

Abstract
Diatoms are highly productive single‐celled algae that form an intricately patterned silica cell wall after every cell division. They take up and utilize silicic acid from seawater via silicon transporter (SIT) proteins. This study examined the evolution of the SIT gene family to identify potential genetic adaptations that enable diatoms to thrive in the modern ocean. By searching for sequence homologs in available databases, the diversity of organisms found to encode SITs increased substantially and included all major diatom lineages and other algal protists. A bacterial‐encoded gene with homology to SIT sequences was also identified, suggesting that a lateral gene transfer event occurred between bacterial and protist lineages. In diatoms, the SIT genes diverged and diversified to produce five distinct clades. The most basal SIT clades were widely distributed across diatom lineages, while the more derived clades were lineage‐specific, which together produced a distinct repertoire of SIT types among major diatom lineages. Differences in the predicted protein functional domains encoded among SIT clades suggest that the divergence of clades resulted in functional diversification among SITs. Both laboratory cultures and natural communities changed transcription of each SIT clade in response to experimental or environmental growth conditions, with distinct transcriptional patterns observed among clades. Together, these data suggest that the diversification of SITs within diatoms led to specialized adaptations among diatoms lineages, and perhaps their dominant ability to take up silicic acid from seawater in diverse environmental conditions.
Keywords: diatoms, gene family, molecular evolution, nutrients, silicon, transporter
Abbreviations
- BLAST
Basic Local Alignment Search Tool
- GeoMICS
Global‐scale Microbial Interactions across Chemical Survey
- HMM
Hidden Markov Model
- JGI
Joint Genome Institute
- MMETSP
Marine Microbial Eukaryotic Transcriptome Sequencing Project
- NCBI
National Center for Biotechnology Information
- PCA
principal component analysis
- SAR
stramenopiles alveolates rhizaria
- SIT
silicon transporter
- TM
transmembrane
Many organisms utilize silicon in some capacity; it is required for bone formation in animals and the creation of siliceous structures in a wide variety of plants, algae, protists, and sponges (Simpson and Volcani 1981). The silica cell walls (frustules) produced by diatoms give these single‐celled algae a distinct and influential role in the ecology and biogeochemistry of the oceans. Diatom silicification links the marine carbon and silicon cycles: diatoms are among the most productive organisms on earth, responsible for an estimated 20% of global primary production, and a corresponding 240 Tmol of biogenic silica precipitation annually (Nelson et al. 1995). Diatoms are also among the most diverse phytoplankton groups in the ocean, with over 20,000 described species (Round and Crawford 1990, Julius 2007) that are most frequently identifiable by distinct silica frustule morphologies. The utilization of silicon by diatoms therefore appears to be strongly linked to their evolution and has resulted in a unique ecological and biogeochemical role for diatoms in the ocean.
Silicon, in the form of silicic acid, is transported into the diatom cell from seawater by silicon transporter (SIT) proteins (Hildebrand et al. 1997, Curnow et al. 2012, Shrestha and Hildebrand 2015). SITs were first identified by Hildebrand et al. (1997) in the pennate diatom Cylindrotheca fusiformis Reimann & J.C.Lewin and were soon thereafter recognized to be encoded by a multi‐copy gene family (Hildebrand et al. 1998). Hildebrand et al. (1998) hypothesized that the SIT proteins encoded by this gene family play different roles in the uptake of silicic acid with different cellular localizations, Si binding affinities, and transport rates. Early phylogenies of SIT genes generally reflected the phylogeny of diatom species, although discrepancies were noted (Thamatrakoln et al. 2006). As additional diatom sequences became available, a pattern of clade divergence became evident: gene homologs were identified among species and paralogs identified within species (Durkin et al. 2012). Five distinct clades were distinguished and named A through E. The SIT genes encoded by diatoms are not homologous to the silicon transporters in sponges (sodium bicarbonate symporters) or plants (aquaporins and efflux transporters; Schröder et al. 2004, Ma et al. 2006, 2007), suggesting that SITs are a distinctly algal adaptation.
Diatoms are stramenopiles and belong to the SAR supergroup of protists (stramenopiles, alveolates, and rhizaria; Sorhannus 2007, Adl et al. 2012). Silicification occurs in all three SAR clades, but is not the rule (Preisig 1994). Moreover, silica precipitation also occurs in the haptophytes (Yoshida et al. 2006), an algal lineage phylogenetically distinct from the SAR group. Among these algal lineages, SIT genes were identified in silicifying stramenopiles (chrysophytes and diatoms) and more recently in haptophytes (Likhoshway et al. 2006, Durak et al. 2016). It is unclear whether the precipitation of silica has a common origin among these algal lineages or whether the trait evolved independently among diverged lineages. The evolutionary history and diversification of diatom SIT genes may help explain how diatoms became the dominant silicifying algae in the modern ocean.
Diatom frustules first appeared in the fossil record 140 million years ago (Gersonde and Harwood 1990) when the hypothesized concentration of silicic acid in the ocean was around 1,000 μM, orders of magnitude greater than that found in today's oceans (Siever 1991, Tréguer et al. 1995, Maldonado et al. 1999). At concentrations greater than 30 μM, silicic acid freely diffuses across the cell membrane (Thamatrakoln and Hildebrand 2008). In the modern ocean, silicic acid concentrations in the surface waters are typically less than 30 μM (Tréguer et al. 1995) and SITs are required to transport silicic acid into diatom cells (Thamatrakoln and Hildebrand 2008). SITs were likely encoded by early diatoms, a hypothesis supported by the presence of SITs in each of the three major diatom lineages (radial centric, multipolar centric, and pennate diatoms). This suggests that early diatoms had the ability to actively transport silicic acid across cell membranes even in a diffusion‐mediated environment. It is unclear what adaptive advantage SITs may have provided in early diatoms, whereas in modern oceans, SITs enable diatoms to utilize silicic acid at reduced concentrations.
Diatom cells respond to changes in silicic acid availability and environmental conditions in part by altering the expression of SIT genes and proteins. SIT homologs encoded by laboratory isolates are differentially transcribed across the cell cycle and in response to silicic acid starvation, suggesting that homologs evolved different functions (Thamatrakoln and Hildebrand 2007, Mock et al. 2008, Sapriel et al. 2009, Durkin et al. 2012, Shrestha and Hildebrand 2015). Notably, the abundance of SIT transcripts and proteins are not correlated, indicating that they are subject to multiple levels of regulation (Thamatrakoln and Hildebrand 2007, Shrestha and Hildebrand 2015). Even so, the large variation in SIT transcript abundance often observed in cultures and in natural communities has been used as an indicator of a cellular response to conditions affecting silicon‐related processes. Durkin et al. (2012) observed changes in the transcription of SIT gene clades in natural communities that were attributed to nutrient availability and community composition. However, the limited reference phylogeny could not resolve the species‐specific responses.
Here, current knowledge about SIT proteins is placed within an evolutionary context to better characterize how SITs may influence present‐day ecology and biogeochemistry. We use large‐scale sequence searches as well as gene expression, phylogenetic, and metatranscriptome analyses to propose a model for SIT evolution in phytoplankton. The continued diversification of these proteins within diatoms has potentially led to specialized adaptations among diatom lineages and perhaps their dominant ability to take up silicic acid from seawater in diverse environmental conditions.
Methods
Identification of transcribed, full‐length SIT sequences
Gene models previously predicted to encode SITs were verified or extended for sequences encoded by Thalassiosira pseudonana Hasle & Heimdal, Pseudo‐nitzschia multiseries Hasle, and Fragilariopsis cylindrus (Grunow) Helmcke & Krieger by mapping transcriptome sequence reads from each species to the predicted SIT‐encoding regions of their respective genomes. Mapped sequence reads originated from transcriptomes of triplicate cultures of each species grown in artificial seawater and harvested during mid‐exponential growth, the onset of silicic acid starvation, or the onset of nitrate starvation, as described in detail by Bender et al. (2014). In brief, diatom isolates were cultured in 100 μmol photons · m−2 · s−1 light at optimal temperatures (T. pseudonana: 20°C, P. multiseries: 13°C, F. cylindrus: 4°C). Cultures were grown in artificial seawater with replete nutrients (882 μM NaNO3, 106 μM Na2SiO3, 36.2 μM NaH2PO4), low Si (1,764 μM NaNO3, 53 μM Na2SiO3, 72.4 μM NaH2PO4) or low nitrate (55 μM NaNO3, 212 μM Na2SiO3, 72.4 μM NaH2PO4). Nutrient replete cultures were harvested during mid‐exponential growth and cultures with low silicic acid or low nitrate were harvested when growth was no longer exponential. The presumed growth‐limiting macronutrient was added back to the remaining culture after harvesting to confirm the identity of the growth‐limiting nutrient. Transcriptome reads are accessible in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo, # GSE56132). Reads were mapped to the diatom genomes using SEAStAR software (Iverson et al. 2012) and the transcript fold change of SIT sequences in nutrient starved relative to nutrient replete growth conditions was determined as described by Bender et al. (2014). To identify full‐length transcribed SIT gene sequences, transcript read coverage from all nine transcriptomes was extracted and combined for each SIT gene using the SEAStAR tool ref_select. Positional coverage of pooled reads across each sequence was determined using a custom python script. The coverage of reads that mapped to each SIT sequence was visualized in R and compared to the gene model. Intron locations predicted by Joint Genome Institute (JGI) gene models were assumed to be correct if coverage in a region dropped to ≤10% of the average coverage across the predicted exons. If coverage did not decrease across an open reading frame with a predicted intron, it was considered part of the open reading frame. Gene models were extended to the first in‐frame methionine and the last in‐frame stop codon within the transcribed region when coverage continued up‐ or downstream of the original gene model. The transcribed regions of F. cylindrus protein ID 246124 and P. multiseries protein ID 311393 could not be confirmed because the original gene models spanned gaps in the genome assemblies.
The predicted protein domains were determined from all available full‐length SIT sequences using InterproScan (Jones et al. 2014). Transmembrane (TM)‐spanning domains were predicted by TMHMM (Krogh et al. 2001) (posterior probability ≥0.1) and coiled‐coil motifs were identified by MARCOILS (Delorenzi and Speed 2002) (prediction threshold ≥10%). The serine percentage of the predicted protein sequences was calculated in a sliding window of 20 amino acids and proteins were defined as serine‐enriched if serine content within a window was greater than 30%.
Phylogeny of SIT gene homologs
A combination of search tools and publicly available databases were used to identify SIT homologs. Basic Local Alignment Search Tool (BLAST) was used to search protein and translated nucleotide sequences from the NCBI nonredundant databases (GenBank) and sequences were aligned to the translated full‐length SIT protein sequences from F. cylindrus, P. multiseries, and T. pseudonana using MAFFT (method: L‐INS‐I; Katoh et al. 2005). This alignment included full‐length and partial gene sequences of SITs, and is referred to hereafter as the reference alignment. A Hidden Markov Model (HMM) profile of the reference alignment was created using hmmbuild in HMMer (hmmer.org). Homologous sequences were identified in the Marine Microbial Eukaryotic Transcriptome Sequencing Project (MMETSP, Keeling et al. 2014) using hmmsearch against the protein sequences that were predicted as part of the MMETSP project from de novo assembled cDNA contigs using ESTScan (Iseli et al. 1999). To confirm that the predicted amino acid sequences were correct, the corresponding nucleotide sequences were individually extracted from the MMETSP nucleotide data sets and translated into six reading frames using the Biopython tools for Python. Potential SIT homologs were identified by hmmsearch using the hmm profile of the reference alignment and extracting the longest open reading frame for each identified MMETSP sequence using a custom script written in R.
Typically, diatom SITs encode 10 TM domains; therefore, topology of all sequences was predicted using the program TMHMM (Krogh et al. 2001). The reference alignment contained a well‐curated set of partial sequences from the Thalassiosirales amplified with PCR that encode seven TM domains (Alverson 2007). Sequences were considered homologs of diatom SITs if the protein fragment they encoded contained at least seven predicted TM domains that aligned with sequences in the reference alignment. Sequences encoding more than 10 TM domains were split into 10 TM domain segments.
SIT protein sequences present in the reference SIT alignment and in the MMETSP data sets were aligned by MAFFT (method: L‐INS‐I) and the aligned sequences were trimmed at the N‐ and C‐terminal to include the region between the first and tenth predicted TM domains. Sequences that spanned a region encoding at least seven TM domains were also included in the alignment. Identical sequences originating from the same species were removed from the alignment. Due to the extraction of sequences from the MMETSP data set by their longest open reading frame, mismatches from the alignment occurred at the N‐ and/or C‐terminal ends in several individual contigs, and these ends were trimmed. The lengths of these sequence ends were typically less than 100 amino acids in length.
A phylogenetic tree was calculated from the trimmed protein alignment using RAXML (options: ‐f a ‐m PROTGAMMAWAGF –x 2584 ‐N 100 ‐p 4568) (Stamatakis 2006). Branch support was calculated from 100 bootstrap trees. The tree was visualized and analyzed using tools in the python program ETE2 (Huerta‐Cepas et al. 2010).
To display the phylogenetic relationships among organisms encoding SIT gene homologs, an 18s rRNA phylogenetic tree was created. The 18s rRNA sequence from one representative species within each SIT‐encoding genus was obtained from the MMETSP 18s rRNA reference sequences (available from http://data.imicrobe.us) or the NCBI nonredundant database. Sequences were aligned using MAFFT (method: G‐INS‐I) and trimmed at the 5′ and 3′ ends so that all sequences covered the same region, resulting in a total alignment length of 1,947 bp. The phylogenetic tree was calculated by RAxML (options: ‐m GTRGAMMA ‐p 58290) and visualized using the program Figtree (http://tree.bio.ed.ac.uk/).
Identifying the molecular origin of diatom SITs
Genes with a distant relationship to SIT sequences were identified in GenBank using a psi‐BLAST search with the SIT1 sequence encoded by T. pseudonana (protein ID 268895) as a query and an e‐value threshold of 10−5 for each search iteration. Each full‐length SIT protein sequence was split into the two (five TM) homologous segments, each defined as a SIT monomer. The potential ancestral homologs identified by psi‐BLAST were aligned and trimmed to the beginning and end of the five TM domain regions. The phylogenetic relationship among diatom SIT monomers and potential ancestral sequences was determined using RAXML (options: ‐f a ‐m PROTGAMMAWAGF –x 2584 ‐N 100 ‐p 4568; Stamatakis 2006). Branch support was calculated from 100 bootstrap trees.
Transcription of SITs in mixed field communities
SIT sequence transcripts were identified in mixed plankton communities sampled during the Global‐scale Microbial Interactions across Chemical Survey (GeoMICS) cruise in the eastern Pacific Ocean crossing the North American shelf break at stations P1, P4, P6, and P8 of the Line P transect time series (http://www.pac.dfo-mpo.gc.ca/science/oceans/data-donnees/line-p/index-eng.html). Surface seawater (5 m) was collected at each station using a Lutz double diaphragm pump. Phytoplankton in whole seawater samples from each station were preserved in 1% Lugol's solution (Throndsen 1978) and a 100 mL aliquot was settled in an Utermöhl chamber to enumerate cell abundance. Settled samples were quantified and identified to the genus level on a Nikon (Melville, New York, USA) 80i microscope at 200× magnification. To evaluate similarity of the community composition among locations, a Bray–Curtis dissimilarity matrix was created from the proportional abundance of all genera at each location. One liter of seawater was filtered through a 2 μm polycarbonate filter and biogenic silica was quantified by the ammonium molybdate method (Strickland and Parsons 1977, Brzezinski and Nelson 1989). Seawater was filtered through a 0.2 μm cellulose acetate syringe filter and dissolved macronutrients were analyzed in the filtrate with a Technicon autoanalyzer II system by the University of Washington Marine Chemistry Laboratory.
To collect samples for metatranscriptome analyses, 4–6 L of water was prefiltered through a 53 μm Nitex filter and plankton were captured on a 2 μm, 142 mm polycarbonate filter in duplicate. Filters from the duplicate samples collected at each station were flash frozen and stored at −80°C until RNA extraction and sequencing of the eukaryotic metatranscriptome. RNA was extracted from cells on frozen filters using the Totally RNA extraction kit (Invitrogen, Carlsbad, California, USA). The RNA was incubated with DNAse I (Ambion) at 37°C for 1 h; DNAse activity was terminated by the DNAse inactivation reagent (Ambion, Foster City, California, USA). The RNA was then purified by ethanol precipitation, and ribosomal RNA was removed using the Poly(A)Purist Kit (Ambion). Poly(A)‐selected samples were purified by ethanol precipitation, and amplified using the MessageAmp II aRNA Amplification Kit (Ambion). Samples were quantified on the NanoDrop and Qubit spectrophotometers. Between 2.5 and 5 μg of amplified polyA‐selected RNA was converted to double‐stranded cDNA using the SuperScript III First Strand Synthesis System (Invitrogen), and the NEBNext mRNA Second Strand Synthesis Module (New England BioLabs, Ipswich, Massachusetts, USA). Following second‐strand synthesis, 2 units of T4 DNA polymerase per μg of input RNA (New England BioLabs) was added to the reaction. Double‐stranded cDNA samples were purified using the PureLink PCR Purification kit (Life Technologies, Carlsbad, California, USA), followed by an ethanol precipitation step, and resuspended in nuclease‐free water. For Illumina MiSeq and HiSeq runs, the cDNA was sheared ultrasonically to 200–250 base pair fragments (Georgia Genomics Facility at University of Georgia), and TruSeq libraries (Illumina Inc., San Diego, CA, USA) were constructed for paired‐end (2 × 150 base pairs) sequencing. Libraries from duplicate samples collected at Stations P1 and P8 were sequenced on an Illumina MiSeq system in three runs for each library and the data from each run were combined into a single data set per sample. Libraries from duplicate samples collected at stations P4 and P6 were sequenced on an Illumina HiSeq system in one run per sample.
Following sequencing, paired‐end Illumina sequences were filtered using the program Trimmomatic v0.33 (Bolger et al. 2014): the adapters were removed, the maxinfo method was used to trim sequence ends, the three leading and trailing nucleotides were removed if their quality score was below 3, sequences shorter than 60 base pairs were discarded, and sequences with average quality scores less than 20 were discarded. Overlapping trimmed read pairs were merged using FLASH v1.2.11 (Magǒ & Salzberg 2011). Any leading T or trailing A residues were removed from reads using a custom python script. Merged sequences that were at least 100 bp in length were included in subsequent analysis. Sequences were deposited in NCBI BioProject (accession no. PRJNA272345).
The metatranscriptome sequences were translated into six reading frames and searched (hmmsearch) using an HMM profile (constructed with hmmbuild) containing aligned SIT homologs from genomes, GenBank, and the MMETSP data sets. Environmental SIT sequences were placed on the SIT phylogenetic tree by pplacer (v1.1.alpha14; options: ‐p –keep‐at‐most 1 –no‐pre‐mask; Matsen et al. 2010). The tree was exported by the related program guppy (option: tog). The tree reference sequence closest to each environmental sequence was used to classify the environmental sequence by lineage and genus using tools in the ETE2 python program. The environmental sequence was defined as unclassified if it was sister to a group of sequences that did not all have the same classification or genus.
The percent contribution of each SIT clade to total number of SIT transcripts was calculated for each major diatom lineage and select genera. Counting uncertainty was estimated as the square root of the number of counts per clade, which assumes that the probability of detecting a given number of reads per clade is Poisson distributed. Replicate samples collected at each station were used to assess variability in the observed proportion of SIT clades transcribed by major diatom lineages. Because counting uncertainty was larger in the quantification of SIT reads at the genus level, in which fewer reads were observed, data from the sample replicates were combined before calculating the percent of transcripts from each SIT clade. Only genera with at least 100 total sequences observed among all stations were included in further analysis. To identify the SIT clades driving variability among genera at each station, a principal component analysis (PCA) of the genus‐level transcript data was performed using tools in R.
Results
Predicted functional diversification of SIT gene clades
Protein domains of SITs (Fig. 1) were predicted from translated full‐length sequences reported previously (C. fusiformis: Hildebrand et al. 1998, Nitzschia alba J.C.Lewin & R.A.Lewin and Skeletonema costatum (Greville) Cleve: Thamatrakoln et al. 2006, Phaeodactylum tricornutum Bohlin: Sapriel et al. 2009, Stephanoeca diplocostata W.N.Ellis: Marron et al. 2013) and sequences whose full length was determined by mapping SOLiD transcriptome reads to previously sequenced genomes (Fig. S1 in the Supporting Information). Mapping of available RNAseq data confirmed that the transcribed open reading frames of the three SIT genes in T. pseudonana each extended upstream of the gene models in a manner consistent with the full‐length sequences determined previously by cDNA sequencing (Thamatrakoln et al. 2006). Although transcript coverage across the intron of the T. pseudonana SIT 3 gene (JGI protein ID 35133) was 20% of the exon coverage level (above our 10% threshold), the presence of this intron was previously confirmed by Thamatrakoln et al. (2006) and we therefore incorporated it in our model. In P. multiseries, the transcribed region of the gene model for protein ID 297536 was extended upstream and a predicted intron was removed, resulting in a single exon. The transcribed region of the gene model for protein ID 338018 was extended downstream due to identification of a frame‐shift sequencing error in a poly‐T region. The coding regions of F. cylindrus gene models for protein IDs 157255 and 138651 were extended in the 5′ and 3′ directions; the intron in 157255 and the intron in 263777 were removed. A frame‐shift sequence error was identified in the gene model for protein ID 264054 resulting in removal of a predicted intron.
Figure 1.
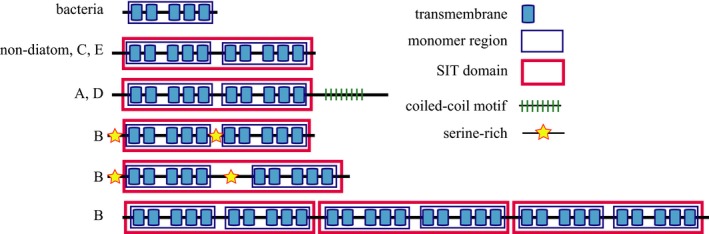
Predicted protein domains determined from full‐length SIT gene sequences. The clade assignment of genes encoding these domains is based on Durkin et al. (2012) and indicated on the left.
All full‐length proteins were predicted to be SITs by InterPro Scan and typically encoded the 10 TM domains (Fig. 1 and Table 1) and conserved amino acid residues identified by Thamatrakoln et al. (2006). The phylogeny and clade assignment of these sequences were previously determined (Durkin et al. 2012) and sequences are referred to here in the context of their clades. One SIT gene encoded by P. multiseries (protein ID 338018) belonging to clade B was predicted to encode 30 TM domains with three concatenated SIT domains. Two clade B sequences (F. cylindrus protein ID 157255 and P. multiseries protein ID 297536) encoded in frame insertions relative to other SITs between the first and second five TM domain monomers that were 50–130 amino acids in length. All five of the clade B SIT genes encoded serine‐enriched regions. Serine enrichment occurred in the N‐terminal region before the first TM domain and/or the fifth loop region, between the fifth and sixth TM domains. SITs from other clades were not serine‐enriched. All 13 of the full‐length clade A and D SIT genes encoded a coiled‐coil motif at the C‐terminal end of the predicted protein.
Table 1.
SIT genes with confirmed full‐length sequences, predicted protein domains, and phylogenetic clade. Dashes indicate genes where data are not applicable
| Organism | Species | ID | Model modified | TM (#) | Coil | S‐enriched | Gene clade |
|---|---|---|---|---|---|---|---|
| Multipolar diatom (Thalassiosirales) | Thalassiosira pseudonana b , c | JGI: 268895 | Yesa | 10 | No | No | E |
| JGI: 41392 | Yesa | 10 | No | No | E | ||
| JGI: 35133 | Yesa | 10 | No | No | E | ||
| Pennate diatom | Fragilariopsis cylindrus b | JGI: 212017 | No | 10 | Yes | No | A |
| JGI: 263777 | Yes | 10 | Yes | No | A | ||
| JGI: 264054 | Yes | 10 | Yes | No | A | ||
| JGI: 157255 | Yes | 10 | No | Yes | B | ||
| JGI: 138651 | Yes | 10 | No | Yes | B | ||
| JGI: 186485 | Yes | 10 | No | No | C | ||
| JGI: 211148 | No | 10 | Yes | No | D | ||
| Pennate diatom | Pseudo‐nitzschia multiseries b | JGI: 338021 | No | 10 | Yes | No | A |
| JGI: 261458 | No | 10 | Yes | No | A | ||
| JGI: 297536 | Yes | 10 | No | Yes | B | ||
| JGI: 338018 | Yes | 30 | No | Yes | B | ||
| JGI: 338017 | No | 10 | Yes | No | D | ||
| Pennate diatom | Phaeodactylum tricornutum d | JGI: 23423 | ‐ | 10 | No | No | C |
| JGI: 48707 | ‐ | 10 | No | No | C | ||
| JGI: 11311 | ‐ | 10 | No | Yes | B | ||
| Pennate diatom | Cylindrotheca fusiformis e | NCBI: AF052458 | ‐ | 10 | Yes | No | A |
| NCBI: AF052454 | ‐ | 10 | Yes | No | A | ||
| NCBI: AF052456 | ‐ | 10 | Yes | No | A | ||
| NCBI: AF052455 | ‐ | 10 | Yes | No | A | ||
| NCBI: AF052457 | ‐ | 10 | Yes | No | A | ||
| Pennate diatom | Nitzschia alba c | NCBI: DQ256058 | ‐ | 10 | Yes | No | A |
| Multipolar diatom (Thalassiosirales) | Skeletonema costatum c | NCBI: DQ256065 | ‐ | 10 | No | No | E |
| Choanoflagellate | Stephanoeca diplocostata f | NCBI: HE981737 | ‐ | 10 | No | No | Non‐diatom |
| NCBI: HE981736 | ‐ | 10 | No | No | Non‐diatom | ||
| NBCI: HE981735 | ‐ | 10 | No | No | Non‐diatom |
Not different from full‐length transcribed sequences determined by Thamatrakoln et al. 2006.
Full‐length sequences identified by this study.
Full‐length sequences identified by Thamatrakoln et al. (2006).
Full‐length sequences identified by Sapriel et al. (2009).
Full‐length sequences identified by Hildebrand et al. (1998).
Full‐length sequences identified by Marron et al. (2013).
Phylogeny of diatom‐like SIT genes
A comprehensive phylogenetic tree of translated SIT genes (Fig. 2) was constructed of SIT sequences from sequenced diatom genomes, the NCBI database, and the MMETSP data sets. This tree contains 424 unique SIT genes, including 45 SIT homologs from nondiatom organisms (Fig. 2; Table S1 in the Supporting Information). The alignment was constrained to sequences encoding at least 7 of the 10 TM domains typical of full‐length SIT proteins, thus some of the previously identified SIT sequences in GenBank and some potential homologs from the MMETSP data sets were excluded. The majority of genes (294 or 70%) were from the MMETSP data sets. Forty‐six of the putative genes encoded more than 10 TM domains, which were trimmed and/or split into 7–10 TM segments in order to align them to the other sequences, resulting in a total of 473 sequences or segmented sequences in the final alignment.
Figure 2.
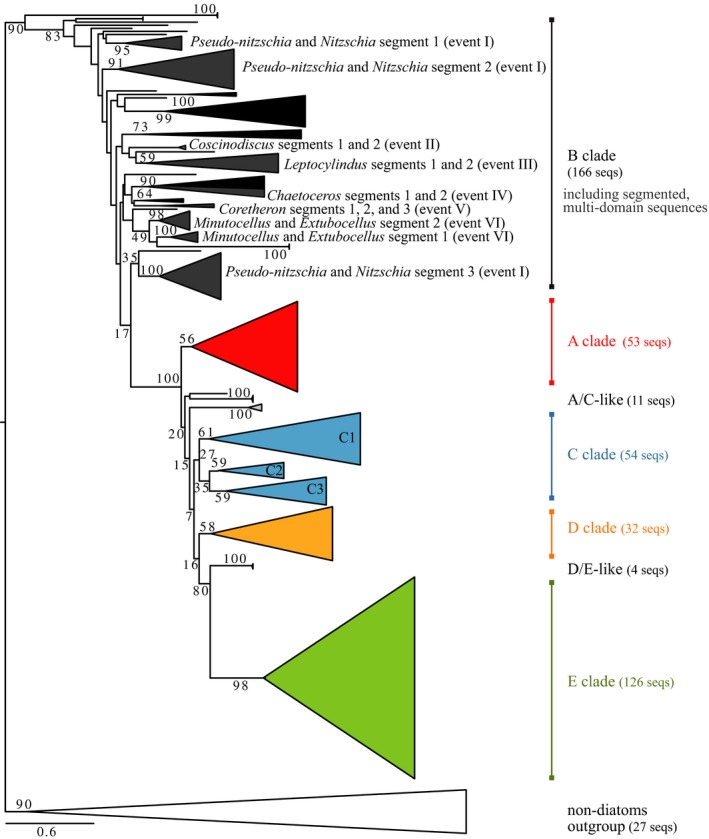
Phylogenetic tree of SIT proteins, including full‐length SIT sequences determined from available diatom genomes, previously published sequences available in GenBank, and the MMETSP data set of marine microbial eukaryotes. Sequence fragments were included in the tree if they encoded at least seven predicted TM domains. Sequences with more than 10 TM domains were segmented and aligned in 10 TM segments, the names of these genera are labeled along with proposed domain duplication events I–VI. The tree is rooted by a clade containing SIT proteins encoded only by non‐diatom organisms, but non‐diatom SIT proteins are also found on other branches of the tree. Branches within a clade with >50% bootstrap support are collapsed for simplicity. Node scores are bootstrap values from 100 bootstrapped trees.
Unlike previous SIT gene phylogenies, this extensive tree was rooted by a monophyletic outgroup of SIT homologs identified in nondiatom protists including haptophytes, chrysophytes, a silicoflagellate, choanoflagellates, ciliates, and a dinoflagellate (Fig. 2). The topology among diatom SITs recapitulated previously defined SIT gene clades A through E (Durkin et al. 2012) at a threshold bootstrap value of 50 (Fig. 2). The full phylogenetic tree, including all labeled branches, is presented in Figure S2 in the Supporting Information and all sequences are presented in Table S1. To resolve the distribution of SIT clades across major diatom lineages, organisms were grouped according to the relationship of their 18S rRNA sequence (Fig. 3).
Figure 3.
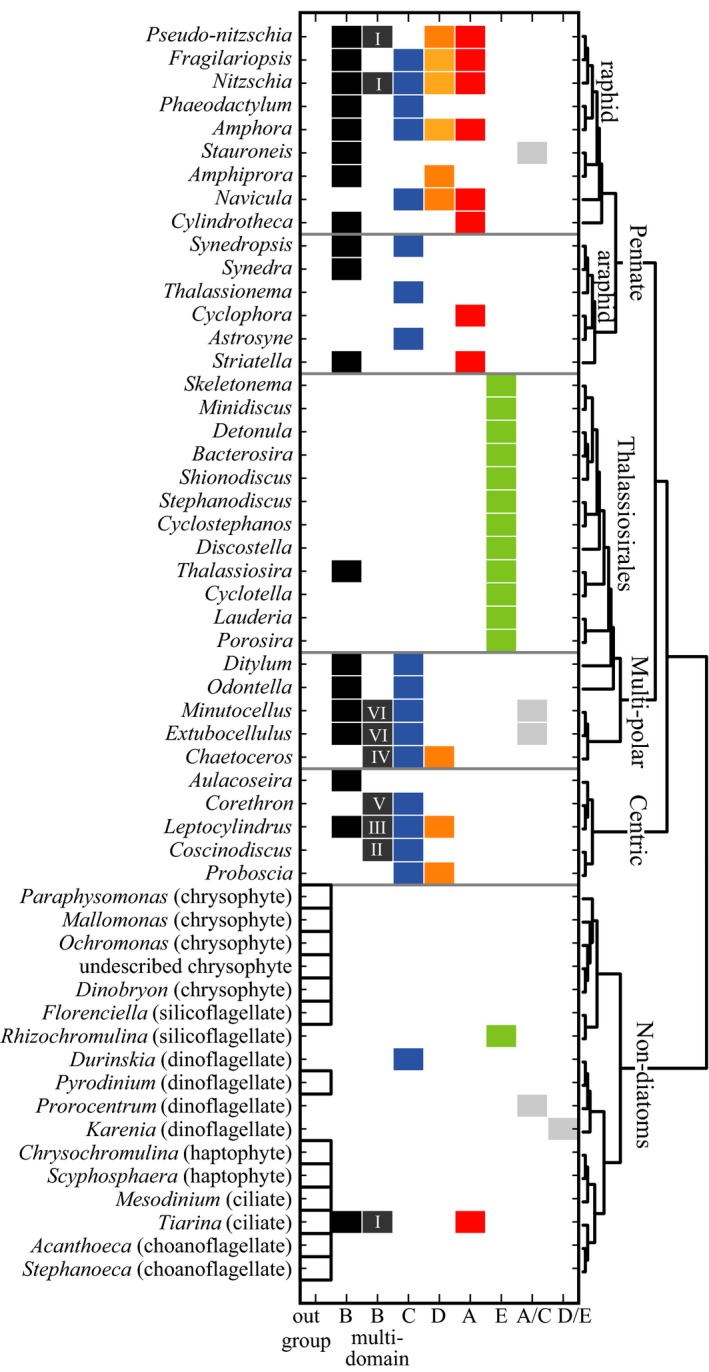
The presence of SIT clades across diatom genera and in non‐diatoms. Columns indicate clades, as defined in Figure 2. Rows indicate genus in order of 18s rRNA genetic relationships (displayed by tree on the right). B clade proteins that contain more than 10 TM domains are separated from B clade sequences encoding ≤10 TM domains. Apparent domain duplication events are labeled as in Figure 2.
Within the diatom portion of the tree, clade B sequences formed the most basal branches (Fig. 2). The radial centric diatom Leptocylindrus encoded the most basal sequences within clade B (see Fig. S2). Clade B included diverse subclades and sequences from all major diatom groups, but not all diatom genera encoded a clade B sequence (Fig. 3). All sequences that encoded more than 10 predicted TM domains belonged to clade B (Fig. 2, dark gray). Segments from different sequences that shared a common ancestor were assumed to have arisen from the same domain duplication event. For example, the presence of two distinct clades containing the first 10 TMs and the second 10 TMs of the segmented SIT sequences encoded by Minutocellus and Extubocellus suggests that the SIT domain was duplicated in a common ancestor of these two genera. In this way, six potentially separate domain duplication or triplication events were identified within the clade B SIT sequences (events I through VI), labeled in Figures 2 and 3.
The remaining four SIT clades were derived from clade B in 100% of the bootstrap trees (Fig. 2). Clade A SIT sequences formed a monophyletic cluster supported by 56% of the bootstrap trees and included only pennate diatom sequences (Fig. 3). One clade A sequence was also identified in the ciliate Tiarina (Fig. 3). Clade C sequences formed a monophyletic clade with three subclades, though bootstrap support was low at the primary node (27%; Fig. 2). Clade C sequences were encoded by centric, multipolar, and pennate diatoms, but were not detected in the order Thalassiosirales, a monophyletic lineage of the multipolar diatoms (Fig. 3). Eleven sequences paraphyletic to clade C (A/C sequences; Fig. 2) were encoded by pennate and multipolar diatoms and the dinoflagellate Prorocentrum. Another monophyletic cluster included sequences from clade D, supported by 58% of the bootstrap trees (Fig. 2). Like clade C, clade D sequences were identified in centric, multipolar, and pennate diatoms, but not the Thalassiosirales. Clade E was supported by 98% of the bootstrap trees and was encoded only by the Thalassiosirales. The dinoflagellate Karenia brevis encoded four sequences basal to the clade E diatom sequences (D/E sequences; Fig. 2). The phylogenetic placement of clades A, C, D, and E relative to one another is inconclusive, with bootstrap scores less than or equal to 20%.
Bacterial SIT homologs
To explore the origin of SITs, sequences with distant homology to diatom SITs were identified. Three of the four identified sequences were encoded by bacteria, including two Synechococcus sp. cyanobacteria isolates KORDI‐100 and CC9616 (NCBI accession numbers WP_038543010.1 and WP_028953550.1) and a Rhodococcus opacus Klatt et al. bacteria isolate (WP_043827568.1). A sequence was also identified in the whole genome sequence of the polychaete worm Capitella teleta Blake et al. (ELT99670.1). The region of the worm genome that encodes the modeled protein is surrounded by gaps in the assembly on both the N‐ and C‐terminal ends, with no other genes predicted to be encoded in this region (JGI genome scaffold 415, nucleotides 15100‐21200, http://genome.jgi-psf.org/Capca1). All four sequences encoded five TM domains homologous to the first and second half of full‐length, 10 TM domain diatom SIT genes (Fig. 1). The bacterial and worm sequences share conserved sequence regions with the five TM SIT monomers: E‐(G/A)‐X‐(Q/L/H) located at the C‐terminal side of the second TM domain and amino acids G‐R‐Q located at the N‐terminal side of the third TM domain (Thamatrakoln et al. 2006).
The phylogeny of full‐length SIT sequences, split into five TM domain monomers, and the bacterial and worm sequences indicate that these sequences share a common ancestor (Fig. 4). All N‐terminal, five TM SIT monomers shared a common ancestral node, as did the C‐terminal of five TM SIT monomers. The five TM monomers from bacteria were sister to both the N‐ and C‐terminal of five TM monomers of all SIT sequences.
Figure 4.
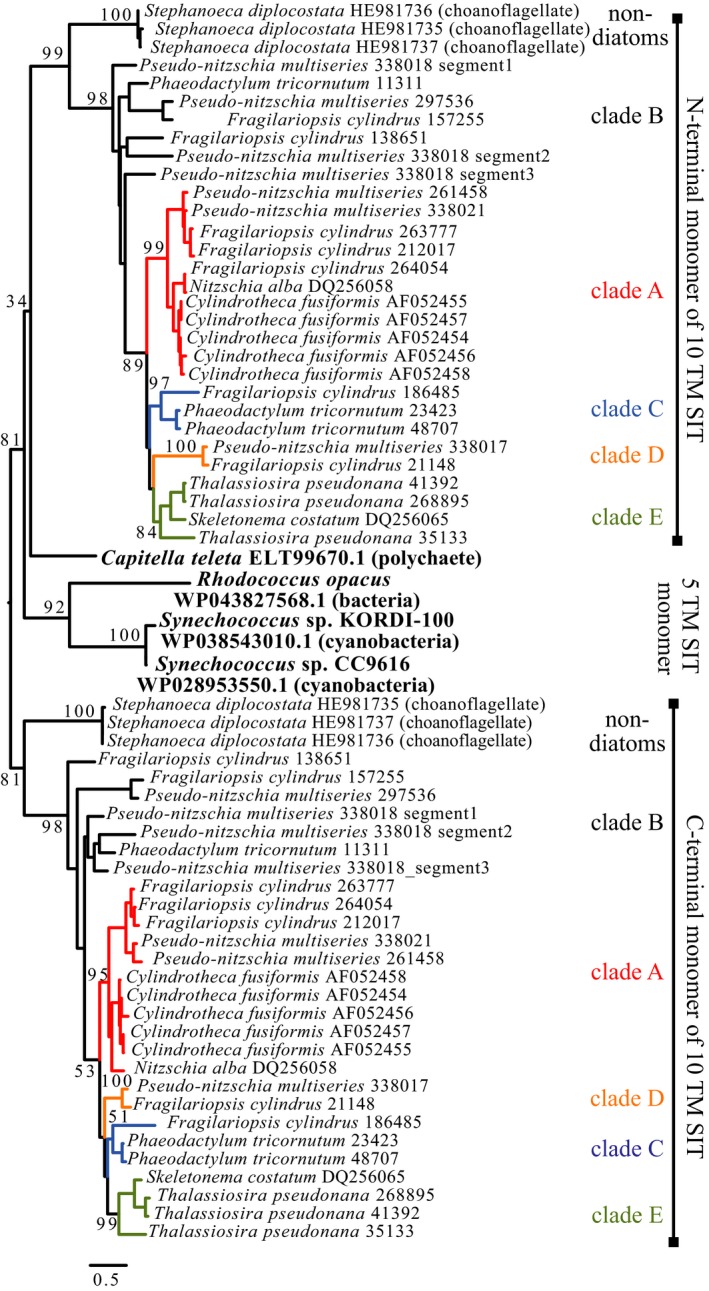
Phylogenetic tree of full‐length amino acid SIT sequences split into five TM domain monomers (N‐terminal and C‐terminal) and homologous sequences encoding five TM domains identified in bacteria and a polychaete worm. Tree branches labeled by clade, as in Figures 2 and 3.
Transcriptional diversification of SIT gene clades
The SIT genes encoded by T. pseudonana, P. multiseries, and F. cylindrus were differentially regulated in cells at the onset of silicic acid or nitrate starvation compared to cells growing in nutrient replete conditions (Fig. S3 in the Supporting Information). The degree to which transcription was up‐ or down‐regulated varied across species, and SIT gene clades were transcribed differently from one another within a species (Fig. 5). SIT genes in clades B, C, and D were either down‐regulated or did not change when silicic acid starvation arrested cell division. A similar pattern was observed when cell division was arrested by nitrate starvation, except that two F. cylindrus SIT B genes were up‐regulated (protein ID 138651 and 157255). Clade A SIT genes were up‐regulated during both nitrate and silicic acid starvation. One P. multiseries SIT A gene was transcribed differently than other SIT A homologs (protein ID 261458; down‐regulated in –Si and no change in –NO3 conditions). The transcript abundance of clade E SITs changed primarily in response to silicic acid starvation. One clade E SIT (protein ID 41492) was slightly down‐regulated in –NO3 conditions.
Figure 5.

Fold change of SIT gene transcripts in Pseudo‐nitzschia multiseries, Fragilariopsis cylindrus, and Thalassiosira pseudonana cultures entering silicic acid starvation (left) or nitrate starvation (right) relative to exponentially growing, nutrient replete cultures. Gray diagonal lines indicate no significant change (false discovery rate‐corrected pair‐wise comparisons, following methods in Bender et al. 2014, P < 0.01).
To observe transcriptional changes among SIT gene clades across more diverse species and growth conditions, the SIT genes transcribed by natural plankton assemblages were identified from samples collected at locations along a coastal section of the Line P transect in the NE Pacific Ocean (Fig. 6 and Table 2). Surface nitrate concentrations were below 1 μM at all four locations and nitrate was likely the primary nutrient limiting growth. Silicic acid concentrations at stations P1, P4, and P8 were ~4 μM and categorized as “low” because silicic acid uptake rates are limited for the average diatom growing in this silicic acid concentration (Martin‐Jézéquel et al. 2000). The concentration of silicic acid at station P6 was about two times higher (8 μM) and was categorized as “high” because the average diatom can take up silicic acid at near maximal rates at this concentration. The concentration of biogenic silica and the abundance of total diatoms were lowest at station P6, the station with the highest dissolved silicic acid concentration (Table 2). The diatom community at station P6 was equally similar to the communities at all other stations (41%–43% similar; Table 3). The diatom communities at stations P1, P4, and P8 were less similar to one another (28%–35%). Between 909 and 2,174 transcripts with homology to SIT sequences were identified at each station by HMM searches of the metatranscriptomes. On average, 8% of the environmental SIT sequences were phylogenetically most related to non‐diatom SIT sequences, according to their placement on the phylogenetic tree by Pplacer. The percent of SITs assigned to specific clades varied across stations (Table 2).
Figure 6.
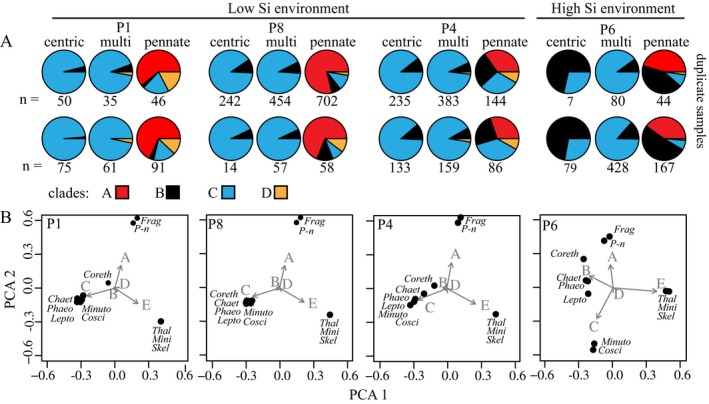
Variability in the SIT genes transcribed by (A) major diatom lineages and (B) the most frequently observed genera in the surface waters at four locations along the Line P transect. (A) Percentage of SIT clades transcribed by centric, multipolar (excluding Thalassiosirales), and pennate diatoms lineages. Upper and lower pie charts are duplicate samples and numbers below the pie charts indicate the total number of SIT transcripts detected in each replicate. (B) Principal component analysis of the SIT clades transcribed at each station by genera with more than 100 detected transcripts. Genera include centric (Coreth = Corethron, Cosci = Coscinodiscus, Lepto = Leptocylindrus), multipolar (Chaet = Chaetoceros, Minuto = Minutocellus), Thalassiosirales (Thal = Thalassiosira, Mini = Minidiscus, Skel = Skeletonema), and pennate (Frag = Fragilariopsis, P‐n = Pseudo‐nitzschia, Phaeo = Phaeodactylum) diatoms.
Table 2.
Locations where natural phytoplankton communities were collected and the associated dissolved nutrient concentrations, BSi concentrations, diatom abundances, and SIT clade transcripts abundances, with percenta of total displayed in parentheses
| Station | Location | Si(OH)4 (μM) | NO3 (μM) | BSi (μM) | Diatoms (106 cells · L−1) | Total SITs | Clade A | Clade B | Clade C | Clade D | Clade E | Nondiatom |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 48.58° N; 125.5° W | 3.5 ± 1.1 | 0.2 ± 0.2 | 2.2 ± 0.1 | 104 | 909 | 90 (10%) | 9 (1%) | 237 (26%) | 22 (2%) | 415 (46%) | 66 (7%) |
| P4 | 48.65° N; 126.67° W | 4.6 ± 1.3 | 0.04 ± 0.04 | 1.5 ± 0.1 | 54 | 1,832 | 76 (4%) | 140 (8%) | 893 (49%) | 31 (2%) | 332 (18%) | 235 (13%) |
| P6 | 48.74° N; 127.67° W | 8.1 ± 2.1 | 0.7 ± 0.1 | 0.3 ± 0.1 | 2 | 1,595 | 87 (5%) | 235 (15%) | 481 (30%) | 4 (0.3%) | 497 (31%) | 114 (7%) |
| P8 | 48.82° N; 128.67° W | 3.4 ± 0.2 | 0.3 ± 0.02 | 0.8 ± 0.2 | 13 | 2,174 | 593 (27%) | 118 (5%) | 782 (36%) | 32 (1%) | 267 (12%) | 64 (3%) |
Percentages at each location do not add to 100% due to the detection of unclassifiable sequences.
BSi, biogenic silica.
Table 3.
Bray–Curtis percent similarity of proportional diatom community compositions observed at four locations along the Line P transect
| Station | P1 | P4 | P6 |
|---|---|---|---|
| P4 | 35 | ||
| P6 | 41 | 43 | |
| P8 | 28 | 33 | 43 |
The identities of environmental SIT transcripts were further assigned to major diatom lineages and genera to better resolve the influence of physiology across stations with different community compositions. The percent of each SIT clade transcribed by major diatom lineages varied across the four stations (Fig. 6A). Variation in the percent transcript abundance indicates a difference in transcription in at least one of the clades. Variation in transcription may not be apparent if all clades are up‐ or down‐regulated equally. The majority of SIT sequences transcribed by centric diatoms (i.e., phylogenetically most related to centric diatom sequences) were from clade C at the stations with low silicic acid concentrations. At the high silicic acid station, the majority of centric diatom SIT transcripts were from clade B (Fig. 6A). The majority of multipolar diatom SIT transcripts were from clade C at all locations, with a minority of transcripts from clades D and B. At the stations with low silicic acid concentrations, pennate diatom SIT transcripts were largely from clade A (stations P1 and P8) or were nearly equally distributed among clades A, B, and C (station P4), with a minority of transcripts from clade D. At the station with higher silicic acid concentrations (P6), 50% of the SIT sequences transcribed by pennate diatoms were from clade B and 41% were from clade A. Duplicate samples from each station identified nearly identical proportions of SIT clades among these lineages, even though the total SIT reads identified sometimes differed substantially between the two replicates (Fig. 6A).
Because replicate samples did not differ substantially in the proportion of SIT clades transcribed by major diatom lineages, replicates were combined in order to quantify the proportion of SIT clades transcribed at the genus level and to reduce the counting uncertainty associated with the data assessed by PCA (Figs. 6B and S4 in the Supporting Information). Most of the variance in the proportion of transcribed SIT clades among genera was accounted for by the first two principal components (stations P1, P4, and P8 >90%; P6 = 74%). Differences among the genera at the stations with low silicic acid concentrations were driven by clade E and C on the first principal component, and by clades A and E on the second principal component. At these locations, three clusters of genera were identified based on their proportion of SIT clade transcripts. The pennate diatoms Pseudo‐nitzschia and Fragilariopsis clustered together due to their transcription of clade A SITs. The Thalassiosirales genera (Thalassiosira, Minidiscus, and Skeletonema) clustered together due to their transcription of clade E SITs. The third cluster was composed of centric (Corethron, Leptocylindrus, Coscinodiscus), multipolar (Chaetoceros, Minutocellus), and pennate (Phaeodactylum) diatoms, which were primarily clustered due to their transcription of clade C SITs. At the station with higher silicic acid concentrations (P6), the genera previously grouped by their transcription of clade C SITs were divided, resulting in four groups of genera. A new group composed of Corethron, Leptocylindrus, Chaetoceros, and Phaeodactylum were defined by an increase in the proportion of SIT B transcripts (Fig. 6B). Other genera remained grouped as at the three other stations.
Discussion
The deep phylogeny of 10 TM SITs highlights the diversification of these proteins across diatom lineages and their presence in non‐diatom protists. Diatoms have five distinct SIT protein types, represented by clades A through E. These clades were previously identified from a phylogeny containing less diverse reference sequences, and their identification was supported by environmental sequences whose taxonomic identity was unknown (Durkin et al. 2012). By leveraging well‐curated data sets from previous studies (e.g., Alverson 2007) and newly available data sets (Keeling et al. 2014), we propose a model of the evolutionary history of SITs (Fig. 7) based on the sequence phylogeny, distribution among taxa, transcriptional variation, and predicted functional domains. Because many of the sequences in the SIT phylogeny originated as expressed mRNA, the absence of a sequence in any individual organism may be due to lack of expression as opposed to absence of the gene in the genome.
Figure 7.
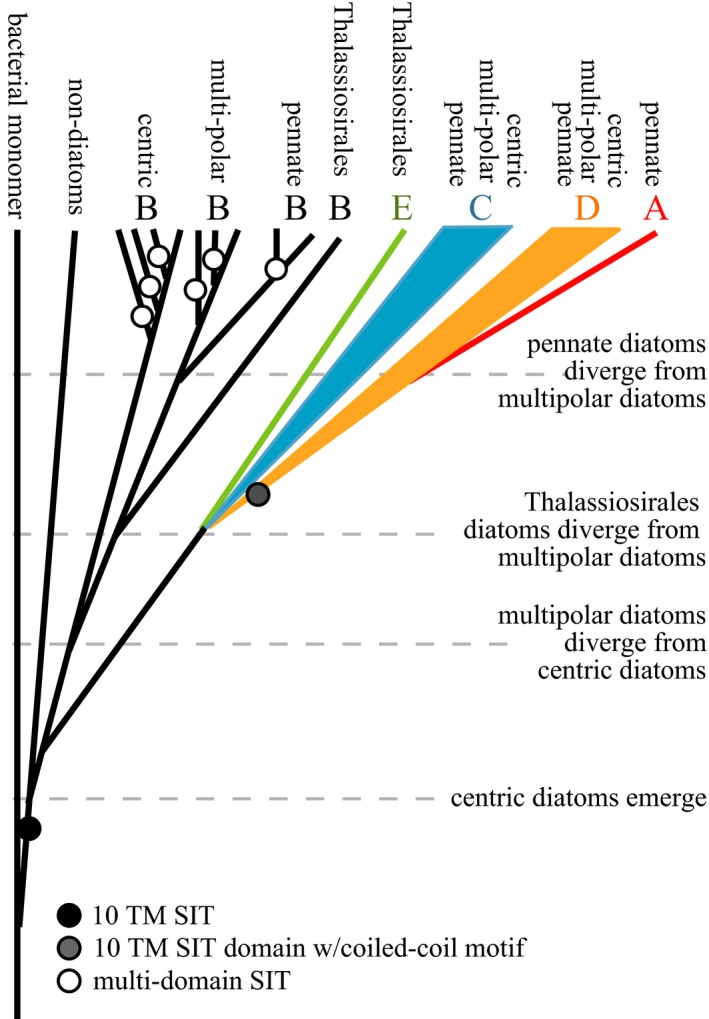
Schematic of hypothesized history of SIT evolution, based on the predicted protein domains in Figure 1, phylogeny in Figure 2, and distribution of SIT clades among lineages in Figure 3. Circles at the base of a branch indicate the evolution of a new domain. The timing of the evolution of different diatom lineages is indicated by horizontal dashed lines, but is not scaled to actual time.
Origin of SITs
SIT sequences likely originated before the emergence of diatoms, in a protist ancestor or bacteria. A gene encoding five TM domains with conserved amino acid sites, homologous to the first and last five TM domains of diatom SITs, was identified in several bacterial strains. Intriguingly, this gene was identified in a strain of Synechococcus, an important marine cyanobacteria that was recently found to accumulate silicon intracellularly (Baines et al. 2012); however, the role of this five TM domain protein in transporting or binding silicon remains unknown. The bacterial sequences were basal to both the N‐ and C‐terminal halves of the 10 TM SITs. In other transporter proteins, whole gene and domain duplications have led to functional proteins with a larger complement of TM domains (Saier 2003). Therefore, we hypothesize that a SIT‐like monomer with five TM domains was transferred from a bacteria into an early protist ancestor, which later underwent a domain duplication to become a 10 TM encoding SIT gene (Fig. 7, black circle). SIT sequences were not identified in any algal groups that arose from the primary endosymbiosis of a heterotrophic protist and a cyanobacterium, even though representative organisms were included in the search databases. The most likely explanation of our results is that the five TM SIT entered eukaryotes near the base of the SAR clade and underwent domain duplication. This hypothesis was also recently proposed by Durak et al. (2016), who identified the SIT‐like monomer in haptophytes.
Genetic descendants from this early SIT gene are present in modern day haptophytes, stramenopiles, and alveolates, referred to in this study as the outgroup SITs. Several of these outgroup organisms (the chrysophyte and silicoflagellate genera) are known to precipitate cellular silica structures (Simpson and Volcani 1981). Outgroup SIT sequences were identified in a prymnesiophyte, ciliates, and dinoflagellates, in which silica precipitation was identified in related genera within their taxonomic class (Bursa 1969, Yoshida et al. 2006, Foissner et al. 2009). The presence of SIT genes in choanoflagellates (not a member of the SAR or haptophyte groups) was proposed to be the result of lateral gene transfer (Marron et al. 2013). The choanoflagellate SITs are most closely related to the haptophyte SIT genes, suggesting that choanoflagellates may have obtained their SITs from a haptophyte‐like alga or a virus carrying haptophyte algal genes.
Emergence of diatom SITs: clade B
When diatoms emerged, they encoded SIT genes divergent from other algal SIT sequences: the clade B SITs. Early diatom cells may have used clade B SITs to actively transport silicic acid even though they likely lived in an ocean where silicic acid concentrations were high enough for passive diffusion to occur (Siever 1991, Maldonado et al. 1999). Clade B SITs appear to play an important role in modern diatoms in sustained, high silicic acid environments. In the laboratory, the majority of SIT B genes encoded by pennates were either up‐regulated or did not respond to low nitrate concentrations that arrested cell division in the presence of elevated silicic acid concentrations. Clade B sequences dominated SIT transcript expression in centric and pennate diatoms in the natural community at Station P6 where the silicic acid concentration was highest (8 μM), but the nitrate concentration (~1 μM) was growth‐limiting. Durkin et al. (2012) also observed an increase in the transcription of SIT B genes in a natural community experiencing high silicic acid concentration and growth‐limiting concentrations of iron. When growth limitation was relieved by the addition of iron, the abundance of SIT B transcripts decreased. The association of SIT B gene expression in environments rich in silicic acid, but limited by other nutrients suggests that the encoded protein may have a unique subfunction among the SITs. Future studies will benefit from the measurement of transcript and protein expression under interacting nutrient conditions.
In high silicic acid environments, diffusion and/or active transport will result in a high concentration of intracellular silicon, which must still be directed in some way to the silica deposition vesicle, possibly by transport across intracellular membranes. The transport and binding of intracellular silicon has not yet been characterized in diatoms, but is considered a vital step in the process of controlled silica precipitation (Hildebrand 2008). The basal position of the B clade and the likely environmental conditions at the time diatoms arose suggests that the original SIT B proteins were not required to bring silicic acid into the cell, but could have been localized within intracellular membranes facilitating the transport of silicic acid to the silica deposition vesicle. This hypothesis could explain the role of these proteins at the high silicic acid concentrations found in both modern and ancient ocean environments.
As clade B SITs evolved within diatoms, the diversification of the primary amino acid sequences did not affect secondary structure delineating TM domains, but may have had ramifications on tertiary structure. Clade B SITs are relatively enriched with serine residues in the N‐terminal and/or 5th loop regions. Serine residues can undergo posttranslational modification by phosphorylation or glycosylation, potentially affecting the function of the protein by altering its structure, enabling interactions with other molecules, or controlling the localization of the protein in the cell. The effect of serine enrichment in SIT B proteins requires further study to determine its functional relevance. Duplication of the SIT B domain occurred independently in different diatom lineages. Interestingly, the native form of SIT3 (clade E) from T. pseudonana appears to be a homotetramer (Curnow et al. 2012), suggesting that the duplicated domains in clade B may have abbreviated the process of assembling the protein quaternary structure.
Divergence of diatom SIT clades A, C, D, and E
The branching pattern among the remaining SIT clades cannot be predicted from the phylogenetic analysis due to the weak bootstrap support at clade‐separating nodes. The SIT gene phylogeny reflects the most recent 18S rRNA phylogeny of diatoms calibrated on fossil data that shows a brief 7 million year period of rapid and sometimes unresolved species radiation that concludes in the advent of the pennate lineage (Sorhannus 2007). It is, therefore, difficult to order the emergence of gene lineages chronologically if they began to diverge before or after the completion of speciation. We hypothesize that clades C, D, and E arose nearly simultaneously from clade B (Fig. 7).
While SIT clades C and D are present in each of the major diatom lineages, clade D SITs diverged independently by their acquisition of a coiled‐coil motif at the C‐terminus of the protein (Fig. 7, gray circle). Hildebrand et al. (1998) first noted a coiled‐coil motif in the SIT sequences encoded by Cylindrotheca fusiformis, and Thamatrakoln et al. (2006) noted its absence in the SIT sequences of T. pseudonana. Coiled‐coil motifs promote protein–protein interactions (Burkhard et al. 2001), suggesting that clade D SITs have properties associated with protein regulation, sensing, signaling, and/or structural stability that differ from the other SITs.
SIT clade E arose in the Thalassiosirales, a divergent subgroup of the multipolar diatom lineage (Medlin et al. 1996, Kaczmarska et al. 2006). Studies of the transcription and cellular localization of clade E SITs indicate that functional divergence within this clade has occurred (Thamatrakoln and Hildebrand 2007, Shrestha and Hildebrand 2015) in the presence of purifying selection within the central portion of the proteins (Alverson 2007). Two clade E SIT proteins of T. pseudonana were localized to the outer membrane (via GFP tagging), while the SIT3 protein remained within internal membranes of the cell (Shrestha and Hildebrand 2015). Clade E SITs appear to eliciting a feedback mechanism that enables cells to sense the environmental silicic acid in anticipation of cell division (Shrestha and Hildebrand 2015) via a measurable response in gene transcription and protein production (Fig. 5; Thamatrakoln and Hildebrand 2007, Mock et al. 2008, Shrestha and Hildebrand 2015). These functionalities would have provided an adaptive advantage when silicic acid concentrations in the ocean were dramatically reduced from the high concentrations available to the earliest diatoms (Siever 1991). Indeed, the transcription of clade C and E genes in the lab and in natural environments (this study), in addition to localization of their proteins to the plasma membrane (Sapriel et al. 2009, Shrestha and Hildebrand 2015), leads us to hypothesize that the divergence of clades C and E from clade B may have been driven by the need to actively transport silicic acid into the cell from seawater.
Like clade E, clade A SITs are lineage‐specific, but they likely diverged from the D clade in an ancestral pennate diatom; a relationship we suggest based on the shared coiled‐coil protein domains between these clades. In spite of their shared protein domains, the pennate‐specific clade A SITs appear to be used differently than the more broadly distributed clade D SITs, as demonstrated by their different transcriptional patterns in both laboratory cultures and field communities. Future experiments are needed to determine whether transcriptional differences translate to functional differences among these clades. Because the transcription of clade A SITs increased in response to both silicic acid and nitrate starvation, these genes may be part of a general response to growth limitation and not silicic acid availability specifically. This contrasts with the transcriptional response of other clades, including clade E genes on which the majority of SIT transcriptional studies have been conducted.
Several SIT genes from clades A through E (diatom clades) were found in the transcriptomes of non‐diatoms. The dinoflagellate Durinskia encodes a pennate diatom‐like SIT sequence and cells of this organism are known to contain a pennate‐like diatom endosymbiont (Pienaar et al. 2007), indicating a possible source for this diatom‐like gene sequence in a non‐diatom organism. The presence of other diatom‐like sequences in non‐diatoms is not as easily explained and may be evidence of the complex history of different endosymbiotic events within of the SAR super group. Likewise, the SIT‐like gene similar to bacterial sequences identified in the genome of the worm Capitella teleta may be the result of lateral gene transfer from a bacteria or misassembly of the genome with bacterial DNA. Further sequencing of these transcripts in laboratory isolates and phylogenetic characterization could help explain these phylogenetic peculiarities.
Ecological relevance of SIT evolution
The divergence and diversification of SIT genes during the evolution of diatom lineages has produced different SIT gene repertoires among diatoms that potentially result in distinctly different approaches to the utilization of silicic acid among species. Recent work focusing on whole‐cell metabolic pathways in both laboratory isolates (Bender et al. 2014) and mixed field communities (Alexander et al. 2015) have identified diverse responses among species utilizing the same nutrient resources. Diversity in the transcriptional responses among diatoms may enable different diatom species to co‐occur in the same environment by inhabiting different niches (Alexander et al. 2015). We suggest that evolved diversity among diatom SIT proteins is an example of genetic variation that enables co‐occurring diatoms to utilize the same resource in slightly different ways. These differences among diatoms were illustrated by the expression of SIT genes in natural communities along the Line P transect. Diatom community composition differed among locations (Table 3), but the diverse reference SIT sequence alignment made it possible to compare SIT transcript abundance at the lineage‐ and genus‐specific level (Fig. 6); this resolution in required to tease apart the combined influence of community composition and physiology on the abundance of SIT transcripts detected in mixed communities. The proportion of transcripts from clades A, E, and C differed consistently among major lineages and at the genus level. Additionally, when the concentration of silicic acid was higher, only a subset of the observed genera changed the proportion of transcribed SIT clades, while the remaining genera maintained the same proportion of SIT clade transcripts. The repertoire of SIT genes relied upon to utilize silicic acid differs by genera and condition, suggesting that co‐occurring organisms have diverse approaches for utilizing the shared resource of silicic acid.
How diatoms respond to changing environmental conditions will influence marine biogeochemistry and marine ecology, and the influence of diatoms on these large‐scale processes is intimately tied to their utilization of silicic acid. Placing silicic acid uptake in the context of SIT gene evolution may enable more nuanced understanding of why silicic acid uptake varies across environments and enable predictions of how it may change in the future.
We thank the large community of scientists that contributed to the creation of the MMETSP data sets, which was funded by the Gordon and Betty Moore Foundation (grant #GBMF2637). We thank Chris Berthiaume for helping with and providing advice about the bioinformatics analysis; Stephanie Guida at NCGR for additional information regarding the original analysis pipelines for the MMETSP data; Megan Bernhardt Schatz for providing cell count data from the GeoMICS cruise; Moira Regan for assistance in processing the biogenic silica samples; and Gwenn Hennon and Helena van Tol for help with sample collection at sea. We also thank two anonymous reviewers for improving this manuscript. This work was partially supported by the Gordon and Betty Moore Foundation (grant #GBMF3776 to EVA). Funding was provided to S. Bender through the Woods Hole Oceanographic Institution, CINAR Postdoctoral Scholar Fellowship. The GeoMICS cruise was funded by the University of Washington and the National Science Foundation OCE‐1205233 awarded to EVA.
Supporting information
Figure S1. Confirmation of full‐length expressed SIT genes in the genomes of the diatom Thalassiosira pseudonana, Fragilariopsis cylindrus, and Pseudo‐nitzschia multiseries.
Figure S2. Phylogenetic tree of SIT proteins, as in Figure 2, displaying all individual branches and all sequences labeled by genus, species, and their sequence identifier (NCBI accession number, JGI protein ID, or MMETSP sequence ID).
Figure S3. Growth of laboratory isolates cultured in triplicate in nutrient replete (left), low silicic acid (middle), or low nitrate (right) seawater media.
Figure S4. Percentage of SIT clades transcribed by genera with more than 100 total sequences detected at four stations along the Line P transect.
Table S1. Comma‐separated table of SIT homologs identified in the MMETSP database, including clade assignment, sequence ID, genus, species, nucleotide sequence, and the longest open reading frame of translated nucleotides used in phylogenetic analysis.
The copyright line for this article was changed on 14 October 2016 after original online publication.
References
- Adl, S. M. , Simpson, A. G. B. , Lane, C. E. , Lukeš, J. , Bass, D. , Bowser, S. S. , Brown, M. W. et al. 2012. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 59:429–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, H. , Jenkins, B. D. , Rynearson, T. A. & Dyhrman, S. T. 2015. Metatranscriptome analyses indicate resource partitioning between diatoms in the field. Proc. Natl. Acad. Sci. USA 112:E2182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson, A. J. 2007. Strong purifying selection in the silicon transporters of marine and freshwater diatoms. Limnol. Oceanogr. 52:1420–9. [Google Scholar]
- Baines, S. B. , Twining, B. S. , Brzezinski, M. A. , Krause, J. W. , Vogt, S. , Assael, D. & McDaniel, H. 2012. Significant silicon accumulation by marine picocyanobacteria. Nat. Geosci. 5:886–91. [Google Scholar]
- Bender, S. J. , Durkin, C. A. , Berthiaume, C. T. , Morales, R. L. & Armbrust, E. V. 2014. Transcriptional responses of three model diatoms to nitrate limitation of growth. Front. Mar. Sci. 1:3. [Google Scholar]
- Bolger, A. M. , Lohse, M. & Usadel, B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski, M. A. & Nelson, D. M. 1989. Seasonal changes in the silicon cycle within a Gulf Stream warm‐core ring. Deep Sea Res. Part Oceanogr. Res. Pap. 36:1009–30. [Google Scholar]
- Burkhard, P. , Stetefeld, J. & Strelkov, S. V. 2001. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 11:82–8. [DOI] [PubMed] [Google Scholar]
- Bursa, A. S. 1969. Actiniscus canadensis n. sp., A. pentasterias Ehrenberg v. arcticus n. var., Pseudoactiniscus apentasterias n. gen., n. sp., Marine relicts in Canadian arctic lakes. J. Protozool. 16:411–8. [Google Scholar]
- Curnow, P. , Senior, L. , Knight, M. J. , Thamatrakoln, K. , Hildebrand, M. & Booth, P. J. 2012. Expression, purification, and reconstitution of a diatom silicon transporter. Biochemistry (Mosc.) 51:3776–85. [DOI] [PubMed] [Google Scholar]
- Delorenzi, M. & Speed, T. 2002. An HMM model for coiled‐coil domains and a comparison with PSSM‐based predictions. Bioinformatics 18:617–25. [DOI] [PubMed] [Google Scholar]
- Durak, G. M. , Taylor, A. R. , Walker, C. E. , Probert, I. , de Vargas, C. , Audic, S. , Schroeder, D. et al. 2016. A role for diatom‐like silicon transporters in calcifying coccolithophores. Nat. Commun. 7:10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin, C. A. , Marchetti, A. , Bender, S. J. , Truong, T. , Morales, R. , Mock, T. & Armbrust, E. 2012. Frustule‐related gene transcription and the influence of diatom community composition on silica precipitation in an iron‐limited environment. Limnol. Oceanogr. 57:1619–33. [Google Scholar]
- Foissner, W. , Weissenbacher, B. , Krautgartner, W.‐D. & Lutz‐Meindl, U. 2009. A cover of glass: first report of biomineralized silicon in a ciliate, Maryna umbrellata (Ciliophora: Colpodea). J. Eukaryot. Microbiol. 56:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersonde, R. & Harwood, D. M. 1990. Lower Cretaceous diatoms from ODP Leg 113 Site 693 (Weddell Sea). Part 1. Vegetative cells In Proceedings of the Ocean Drilling Program, Scientific Results. Ocean Drilling Program, College Station, Texas, pp. 365–402. [Google Scholar]
- Hildebrand, M. 2008. Diatoms, biomineralization processes, and genomics. Chem. Rev. 108:4855–74. [DOI] [PubMed] [Google Scholar]
- Hildebrand, M. , Dahlin, K. & Volcani, B. E. 1998. Characterization of a silicon transporter gene family in Cylindrotheca fusiformis: sequences, expression analysis, and identification of homologs in other diatoms. Mol. Gen. Genet. MGG. 260:480–6. [DOI] [PubMed] [Google Scholar]
- Hildebrand, M. , Volcani, B. E. , Gassmann, W. & Schroeder, J. I. 1997. A gene family of silicon transporters. Nature 385:688–9. [DOI] [PubMed] [Google Scholar]
- Huerta‐Cepas, J. , Dopazo, J. & Gabaldon, T. 2010. ETE: a python environment for tree exploration. BMC Bioinformatics 11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseli, C. , Jongeneel, C. V. & Bucher, P. 1999. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell Syst. Mol. Biol. 99:138–48. [PubMed] [Google Scholar]
- Iverson, V. , Morris, R. M. , Frazar, C. D. , Berthiaume, C. T. , Morales, R. L. & Armbrust, E. V. 2012. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335:587–90. [DOI] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H. Y. , Fraser, M. , Li, W. , McAnulla, C. , McWilliam, H. et al. 2014. InterProScan 5: genome‐scale protein function classification. Bioinformatics 30:1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius, M. L. 2007. Perspectives on the evolution and diversification of the diatoms In Starratt S. [Ed.] Pond Scum to Carbon Sink: Geological and Environmental Applications of the Diatoms. The Paleontological Society, 2007, pp. 1–13. [Google Scholar]
- Kaczmarska, I. , Beaton, M. , Benoit, A. C. & Medlin, L. K. 2006. Molecular phylogeny of selected members of the order Thalassiosirales (Bacillariophyta) and the evolution of the fultoportula. J. Phycol. 42:121–38. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , Kuma, K. , Toh, H. & Miyata, T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33:511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling, P. J. , Burki, F. , Wilcox, H. M. , Allam, B. , Allen, E. E. , Amaral‐Zettler, L. A. , Armbrust, E. V. et al. 2014. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12:e1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , von Heijne, G. & Sonnhammer, E. L. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–80. [DOI] [PubMed] [Google Scholar]
- Likhoshway, Y. V. , Masyukova, Y. A. , Sherbakova, T. A. , Petrova, D. P. & Grachev, M. A. 2006. Detection of the gene responsible for silicic acid transport in chrysophycean algae. Dokl. Biol. Sci. 408:256–60. [DOI] [PubMed] [Google Scholar]
- Ma, J. F. , Tamai, K. , Yamaji, N. , Mitani, N. , Konishi, S. , Katsuhara, M. , Ishiguro, M. et al. 2006. A silicon transporter in rice. Nature 440:688–91. [DOI] [PubMed] [Google Scholar]
- Ma, J. F. , Yamaji, N. , Mitani, N. , Tamai, K. , Konishi, S. , Fujiwara, T. , Katsuhara, M. et al. 2007. An efflux transporter of silicon in rice. Nature 448:209–12. [DOI] [PubMed] [Google Scholar]
- Magǒ, T. & Salzberg, S. L. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957‐2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado, M. , Carmona, M. C. , Uriz, M. J. & Cruzado, A. 1999. Decline in Mesozoic reef‐building sponges explained by silicon limitation. Nature 401:785–8. [Google Scholar]
- Marron, A. O. , Alston, M. J. , Heavens, D. , Akam, M. , Caccamo, M. , Holland, P. W. H. & Walker, G. 2013. A family of diatom‐like silicon transporters in the siliceous loricate choanoflagellates. Proc. R. Soc. Lond. Biol. Sci. 280:20122543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Jézéquel, V. , Hildebrand, M. & Brzezinski, M. A. 2000. Silicon metabolism in diatoms: implications for growth. J. Phycol. 36:821–40. [Google Scholar]
- Matsen, F. , Kodner, R. & Armbrust, E. V. 2010. pplacer: linear time maximum‐likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlin, L. , Gersonde, R. , Kooistra, W. & Wellbrock, U. 1996. Evolution of the diatoms (Bacillariophyta) III. Molecular evidence for the origin of the Thalassiosirales. Nova Hedwig. 112:221–34. [Google Scholar]
- Mock, T. , Samanta, M. P. , Iverson, V. , Berthiaume, C. , Robison, M. , Holtermann, K. , Durkin, C. et al. 2008. Whole‐genome expression profiling of the marine diatom Thalassiosira pseudonana identifies genes involved in silicon bioprocesses. Proc. Natl. Acad. Sci. USA 105:1579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, D. M. , Tréguer, P. , Brzezinski, M. A. , Leynaert, A. & Quéguiner, B. 1995. Production and dissolution of biogenic silica in the ocean: revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Global Biogeochem. Cy. 9:359–72. [Google Scholar]
- Pienaar, R. , Sakai, H. & Horiguchi, T. 2007. Description of a new dinoflagellate with a diatom endosymbiont, Durinskia capensis sp. nov. (Peridiniales, Dinophyceae) from South Africa. J. Plant. Res. 120:247–58. [DOI] [PubMed] [Google Scholar]
- Preisig, H. R. 1994. Siliceous structures and silicification in flagellated protists. Protoplasma 181:29–42. [Google Scholar]
- Round, F. E. & Crawford, R. M. 1990. Phylum Bacillariophyta In Margulis L., Corliss J. O., Melkonian M. & Chapman D. J. [Ed.] Handbook of Protoctista. Jones and Bartlett, Boston, pp. 574–96. [Google Scholar]
- Saier, M. H. 2003. Tracing pathways of transport protein evolution. Mol. Microbiol. 48:1145–56. [DOI] [PubMed] [Google Scholar]
- Sapriel, G. , Quinet, M. , Heijde, M. , Jourdren, L. , Tanty, V. , Luo, G. , Le Crom, S. et al. 2009. Genome‐wide transcriptome analyses of silicon metabolism in Phaeodactylum tricornutum reveal the multilevel regulation of silicic acid transporters. PLoS ONE 4:e7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, H.C. , Perović‐Ottstadt, S. , Rothenberger, M. , Wiens, M. , Schwertner, H. , Batel, R. , Korzhev, M. et al. 2004. Silica transport in the demosponge Suberites domuncula: fluorescence emission analysis using the PDMPO probe and cloning of a potential transporter. Biochem. J. 381:665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha, R. P. & Hildebrand, M. 2015. Evidence for a regulatory role of diatom silicon transporters in cellular silicon responses. Eukaryot. Cell 14:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siever, R. 1991. Silica in the oceans: biological‐geochemical interplay In Schneider S.H. & Boston P.J. [Ed.] Scientists on Gaia. MIT Press, Boston, pp. 287–95. [Google Scholar]
- Simpson, T. L. & Volcani, B. E. 1981. Silicon and Siliceous Structures in Biological Systems. Springer‐Verlag, New York. [Google Scholar]
- Sorhannus, U. 2007. A nuclear‐encoded small‐subunit ribosomal RNA timescale for diatom evolution. Mar. Micropaleontol. 65:1–12. [Google Scholar]
- Stamatakis, A. 2006. RAxML‐VI‐HPC: maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–90. [DOI] [PubMed] [Google Scholar]
- Strickland, J. D. H. & Parsons, T. R. 1977. A Practical Handbook of Sea Water Analysis. Fisheries Research Board of Canada, Ottawa, Ontario. [Google Scholar]
- Thamatrakoln, K. , Alverson, A. J. & Hildebrand, M. 2006. Comparative sequence analysis of diatom silicon transporters: toward a mechanistic model of silicon transport. J. Phycol. 42:822–34. [Google Scholar]
- Thamatrakoln, K. & Hildebrand, M. 2007. Analysis of Thalassiosira pseudonana silicon transporters indicates distinct regulatory levels and transport activity through the cell cycle. Eukaryot. Cell 6:271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thamatrakoln, K. & Hildebrand, M. 2008. Silicon uptake in diatoms revisited: a model for saturable and nonsaturable uptake kinetics and the role of silicon transporters. Plant Physiol. 146:1397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throndsen, J. 1978. Preservation and storage. In Sournia A. [Ed.] Phytoplankton Manual. Museum National d'Histoire Natuelle, Paris, 69–74. [Google Scholar]
- Tréguer, P. , Nelson, D. M. , Van Bennekom, A. J. , DeMaster, D. J. , Leynaert, A. & Quéguiner, B. 1995. The silica balance in the world ocean: a reestimate. Science 268:375–9. [DOI] [PubMed] [Google Scholar]
- Yoshida, M. , Noël, M. H. , Nakayama, T. , Naganuma, T. & Inouye, I. 2006. A Haptophyte bearing siliceous scales: ultrastructure and phylogenetic position of Hyalolithus Neolepis gen. et sp. nov. (Prymnesiophyceae, Haptophyta). Protist 157:213–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Confirmation of full‐length expressed SIT genes in the genomes of the diatom Thalassiosira pseudonana, Fragilariopsis cylindrus, and Pseudo‐nitzschia multiseries.
Figure S2. Phylogenetic tree of SIT proteins, as in Figure 2, displaying all individual branches and all sequences labeled by genus, species, and their sequence identifier (NCBI accession number, JGI protein ID, or MMETSP sequence ID).
Figure S3. Growth of laboratory isolates cultured in triplicate in nutrient replete (left), low silicic acid (middle), or low nitrate (right) seawater media.
Figure S4. Percentage of SIT clades transcribed by genera with more than 100 total sequences detected at four stations along the Line P transect.
Table S1. Comma‐separated table of SIT homologs identified in the MMETSP database, including clade assignment, sequence ID, genus, species, nucleotide sequence, and the longest open reading frame of translated nucleotides used in phylogenetic analysis.