laminin, biglycan, decorin, and versican, have been reported to influence cell proliferation. Collagen I and fibronectin promote ASM cell proliferation, whereas laminin chains induce apoptosis 95, 96. The core protein of biglycan and decorin binds cytokines such as TGF‐α 97, but the fine structures of the side chains of PGs also have a great impact, owing to their potential to interact with cytokines and growth factors. For example, glycosaminoglycan side chains rich in l‐iduronate have been found to inhibit the proliferation of fibroblasts 98 by potentially binding growth factors 99. Similarly, heparan sulphate PG (HSPG) is known for its antiproliferative effect; however, treating fibroblasts with HSPG together with platelet‐derived growth factor‐BB enhances mitogenicity. The ECM derived from cells isolated from patients with asthma induced a greater proliferative capacity than that from cells from people without asthma 9, and these matrices have been shown to have defined differences in ECM proteins that have previously been reported to be proproliferative 9, 42. These complex effects indicate that the ECM itself may regulate cell proliferation by affecting the expression of receptors for cytokines and growth factors, which may initiate both stimulatory and inhibitory signals 100.
Proof of concept in disease
There are great differences in distal versus central lung airways in several different lung disorders, in terms of both the composition of the ECM and the phenotypic differences in mesenchymal cells, including proliferation and ECM production 19, 56. Centrally derived fibroblasts are larger and have more projections than distal fibroblasts 101. In addition, distal fibroblasts have a higher proliferative potential than central fibroblasts from healthy subjects 56 or asthma patients 101, but not than those from COPD patients, which, intriguingly, had a lower proliferation potential than those from control subjects. In contrast, ASM cells from asthma patients proliferate at a higher rate than those from healthy subjects 102, 103. In pulmonary fibrosis patients, fibroblast populations that are high producers of hyaluronan and decorin and have lower proliferative potential may play a crucial role in the pathogenesis of fibrosis 104. Also after lung transplantation, the production of perlecan and decorin has been noted to negatively correlate with fibroblast proliferation, which may represent early signs of ongoing remodelling and potential markers for Bronchiolitis Obliterans Syndrome (BOS) 55.
Cellular responses to the stiffness of the microenvironment
Alterations in collagen fibre structure and stiffness are of great importance for tissue structure and elasticity, but also for fibroblast responses. Indeed, stiffening of the ECM during remodelling and the development of fibrosis affects cell adhesion and migration, so that cells migrate towards a stiffer ECM 105. Fibroblasts increase their spread area, increase actin stress fibre formation and form larger focal adhesion complexes as the culture substrate stiffness increases. Cell proliferation is also increased with increased substrate stiffness. These changes require localization of the nuclear transcriptional regulator Yes‐associated protein (YAP) in the cell nucleus 106, and this event is tightly coupled to larger traction force generation. In culture, YAP accumulates in the nuclei of fibroblasts grown on pathologically stiff matrices but not on physiologically compliant matrices, and may therefore be heavily involved in amplifying and sustaining pathological fibrosis 107.
Proof of concept in disease
Pathological fibrosis is both a cause and a consequence of fibroblast activation. Interestingly, not only the ECM but also cells themselves become stiffer in lung fibrotic diseases. In scleroderma, atomic force microscopy showed that fibroblasts were stiffer and had lower elasticity than fibroblasts from healthy controls 108. In bleomycin‐induced lung fibrosis, locally induced tissue stiffness was accompanied by a change in fibroblast phenotype from quiescence to progressively increasing proliferation and matrix production. The increased matrix stiffness also strongly suppressed fibroblast production of cyclooxygenase‐2 and prostaglandin E2 (PGE2), an autocrine inhibitory mechanism opposing fibrogenesis 109. Differences in proliferation and contraction, along with responsiveness to PGE2, have been demonstrated between fibroblasts from IPF patients and those from healthy controls, when fibroblasts were grown on stiff matrices. Promisingly, these differences were ablated when fibroblasts were grown on soft matrices, even though IPF fibroblasts remained relatively resistant to PGE2 110. De‐cellularized lung matrices from IPF patients are stiffer than matrices from healthy controls 82, and induce fibroblasts to pathologically remodel the ECM 39. Therefore, the stiffer matrix has profound effects on cellular behaviour in disease, providing a mechanism to enhance myofibroblast differentiation, ECM synthesis, and cross‐linking, and thereby driving fibrosis progression. These data give new hope for targeting mechanical cues and pathways triggered by the ECM stiffness when cells are unresponsive to anti‐fibrotic agents.
Little is known about disease‐specific changes in cellular behaviours that are driven by the ECM. This is an area where further research may provide significant clues about mechanisms that may be targeted in disease‐specific manners as we strive to develop personalized medicine approaches for the management of patients with lung diseases.
Why is it important to understand the contribution of the ECM in lung diseases?
The course of many chronic lung diseases is associated with changes in the composition, content and structural ordering of the ECM components. Such changes vary greatly between the different pathologies, with massive deposition being seen in diseases such as IPF. In contrast, destruction predominates in others, such as in the lung tissue of COPD patients, but here there is fibrosis in the airway wall. To date, there are few therapeutic regimens aimed at specifically treating ECM remodelling. Changes in the ECM, which are driven by multiple cell types in the lung, affect lung function and cell biology, but, more importantly, dysregulation of the ECM seems to provide a positive feedback loop to drive fibrosis progression (Figure 5).
Figure 5.
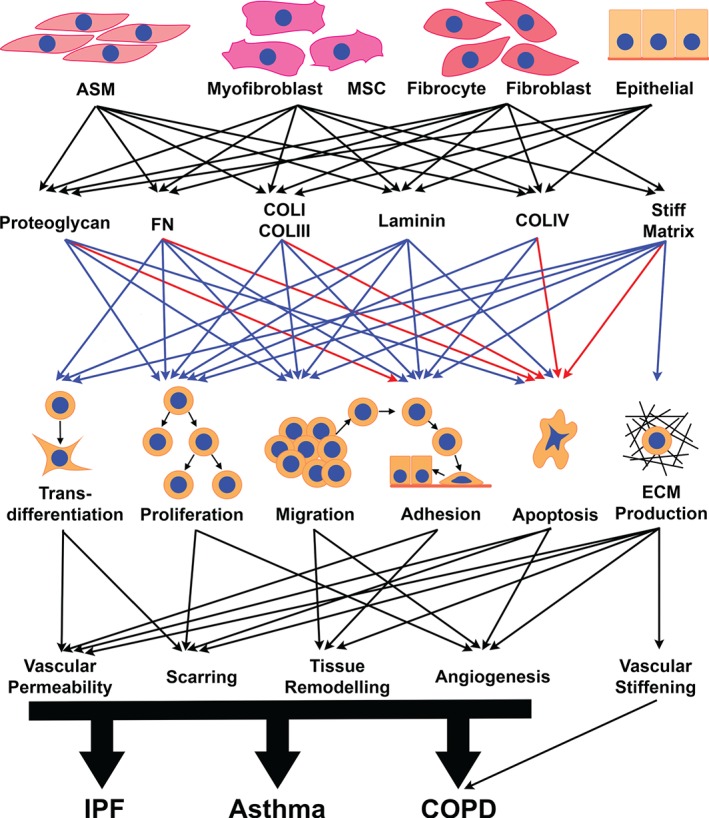
The remodelled ECM in the lungs is produced by multiple cell types, and in turn modulates the behaviour of the cells to impact on the perpetuation of disease pathology. Blue arrows indicate a positive/promoting effect, and red arrows indicate a negative/inhibitory effect. COLI, collagen I; COLIII, collagen III; COLIV, collagen IV; FN, Fibronectin.
We therefore need to understand much more about the mechanisms underlying these changes and the resultant in vivo processes. The positive results of the pirfenidone 111 and nintedanib 112 phase III trials, demonstrating that agents targeting the different pathways that drive lung fibrosis can reduce the progression of IPF, provide excellent examples of why this understanding is important 113. It is also essential to understand how currently available therapies interfere with ECM elements, such as the substitution of ASM with ‘fibrotic’ tissue by bronchial thermoplasty 114, or the prolonged use of corticosteroids or combination therapy in asthma or COPD. Recognizing and investigating the under‐recognized elements of ECM remodelling in lung pathologies provides an exciting opportunity to obtain the next, urgently needed breakthrough to advance our ability to reduce the mortality resulting from lung diseases.
Author contributions statement
The authors contributed in the following way: JKB, TM, GW‐T: conceived the concept and designed the review; GT: generated the second harmonics images and the cell types contribution figure; TM: collected and analysed the images of Verhoeff–Masson‐stained tissue; GW‐T, JK: designed the mesenchymal stromal cells schematic and the summary figure; JB: designed and compiled the photomicrograph figures; all authors: contributed equally to the literature search and writing.
SUPPLEMENTARY MATERIAL ONLINE.
Movie S1. 3‐dimensional SHG signals in a section showing an airway in an asthma patient's lung tissue.
Movie S2. 3‐dimensional SHG signals in a section showing an airway in a COPD patient's lung tissue.
Movie S3. 3‐dimensional SHG signals in a section showing an airway in an IPF patient's lung tissue.
Movie S4. 3‐dimensional SHG signals in a section showing alveoli in an IPF patient's lung tissue.
Movie S5. 3‐dimensional SHG signals in a section showing an airway in non‐diseased lung tissue.
Movie S6. 3‐dimensional SHG signals in a section showing alveoli in non‐diseased lung tissue.
Supporting information
Movie S1. 3‐dimensional SHG signals in a section showing an airway in an asthma patient's lung tissue.
Movie S2. 3‐dimensional SHG signals in a section showing an airway in a COPD patient's lung tissue.
Movie S3. 3‐dimensional SHG signals in a section showing an airway in an IPF patient's lung tissue.
Movie S4. 3‐dimensional SHG signals in a section showing alveoli in an IPF patient's lung tissue.
Movie S5. 3‐dimensional SHG signals in a section showing an airway in non‐diseased lung tissue.
Movie S6. 3‐dimensional SHG signals in a section showing alveoli in non‐diseased lung tissue.
Acknowledgements
JB was funded by National Health and Medical Research Council (NH&MRC) Australia Fellowship #1032695 and a Rosalind Franklin Fellowship, co‐funded by European Union and University of Groningen, University Medical Centre Groningen. TM was funded by the Brazilian Research National Council (CNPq). GW‐T was supported by the Swedish Foundation for Strategic Research, the Swedish Medical Research Council (11550), the Swedish Heart‐Lung Foundation, the Medical Faculty at Lund University, and ALF (a government public health grant).
No conflicts of interest were declared.
References
- 1. Huber HL, Koessler KK. The pathology of bronchial asthma. Arch Intern Med 1922; 30 : 689–760. [Google Scholar]
- 2. Roche WR, Beasley R, Williams JH, et al. Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1989; 1: 520–524. [DOI] [PubMed] [Google Scholar]
- 3. Jeffery PK, Wardlaw AJ, Nelson FC, et al. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am Rev Respir Dis 1989; 140: 1745–1753. [DOI] [PubMed] [Google Scholar]
- 4. Broekema M, Timens W, Vonk JM, et al. Persisting remodeling and less airway wall eosinophil activation in complete remission of asthma. Am J Respir Crit Care Med 2011; 183: 310–316. [DOI] [PubMed] [Google Scholar]
- 5. Payne DN, Rogers AV, Adelroth E, et al. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med 2003; 167: 78–82. [DOI] [PubMed] [Google Scholar]
- 6. Saglani S, Molyneux C, Gong H, et al. Ultrastructure of the reticular basement membrane in asthmatic adults, children and infants. Eur Respir J 2006; 28: 505–512. [DOI] [PubMed] [Google Scholar]
- 7. Mauad T, Bel EH, Sterk PJ. Asthma therapy and airway remodeling. J Allergy Clin Immunol 2007; 120: 997–1009; quiz 1010–1001. [DOI] [PubMed] [Google Scholar]
- 8. Chan V, Burgess JK, Ratoff JC, et al. Extracellular matrix regulates enhanced eotaxin expression in asthmatic airway smooth muscle cells. Am J Respir Crit Care Med 2006; 174: 379–385. [DOI] [PubMed] [Google Scholar]
- 9. Johnson PR, Burgess JK, Underwood PA, et al. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J Allergy Clin Immunol 2004; 113: 690–696. [DOI] [PubMed] [Google Scholar]
- 10. James A, Mauad T, Abramson M, et al. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med 2012 ; 186: 568–569. [DOI] [PubMed] [Google Scholar]
- 11. Araujo BB, Dolhnikoff M, Silva LF, et al. Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur Respir J 2008; 32: 61–69. [DOI] [PubMed] [Google Scholar]
- 12. Pini L, Hamid Q, Shannon J, et al. Differences in proteoglycan deposition in the airways of moderate and severe asthmatics. Eur Respir J 2007; 29: 71–77. [DOI] [PubMed] [Google Scholar]
- 13. Yick CY, Ferreira DS, Annoni R, et al. Extracellular matrix in airway smooth muscle is associated with dynamics of airway function in asthma. Allergy 2012; 67: 552–559. [DOI] [PubMed] [Google Scholar]
- 14. de Medeiros Matsushita M, da Silva LF, dos Santos MA, et al. Airway proteoglycans are differentially altered in fatal asthma. J Pathol 2005; 207: 102–110. [DOI] [PubMed] [Google Scholar]
- 15. Dolhnikoff M, da Silva LF, de Araujo BB, et al. The outer wall of small airways is a major site of remodeling in fatal asthma. J Allergy Clin Immunol 2009; 123: 1090–1097. [DOI] [PubMed] [Google Scholar]
- 16. Mauad T, Silva LF, Santos MA, et al. Abnormal alveolar attachments with decreased elastic fiber content in distal lung in fatal asthma. Am J Respir Crit Care Med 2004; 170: 857–862. [DOI] [PubMed] [Google Scholar]
- 17. Benayoun L, Druilhe A, Dombret MC, et al. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med 2003; 167: 1360–1368. [DOI] [PubMed] [Google Scholar]
- 18. Chakir J, Shannon J, Molet S, et al. Airway remodeling‐associated mediators in moderate to severe asthma: effect of steroids on TGF‐beta, IL‐11, IL‐17, and type I and type III collagen expression. J Allergy Clin Immunol 2003; 111: 1293–1298. [DOI] [PubMed] [Google Scholar]
- 19. Weitoft M, Andersson C, Andersson‐Sjoland A, et al. Controlled and uncontrolled asthma display distinct alveolar tissue matrix compositions. Respir Res 2014; DOI: 10.1186/1465-9921-15-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pretolani M, Dombret MC, Thabut G, et al. Reduction of airway smooth muscle mass by bronchial thermoplasty in patients with severe asthma. Am J Respir Crit Care Med; 190: 1452–1454. [DOI] [PubMed] [Google Scholar]
- 21. Shifren A, Mecham RP. The stumbling block in lung repair of emphysema: elastic fiber assembly. Proc Am Thorac Soc 2006; 3: 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukuda Y, Masuda Y, Ishizaki M, et al. Morphogenesis of abnormal elastic fibers in lungs of patients with panacinar and centriacinar emphysema. Hum Pathol 1989; 20: 652–659. [DOI] [PubMed] [Google Scholar]
- 23. Black PN, Ching PS, Beaumont B, et al. Changes in elastic fibres in the small airways and alveoli in COPD. Eur Respir J 2008; 31: 998–1004. [DOI] [PubMed] [Google Scholar]
- 24. Annoni R, Lancas T, Yukimatsu Tanigawa R, et al. Extracellular matrix composition in COPD. Eur Respir J 2012; 40: 1362–1373. [DOI] [PubMed] [Google Scholar]
- 25. Deslee G, Woods JC, Moore CM, et al. Elastin expression in very severe human COPD. Eur Respir J 2009; 34: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brandsma CA, van den Berge M, Postma DS, et al. A large lung gene expression study identifying fibulin‐5 as a novel player in tissue repair in COPD. Thorax 2015; 70: 21–32. [DOI] [PubMed] [Google Scholar]
- 27. Vlahovic G, Russell ML, Mercer RR, et al. Cellular and connective tissue changes in alveolar septal walls in emphysema. Am J Respir Crit Care Med 1999; 160: 2086–2092. [DOI] [PubMed] [Google Scholar]
- 28. Gosselink JV, Hayashi S, Elliott WM, et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181: 1329–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merrilees MJ, Ching PS, Beaumont B, et al. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir Res 2008; DOI: 10.1186/1465-9921-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Straaten JF, Coers W, Noordhoek JA, et al. Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod Pathol 1999; 12: 697–705. [PubMed] [Google Scholar]
- 31. Abraham T, Hogg J. Extracellular matrix remodeling of lung alveolar walls in three dimensional space identified using second harmonic generation and multiphoton excitation fluorescence. J Struct Biol 2010; 171: 189–196. [DOI] [PubMed] [Google Scholar]
- 32. Tjin G, Xu P, Kable SH, et al. Quantification of collagen I in airway tissues using second harmonic generation. J Biomed Opt 2014; 19: 36005. [DOI] [PubMed] [Google Scholar]
- 33. Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012; 380: 680–688. [DOI] [PubMed] [Google Scholar]
- 34. Kuhn C, 3rd , Boldt J, King TE, Jr , et al. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis 1989; 140: 1693–1703. [DOI] [PubMed] [Google Scholar]
- 35. Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med 2012; 18: 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bensadoun ES, Burke AK, Hogg JC, et al. Proteoglycan deposition in pulmonary fibrosis. Am J Respir Crit Care Med 1996; 154: 1819–1828. [DOI] [PubMed] [Google Scholar]
- 37. Estany S, Vicens‐Zygmunt V, Llatjos R, et al. Lung fibrotic tenascin‐C upregulation is associated with other extracellular matrix proteins and induced by TGFbeta1. BMC Pulm Med 2014; DOI: 10.1186/1471-2466-14-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Enomoto N, Suda T, Kono M, et al. Amount of elastic fibers predicts prognosis of idiopathic pulmonary fibrosis. Respir Med 2013; 107: 1608–1616. [DOI] [PubMed] [Google Scholar]
- 39. Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 2014; 124: 1622–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Kluijver J, Schrumpf JA, Evertse CE, et al. Bronchial matrix and inflammation respond to inhaled steroids despite ongoing allergen exposure in asthma. Clin Exp Allergy 2005; 35: 1361–1369. [DOI] [PubMed] [Google Scholar]
- 41. Kunz LI, Strebus J, Budulac SE, et al. Inhaled steroids modulate extracellular matrix composition in bronchial biopsies of COPD patients: a randomized, controlled trial. PLoS One 2013; 8: e63430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson PR, Black JL, Carlin S, et al. The production of extracellular matrix proteins by human passively sensitized airway smooth‐muscle cells in culture: the effect of beclomethasone. Am J Respir Crit Care Med 2000; 162: 2145–2151. [DOI] [PubMed] [Google Scholar]
- 43. Poiani GJ, Tozzi CA, Thakker‐Varia S, et al. Effect of glucocorticoids on collagen accumulation in pulmonary vascular remodeling in the rat. Am J Respir Crit Care Med 1994; 149: 994–999. [DOI] [PubMed] [Google Scholar]
- 44. Graham MF, Willey A, Adams J, et al. Corticosteroids increase procollagen gene expression, synthesis, and secretion by human intestinal smooth muscle cells. Gastroenterology 1995; 109: 1454–1461. [DOI] [PubMed] [Google Scholar]
- 45. Warshamana GS, Martinez S, Lasky JA, et al. Dexamethasone activates expression of the PDGF‐alpha receptor and induces lung fibroblast proliferation. Am J Physiol 1998; 274: L499–L507. [DOI] [PubMed] [Google Scholar]
- 46. Burgess JK, Oliver BGG, Poniris MH, et al. A phosphodiesterase 4 inhibitor inhibits matrix protein deposition in airways in vitro. J Allergy Clin Immunol 2006; 118: 649–657. [DOI] [PubMed] [Google Scholar]
- 47. Goulet S, Bihl MP, Gambazzi F, et al. Opposite effect of corticosteroids and long‐acting beta(2)‐agonists on serum‐ and TGF‐beta(1)‐induced extracellular matrix deposition by primary human lung fibroblasts. J Cell Physiol 2007; 210: 167–176. [DOI] [PubMed] [Google Scholar]
- 48. Chen G, Khalil N. In vitro wounding of airway smooth muscle cell monolayers increases expression of TGF‐beta receptors. Respir Physiol Neurobiol 2002; 132: 341–346. [DOI] [PubMed] [Google Scholar]
- 49. Tomic R, Lassiter CC, Ritzenthaler JD, et al. Anti‐tissue remodeling effects of corticosteroids: fluticasone propionate inhibits fibronectin expression in fibroblasts. Chest 2005; 127: 257–265. [DOI] [PubMed] [Google Scholar]
- 50. Profita M, Gagliardo R, Di Giorgi R, et al. In vitro effects of flunisolide on MMP‐9, TIMP‐1, fibronectin, TGF‐beta1 release and apoptosis in sputum cells freshly isolated from mild to moderate asthmatics. Allergy 2004; 59: 927–932. [DOI] [PubMed] [Google Scholar]
- 51. Westergren‐Thorsson G, Larsen K, Nihlberg K, et al. Pathological airway remodelling in inflammation. Clin Respir J 2010; 4(suppl 1): 1–8. [DOI] [PubMed] [Google Scholar]
- 52. Wright DB, Meurs H, Dekkers BG. Integrins: therapeutic targets in airway hyperresponsiveness and remodelling? Trends Pharmacol Sci 2014; 35: 567–574. [DOI] [PubMed] [Google Scholar]
- 53. Duffield JS, Lupher M, Thannickal VJ, et al. Host responses in tissue repair and fibrosis. Annu Rev Pathol 2013; 8: 241–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Larsen K, Malmstrom J, Wildt M, et al. Functional and phenotypical comparison of myofibroblasts derived from biopsies and bronchoalveolar lavage in mild asthma and scleroderma. Respir Res 2006; DOI: 10.1186/1465-9921-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Andersson‐Sjoland A, Thiman L, Nihlberg K, et al. Fibroblast phenotypes and their activity are changed in the wound healing process after lung transplantation. J Heart Lung Transplant 2011; 30: 945–954. [DOI] [PubMed] [Google Scholar]
- 56. Hallgren O, Nihlberg K, Dahlback M, et al. Altered fibroblast proteoglycan production in COPD. Respir Res 2010; DOI: 10.1186/1465-9921-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Larsen K, Tufvesson E, Malmstrom J, et al. Presence of activated mobile fibroblasts in bronchoalveolar lavage from patients with mild asthma. Am J Respir Crit Care Med 2004; 170: 1049–1056. [DOI] [PubMed] [Google Scholar]
- 58. Dulauroy S, Di Carlo SE, Langa F, et al. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med 2012; 18: 1262–1270. [DOI] [PubMed] [Google Scholar]
- 59. Rolandsson S, Andersson Sjoland A, Brune JC, et al. Primary mesenchymal stem cells in human transplanted lungs are CD90/CD105 perivascularly located tissue‐resident cells. BMJ Open Respir Res 2014; 1: e000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kramann R, Schneider RK, DiRocco DP, et al. Perivascular gli1(+) progenitors are key contributors to injury‐induced organ fibrosis. Cell Stem Cell 2015; 16: 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lekkerkerker AN, Aarbiou J, van Es T, et al. Cellular players in lung fibrosis. Curr Pharm Des 2012; 18: 4093–4102. [DOI] [PubMed] [Google Scholar]
- 62. Andersson‐Sjoland A, de Alba CG, Nihlberg K , et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 2008; 40: 2129–2140. [DOI] [PubMed] [Google Scholar]
- 63. Mehrad B, Burdick MD, Strieter RM. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int J Biochem Cell Biol 2009; 41: 1708–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andersson‐Sjoland A, Nihlberg K, Eriksson L, et al. Fibrocytes and the tissue niche in lung repair. Respir Res 2011; DOI: 10.1186/1465-9921-12-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nihlberg K, Larsen K, Hultgardh‐Nilsson A, et al. Tissue fibrocytes in patients with mild asthma: a possible link to thickness of reticular basement membrane? Respir Res 2006; DOI: 10.1186/1465-9921-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zeisberg M, Neilson EG. Biomarkers for epithelial–mesenchymal transitions. J Clin Invest 2009; 119: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nguyen Hoang AT, Chen P, Bjornfot S, et al. Technical advance: live‐imaging analysis of human dendritic cell migrating behavior under the influence of immune‐stimulating reagents in an organotypic model of lung. J Leukoc Biol 2014; 96: 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Petersen TH, Calle EA, Zhao L, et al. Tissue‐engineered lungs for in vivo implantation. Science 2010; 329: 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nguyen Hoang AT, Chen P, Juarez J, et al. Dendritic cell functional properties in a three‐dimensional tissue model of human lung mucosa. Am J Physiol Lung Cell Mol Physiol 2012; 302: L226–L237. [DOI] [PubMed] [Google Scholar]
- 70. Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol 2014; 15: 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci 2010; 123: 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Laurent GJ. Dynamic state of collagen: pathways of collagen degradation in vivo and their possible role in regulation of collagen mass. Am J Physiol 1987; 252: C1–C9. [DOI] [PubMed] [Google Scholar]
- 73. Mays PK, Mcanulty RJ, Laurent GJ. Age‐related‐changes in lung collagen‐metabolism – a role for degradation in regulating lung collagen production. Am Rev Respir Dis 1989; 140: 410–416. [DOI] [PubMed] [Google Scholar]
- 74. Lohmander S. Turnover of proteoglycans in guinea‐pig costal cartilage. Arch Biochem Biophys 1977; 180: 93–101. [DOI] [PubMed] [Google Scholar]
- 75. Rada JA, Achen VR, Rada KG. Proteoglycan turnover in the sclera of normal and experimentally myopic chick eyes. Invest Ophthalmol Vis Sci 1998; 39: 1990–2002. [PubMed] [Google Scholar]
- 76. Shapiro SD, Endicott SK, Province MA, et al. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D‐aspartate and nuclear weapons‐related radiocarbon. J Clin Invest 1991; 87: 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Andreotti L, Bussotti A, Cammelli D, et al. Connective‐tissue in aging lung. Gerontology 1983; 29: 377–387. [DOI] [PubMed] [Google Scholar]
- 78. Houghton AM. Matrix metalloproteinases in destructive lung disease. Matrix Biol 2015; 44–46: 167–174. [DOI] [PubMed] [Google Scholar]
- 79. Sand JMB, Knox AJ, Lange P, et al. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir Res 2015; DOI: 10.1186/s12931-015-0225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Senior RM, Bielefeld DR, Abensohn MK. The effects of proteolytic enzymes on the tensile strength of human lung. Am Rev Respir Dis 1975; 111: 184–188. [DOI] [PubMed] [Google Scholar]
- 81. White ES. Lung extracellular matrix and fibroblast function. Ann Am Thorac Soc 2015; 12(suppl 1): S30–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Booth AJ, Hadley R, Cornett AM, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med 2012; 186: 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Parameswaran H, Majumdar A, Suki B. Linking microscopic spatial patterns of tissue destruction in emphysema to macroscopic decline in stiffness using a 3D computational model. PLoS Comput Biol 2011; 7: e1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Annoni R, Lancas T, Tanigawa RY, et al. Extracellular matrix composition in chronic obstructive pulmonary disease. Eur Respir J 2012; 40: 1362–1373. [DOI] [PubMed] [Google Scholar]
- 85. Shi M, Zhu J, Wang R, et al. Latent TGF‐beta structure and activation. Nature 2011; 474: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wipff PJ, Rifkin DB, Meister JJ, et al. Myofibroblast contraction activates latent TGF‐beta1 from the extracellular matrix. J Cell Biol 2007; 179: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Karsdal MA, Nielsen MJ, Sand JM, et al. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol 2013; 11: 70–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Noordhoek JA, Postma DS, Chong LL, et al. Different modulation of decorin production by lung fibroblasts from patients with mild and severe emphysema. COPD 2005; 2: 17–25. [DOI] [PubMed] [Google Scholar]
- 89. Barry‐Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase‐like‐2 impedes the development of a pathologic microenvironment. Nat Med 2010; 16: 1009–1017. [DOI] [PubMed] [Google Scholar]
- 90. Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science 2003; 302: 1704–1709. [DOI] [PubMed] [Google Scholar]
- 91. Charras G, Sahai E. Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol 2014; 15: 813–824. [DOI] [PubMed] [Google Scholar]
- 92. Gershlak JR, Black LD, 3rd. Beta 1 integrin binding plays a role in the constant traction force generation in response to varying stiffness for cells grown on mature cardiac extracellular matrix. Exp Cell Res 2015; 330: 311–324. [DOI] [PubMed] [Google Scholar]
- 93. Tufvesson E, Westergren‐Thorsson G. Biglycan and decorin induce morphological and cytoskeletal changes involving signalling by the small GTPases RhoA and Rac1 resulting in lung fibroblast migration. J Cell Sci 2003; 116: 4857–4864. [DOI] [PubMed] [Google Scholar]
- 94. Scheja A, Larsen K, Todorova L, et al. BALF‐derived fibroblasts differ from biopsy‐derived fibroblasts in systemic sclerosis. Eur Respir J 2007; 29: 446–452. [DOI] [PubMed] [Google Scholar]
- 95. Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol 2000; 23: 335–344. [DOI] [PubMed] [Google Scholar]
- 96. Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol 2001; 25: 569–576. [DOI] [PubMed] [Google Scholar]
- 97. Tufvesson E, Westergren‐Thorsson G. Tumour necrosis factor‐alpha interacts with biglycan and decorin. FEBS Lett 2002; 530: 124–128. [DOI] [PubMed] [Google Scholar]
- 98. Westergren‐Thorsson G, Onnervik PO, Fransson LA, et al. Proliferation of cultured fibroblasts is inhibited by L‐iduronate‐containing glycosaminoglycans. J Cell Physiol 1991; 147: 523–530. [DOI] [PubMed] [Google Scholar]
- 99. Westergren‐Thorsson G, Persson S, Isaksson A, et al. L‐iduronate‐rich glycosaminoglycans inhibit growth of normal fibroblasts independently of serum or added growth factors. Exp Cell Res 1993; 206: 93–99. [DOI] [PubMed] [Google Scholar]
- 100. Malmstrom J, Westergren‐Thorsson G. Heparan sulfate upregulates platelet‐derived growth factor receptors on human lung fibroblasts. Glycobiology 1998; 8: 1149–1155. [DOI] [PubMed] [Google Scholar]
- 101. Kotaru C, Schoonover KJ, Trudeau JB, et al. Regional fibroblast heterogeneity in the lung: implications for remodeling. Am J Respir Crit Care Med 2006; 173: 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Johnson PRA, Roth M, Tamm M, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med 2001; 164: 474–477. [DOI] [PubMed] [Google Scholar]
- 103. Trian T, Benard G, Begueret H, et al. Bronchial smooth muscle remodeling involves calcium‐dependent enhanced mitochondrial biogenesis in asthma. J Exp Med 2007; 204: 3173–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Westergren‐Thorsson G, Sime P, Jordana M, et al. Lung fibroblast clones from normal and fibrotic subjects differ in hyaluronan and decorin production and rate of proliferation. Int J Biochem Cell Biol 2004; 36: 1573–1584. [DOI] [PubMed] [Google Scholar]
- 105. Plotnikov SV, Waterman CM. Guiding cell migration by tugging. Curr Opin Cell Biol 2013; 25: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Yuan Y, Zhong W, Ma G, et al. Yes‐associated protein regulates the growth of human non‐small cell lung cancer in response to matrix stiffness. Mol Med Rep 2015; 11: 4267–4272. [DOI] [PubMed] [Google Scholar]
- 107. Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 2015; 308: L344–L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Reich A, Meurer M, Eckes B, et al. Surface morphology and mechanical properties of fibroblasts from scleroderma patients. J Cell Mol Med 2009; 13: 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu F, Mih JD, Shea BS, et al. Feedback amplification of fibrosis through matrix stiffening and COX‐2 suppression. J Cell Biol 2010; 190: 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Marinkovic A, Liu F, Tschumperlin DJ. Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts. Am J Respir Cell Mol Biol 2013; 48: 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. King TE, Jr , Bradford WZ, Castro‐Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. [DOI] [PubMed] [Google Scholar]
- 112. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. [DOI] [PubMed] [Google Scholar]
- 113. Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis aiming to rein in runaway wound‐healing responses. Am J Respir Crit Care Med 2014; 190: 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kirby M, Ohtani K, Lopez Lisbona RM, et al. Bronchial thermoplasty in asthma: 2‐year follow‐up using optical coherence tomography. Eur Respir J 2015; 46: 859–862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. 3‐dimensional SHG signals in a section showing an airway in an asthma patient's lung tissue.
Movie S2. 3‐dimensional SHG signals in a section showing an airway in a COPD patient's lung tissue.
Movie S3. 3‐dimensional SHG signals in a section showing an airway in an IPF patient's lung tissue.
Movie S4. 3‐dimensional SHG signals in a section showing alveoli in an IPF patient's lung tissue.
Movie S5. 3‐dimensional SHG signals in a section showing an airway in non‐diseased lung tissue.
Movie S6. 3‐dimensional SHG signals in a section showing alveoli in non‐diseased lung tissue.