

Abstract
Aims
To characterize the pharmacology of MEDI0382, a peptide dual agonist of glucagon‐like peptide‐1 (GLP‐1) and glucagon receptors.
Materials and methods
MEDI0382 was evaluated in vitro for its ability to stimulate cAMP accumulation in cell lines expressing transfected recombinant or endogenous GLP‐1 or glucagon receptors, to potentiate glucose‐stimulated insulin secretion (GSIS) in pancreatic β‐cell lines and stimulate hepatic glucose output (HGO) by primary hepatocytes. The ability of MEDI0382 to reduce body weight and improve energy balance (i.e. food intake and energy expenditure), as well as control blood glucose, was evaluated in mouse models of obesity and healthy cynomolgus monkeys following single and repeated daily subcutaneous administration for up to 2 months.
Results
MEDI0382 potently activated rodent, cynomolgus and human GLP‐1 and glucagon receptors and exhibited a fivefold bias for activation of GLP‐1 receptor versus the glucagon receptor. MEDI0382 produced superior weight loss and comparable glucose lowering to the GLP‐1 peptide analogue liraglutide when administered daily at comparable doses in DIO mice. The additional fat mass reduction elicited by MEDI0382 probably results from a glucagon receptor‐mediated increase in energy expenditure, whereas food intake suppression results from activation of the GLP‐1 receptor. Notably, the significant weight loss elicited by MEDI0382 in DIO mice was recapitulated in cynomolgus monkeys.
Conclusions
Repeated administration of MEDI0382 elicits profound weight loss in DIO mice and non‐human primates, produces robust glucose control and reduces hepatic fat content and fasting insulin and glucose levels. The balance of activities at the GLP‐1 and glucagon receptors is considered to be optimal for achieving weight and glucose control in overweight or obese Type 2 diabetic patients.
Keywords: bodyweight, cynomolgus monkeys, diet‐induced obese mice, dual agonist, GLP‐1 receptor knock‐out mice, glucagon, glucagon‐like peptide‐1, glucose tolerance, liraglutide, obesity, type 2 diabetes
1. INTRODUCTION
Analogues of glucagon‐like peptide‐1 (GLP‐1), an incretin hormone released by the L cells of the gut that promotes potentiation of glucose‐stimulated insulin secretion from pancreatic β‐cells and delays gastric emptying, have been used extensively to reduce elevated plasma glucose levels in type 2 diabetic patients.1, 2 GLP‐1 analogues also induce satiety and significantly reduce body weight in animal models of obesity, which translates to a small decrease in excess body weight in humans.3, 4, 5, 6 Glucagon is a hormone secreted by the α‐cells of the pancreas that elevates blood glucose levels by promoting glycogen breakdown and glucose release from hepatocytes.7 Glucagon also reduces food intake in man, an observation first reported over 50 years ago.8 Studies in rats indicated that this effect of glucagon is due to increased satiety, by acting directly in the portal venous/hepatic area to decrease meal size.9 The endogenous dual GLP‐1/glucagon receptor agonist, oxyntomodulin, decreased food intake and increased energy expenditure, thus reducing bodyweight by 2.4% following treatment of healthy overweight and obese humans for 4 weeks.10, 11, 12, 13, 14 The half‐life of oxyntomodulin is, however, very short (c. 10 minutes), which precludes its clinical utility.15 Several reports have recently emerged describing the effects of longer half‐life synthetic dual GLP‐1/glucagon receptor agonist peptides in rodent models of obesity and diabetes, following the original report from Day et al. (2009).16, 17, 18 However, none of these novel analogues has been reported to have advanced to clinical studies to date.
Here we report on the pharmacological properties of MEDI0382 (Figure 1), a novel dual GLP‐1/glucagon receptor peptide agonist with a balance of agonism at the GLP‐1 and glucagon receptors designed to facilitate both weight loss and glycaemic control, that is currently being developed to treat overweight or obese patients with type 2 diabetes (T2D).19 The half‐life of MEDI0382 has been extended by enhancing stability to peptidase degradation and by palmitoylation to facilitate reversible binding to plasma albumin.20, 21 MEDI0382 was designed and synthesized as part of a chemistry lead generation and optimization campaign that produced a series of dual GLP‐1/glucagon receptor agonist peptides exhibiting a range of GLP‐1 and glucagon receptor agonist activities. The desired balance of GLP‐1 and glucagon receptor activities for combining weight loss and glucose control in humans was identified on the basis of systems biology modelling of clinical data for GLP‐1 and glucagon analogues and pharmacokinetic‐pharmacodynamic in vivo screening of peptides with different activity ratios in diet‐induced obesity (DIO) mouse models.
Figure 1.
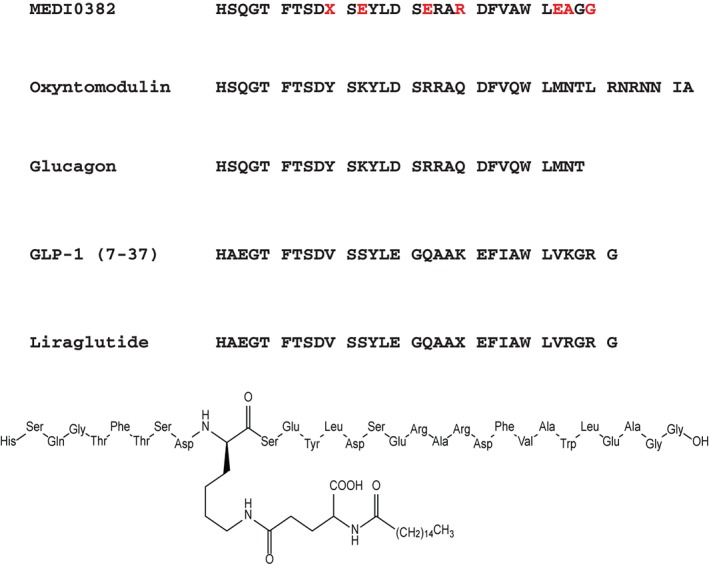
Amino acid sequence and chemical structure of MEDI0382. Sequences of the related peptides oxyntomodulin, glucagon, GLP‐1 (amino acids 7‐37) and liraglutide are also shown. Differences in amino acids from native glucagon and GLP‐1 are denoted in red. X represents lysine linked through gamma glutamate to palmitic acid.
2. MATERIALS AND METHODS
2.1. Synthesis
MEDI0382 (Figure 1) was chemically synthesized. Peptide chain elongation on the resin was performed with the aid of a solid phase peptide synthesizer using manufacturer‐supplied protocols for coupling of Fmoc‐amino acids. Glutamine residues 20 and 24 were substituted with amino acids that were not susceptible to deamidation and arginine residue 17 was replaced with glutamate to reduce susceptibility to proteolysis. The orthogonal protection of the side chain of Lys10 was removed and the free epsilon‐amino group was coupled to gamma‐glutamate, followed by palmitic acid to facilitate reversible binding to albumin to extend plasma half‐life. GLP‐1, oxyntomodulin and glucagon were purchased from Bachem AG (Bubendorf, Switzerland) and liraglutide from PolyPeptide Laboratories (Strasbourg, France).
2.2. Cyclic AMP accumulation assay
Stable Chinese hamster ovary (CHO) cell lines expressing human, mouse, rat or cynomolgus monkey GLP‐1 or glucagon receptor were generated at AstraZeneca or MedImmune using public domain‐ or in‐house‐determined sequences for each receptor. INS‐1 832/3 and EndoC‐βH1 cells were kindly supplied by Prof. Christopher B. Newgard (Duke University, Durham, NC, USA) and Prof. Raphael Scharfmann (Endocells, Paris, France), respectively. Cryopreserved primary rat, mouse and human pooled hepatocytes were purchased from Life Technologies (Carlsbad, CA, USA). Agonist potency determinations (EC50 values) for peptides inducing cAMP production were measured in the presence of 0.1% BSA or human, cynomolgus monkey, rat or mouse serum albumin at physiological concentrations (4.4%, 4.2%, 3.2% and 3.2%, respectively; Sigma Aldrich UK or Equitech Bio Inc, Kerrville, TX, USA) in order to account for the impact that differences among species in plasma protein binding will have on in vivo efficacy. cAMP generation was measured using the CisBio dynamic d2 cAMP HTRF assay kit (CisBio, Codolet, France) according to the manufacturer's guidelines as previously described.22, 23
2.3. Hepatic glucose output (HGO) assay
The effect of MEDI0382, glucagon and oxyntomodulin on glucose output was tested in primary rat hepatocytes. Fresh Sprague Dawley rat hepatocytes were purchased from Triangle Research Labs (Research Triangle Park, NC, USA). Cells (150 000 cells/well) grown on collagen‐coated plates, were washed three times with phosphate‐buffered saline prior to incubation with compounds or vehicle at 37°C for 120 minutes. The supernatant was removed from each well and cells were lysed in RIPA buffer (50 mM Tris HCl, 150 mM NaCl, 1% Triton X‐100, pH 7.4). Supernatants were centrifuged and assayed for glucose content using the Amplex Red Glucose/Glucose Oxidase Assay Kit (Life Technologies). Protein concentration was determined with the Pierce BCA protein assay kit (Thermo Scientific, Waltham, MA, USA). Glucose concentrations were normalized to protein content per well.
2.4. Glucose‐stimulated insulin secretion (GSIS) assay
Rat INS‐1 832/3 insulinoma cells were seeded in a 48‐well plate (250 000 cells/well) for 48 hours and then cultured in the presence of low glucose (2.8 mM) in Krebs Ringer buffer (2.6 mM CaCl2, 98.5 mM NaCl, 4 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 20 mM HEPES, 25.9 mM NaHCO3, 0.2% BSA, pH 7.4) for 1 hour at 37°C prior to incubation with compounds in the presence of high glucose (8.3 mM) for 1 hour. Insulin secretion was quantified using Mesoscale Discovery ECL detection (Rockville, MD, USA).
2.5. Animal studies
Animal studies were conducted at MedImmune, Inc. (USA), the Mouse Metabolic Phenotypic Center at Vanderbilt University (USA), Gubra (Denmark), and Envigo (UK) according to protocols reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the testing facility and in compliance with the applicable national laws and regulations concerning humane care and use of laboratory animals and the AstraZeneca Animal Welfare and Bioethics policies.
C57Bl/6 mice were obtained from Jackson Labs (Bar Harbor, ME, USA) and were maintained on high‐fat diet [HFD; 60% calories derived from fat (D12492, Research Diets, Inc., New Brunswick, NJ, USA)] except where noted otherwise. GLP‐1 receptor knockout (KO) mice and their wildtype C57Bl/6 littermates were obtained from Jackson labs and maintained on chow diet (# 2018) (Harlan Teklad, Madison, WI, USA). Animals were housed on a 12 hour light/12 hour dark cycle at standard temperature and humidity conditions with ad libitum access to water and food (except where noted otherwise, e.g. during fasting prior to glucose tolerance tests, measurement of food intake or collection of samples for metabolic analysis).
Naïve cynomolgus monkeys (Macaca fascicularis) were obtained from a certified supplier, group housed, allowed access to water ad libitum and fed on a pelleted diet for monkeys, supplemented with fresh fruit and biscuits. Supplementary feeding of bread, biscuits and/or malt loaf with jam and peanut butter was instigated during the second or third weeks of treatment to minimize body weight loss. Animals ranged from 33 to 36 months of age (2.6‐4.2 kg) at start of treatment. Access to food was withheld for at least 3 hours prior to sampling for blood glucose or insulin.
2.6. Acute food intake in mice
Acute effects of MEDI0382 on food consumption in DIO mice were assessed by continuous measurement of food intake over a 24 hours period using a BioDAQ food intake monitoring system (Research Diets Inc.). Mice (16‐17 week old male C57Bl/6 fed a 60% HFD for 10‐11 weeks) were housed singly in the BioDAQ system and switched to a 45% kcal HFD diet (D12451, Research Diets Inc.) to facilitate accurate food intake measurements. Mice were acclimatized for 3‐4 weeks in the BioDAQ system. Animals, weighing approximately 37 g, were randomized according to 24 hours food intake and body weight the day before dosing. On the day of the study, mice (n = 8/group) were fasted for 6 hours prior to dosing. Vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5), liraglutide (10 nmol/kg) or MEDI0382 (10 nmol/kg) were administered s.c. approximately 1 hour prior to switching off lights, whereas oxyntomodulin (OXM; 1000 nmol/kg) was administered s.c. 10 minutes prior to lights out. Animals were given access to 45% HFD following lights out and food intake was recorded for 24 hours in the BioDAQ system.
Male GLP‐1 receptor KO mice and their wildtype (WT) littermates (7‐9 weeks of age) were housed one per cage and allowed to acclimate for 5 days. Mice were sham dosed s.c. with vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) for 2 days prior to study start. Mice (mean body weight c. 27‐28 g) were randomized to treatment groups based on body weight and 24 hours food intake. Following an overnight fast, mice (n = 5‐7/group) were injected with vehicle, MEDI0382 (30 or 100 nmol/kg), liraglutide (30 nmol/kg) or the selective glucagon receptor agonist IUB288 (30 or 100 nmol/kg)24 and food was returned 30 minutes following dosing. Food was weighed at 1, 2, 4, 6, 8 and 24 hours time points after return of food.
2.7. Acute intraperitoneal glucose tolerance test (IPGTT) in mice
Acute effects of MEDI0382 on glucose tolerance were assessed in an intraperitoneal (i.p.) glucose tolerance test (IPGTT) in DIO mice, GLP‐1 receptor KO mice or their wildtype littermates. Male C57Bl/6 mice (16‐17 weeks of age) which had been maintained for 10‐11 weeks on a 60% HFD were singly housed and allowed to acclimate for approximately 3 weeks prior to study start with continued access to 60% HFD. Mice (mean body weight c. 40 g; n = 5‐7/group) were sham dosed s.c. with vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) for 2‐3 days to acclimate to dosing and were randomized into groups based on bodyweight. Mice were then fasted overnight prior to i.p. administration of glucose (1.5 g/kg). One hour prior to glucose administration, mice were dosed s.c. with vehicle, MEDI0382 (1 or 10 nmol/kg) or liraglutide (10 or 100 nmol/kg), whereas oxyntomodulin (300 nmol/kg) was administered 10 minutes prior to the glucose challenge (t = 0 minute). Blood glucose measurements from tail snips were performed at −60, 0 (immediately prior to glucose challenge), 10, 30, 60 and 120 minute time points. Blood for plasma insulin measurement was collected at −60, 0, 10 and 30 minutes.
Male GLP‐1 receptor KO mice and their wildtype littermates (12‐15 weeks of age) housed one per cage for 5 days were randomized according to body weight (mean bodyweight c. 27‐29 g; n = 6/group) and sham dosed s.c. with vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) for 2 days prior to study. Mice fasted for 6 hours were then injected s.c. with vehicle, MEDI0382 (3 nmol/kg) or liraglutide (3 nmol/kg) at 1 hour prior to an IPGTT (2 g/kg glucose; t = 0 minute). Blood glucose from tail snips was measured at 0, 15, 30, 45, 60, 90 and 120 minutes post glucose challenge.
2.8. Acute glucose control in 2‐hours fasted mice
Male GLP‐1 receptor KO mice and their wildtype littermates (9‐11 weeks of age) were housed singly for 10 days, and sham dosed s.c. with vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) for 3 days prior to study start. Mice were randomized by body weight, (weighing c. 27‐29 g; n = 6/group) fasted for 2 hours and injected with somatostatin (10 mg/kg, s.c.) to prevent release of glucagon and insulin from pancreas. Thirty minutes later (t = 0 hour) mice were injected s.c. with vehicle, MEDI0382 (3, 10 or 30 nmol/kg) or IUB288 (30 nmol/kg). Blood glucose from tail snips was measured at −0.5, 0, 0.25, 0.5, 1, 1.5, 2 and 3 hours time points.
2.9. Effects in DIO mice following repeated administration
Male mice (C57Bl/6) were fed a 60% HFD for 14‐16 weeks and were approximately 25‐27 weeks of age (mean body weight: 43 g) at start of study. Animals (n = 11‐12/group) received daily s.c. doses of vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5), MEDI0382 (10 or 30 nmol/kg) or liraglutide (40 nmol/kg). Food consumption and body weight were recorded daily. On day 21, an i.p. glucose bolus (1 g glucose/kg) was administered to mice fasted for 4 hours and blood glucose levels were measured at −45, 0, 30, 60, 90, 120 and 180 minutes relative to glucose administration (t = 0 minute). Blood samples for preparation of plasma were taken from the tail vein for measurement of MEDI0382 and liraglutide exposure at 1, 4, 8 and 24 hours post‐dose on the first and last days of dosing (Day 1 and 27, respectively).
2.10. Energy expenditure in DIO mice
Male DIO mice, approximately 20 weeks of age, individually housed with access to high‐fat diet (60% kcal/fat, D12492, Research Diets Inc.; previously on HFD for c. 16 weeks) were randomized to one of three groups based on body weight (mean body weight 41 g): vehicle (fed ad libitum; n = 4), MEDI0382‐treated (n = 8) or vehicle‐treated but pair‐fed with the same amount of food as consumed by MEDI0382‐treated mice (n = 8). Vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) or MEDI0382 (10 nmol/kg) were administered s.c. daily just prior to lights‐off for 21 days. Body weight and food intake were recorded daily. Body composition was determined by NMR (Bruker minispec, Bruker, Billerica, MA, USA) on days 0, 16 and 21. On days 16‐21, mice were housed in an indirect calorimetry system (Promethion, Sable Systems, Las Vegas, NV, USA) for energy expenditure assessment. Oxygen consumption and carbon dioxide production were measured for each mouse at 5 minute intervals for 30 seconds, to calculate rate of oxygen consumption (VO2), a proxy for metabolic rate. Respiratory exchange ratio (RER) was calculated as the ratio of rate of CO2 production (VCO2) over VO2. Ambulatory activity was measured every second with XYZ beams (BXYZ‐R, Sable Systems, NV, USA). Data acquisition and instrument control were coordinated by MetaScreen v2.2.18 and raw data were processed using ExpeData v1.7.30 (Sable Systems). The first 24 hours of data were discarded and three full diurnal cycles of data were averaged to calculate the mean 24 hours VO2 and RER. VO2 was normalized to mean fat‐free mass determined via NMR either side of indirect calorimetry to account for differences in body mass between treatment groups.
2.11. Effects in cynomolgus monkeys following repeated administration
Effects of MEDI0382 on food consumption, bodyweight and fasting glucose were assessed in healthy cynomolgus monkeys as part of 4‐ and 8‐week toxicity studies and, in addition, effects on fasting insulin were measured as part of a 4‐week toxicity study. In the 4‐week study, groups of three male monkeys received daily s.c. doses of vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) or MEDI0382 at dose levels of 4, 8 or 27 nmol/kg/d for 29 days. In the 8‐week study, groups of three male and three female monkeys received daily s.c. doses of vehicle (1.85% w/v propylene glycol and 0.04% v/v Tween 80 in 50 mM phosphate buffer, pH 7.5) or MEDI0382 at dose levels of 8, 16 or 27 nmol/kg/d for 57 days. Additional groups of two male and two female cynomolgus monkeys were similarly treated at the same dose levels for 8 weeks but were retained for an additional 4‐week treatment‐free period to assess reversibility of bodyweight changes. Food consumption and body weights were recorded daily and blood samples were collected for measurement of MEDI0382, glucose and insulin (4‐week study only) prior to dosing and at 1, 4, 8, 12, 18 and 24 hours after dosing on Day 1 and 29 (4‐week study) or 57 (8‐week study).
2.12. Measurement of peptides
Blood was collected into tubes containing EDTA anticoagulant, centrifuged at 2000 × g for 10 minutes at ambient temperature and plasma immediately frozen prior to measurement of MEDI0382 or liraglutide by LC‐MS/MS after protein precipitation extraction with either ice‐cold 75% acetonitrile or 95% ethanol. Following mixing and centrifugation, supernatant was injected on an Acquity BEH C8 or C18 UPLC column (Waters corp., MA, USA) and eluted using a gradient mobile‐phase (Mobile phase A: 0.2% formic acid in water or 0.2% formic acid and 2% acetonitrile in water, Mobile phase B: 0.2% formic acid in acetonitrile) using a Waters Acquity LC system and Applied Biosystems 5500 mass spectrometer.
2.13. Measurement of glucose and insulin
Blood was sampled from a tail vein or tail snips in mice and a peripheral vein in monkeys. Blood glucose from tail snips was determined at the specified time points using the Ascensia Breeze 2 blood glucose monitoring system (Bayer Healthcare LLC, Mishawaka, IN, USA) in the acute mouse studies. Blood glucose from venous samples was measured with a BIOSEN c‐Line glucose meter (EKF‐diagnostics, Barleben, Germany) in the repeat dose DIO mouse study or with an ACCU‐CHEK glucose meter (La Roche AG, Basel, Switzerland) in monkeys. Blood was collected into tubes containing EDTA anticoagulant, centrifuged at 2000 × g for 10 minutes at ambient temperature and plasma immediately frozen prior to measurement of insulin by immunoassay using a Mesoscale rat/mouse insulin kit (K152BZC‐1, Rockville, MD, USA) in the mouse acute IPGTT study, an AlphaLisa kit (Perkin Elmer, Waltham, MA, USA) in the repeat dose DIO mouse study and an EMD Millipore kit (Nottingham, UK) in monkeys. Access to food was withheld for monkeys for at least 3 hours prior to sampling for glucose and insulin.
2.14. Statistical methods
Graphical presentations, calculations and statistical analyses were carried out with GraphPad software (San Diego, CA, USA). Results are expressed as mean ± standard error of the mean (SEM) unless otherwise stated. In vivo data were analysed with two‐way ANOVA followed by Dunnett's post‐hoc test (food intake, body weight and IPGTT glucose challenge data) or a one‐way ANOVA followed by Dunnett's or Bonferroni's post‐hoc test (interval food intake, IPGTT AUC and terminal fat and clinical chemistry data). Longitudinal food intake and body weight data in the energy expenditure study were analysed by two‐way repeated measures ANOVA and Tukey's multiple comparison post‐hoc test. Body composition and mean 24 hours VO2, mean 24 hours RER and physical activity data were analysed using one‐way ANOVA and Tukey's multiple comparison test. In all cases, p < .05 was considered significant. Average steady state plasma exposure (Css, average) was calculated using a non‐linear mixed effect pharmacokinetic modelling approach using the FOCE Extended Least Squares estimation method as implemented in Phoenix WinNonlin 6.4, NMLE 1.3 (Certara L.P., Princeton, NJ, USA).
3. RESULTS
3.1. MEDI0382 is a potent and selective GLP‐1 and glucagon receptor agonist in vitro
The potency (EC50) values for MEDI0382 as measured by cAMP generation in CHO cells over‐expressing human recombinant GLP‐1 or glucagon receptors in the presence of 0.1% BSA were 6.9 and 10.2 pM, respectively, which were within 10‐fold of the native ligands (Figure 2 and Table 1). These potencies were decreased by protein binding when assays were performed in the presence of physiological concentrations of plasma albumin. The potency relative to the native ligands GLP‐1 and glucagon is higher for MEDI0382 at the human GLP‐1R (1%) than at the human glucagon receptor (0.2%) and this relationship is maintained for mouse, rat and cynomolgus monkey GLP‐1 and glucagon receptors (Table 2). MEDI0382 stimulated a concentration‐dependent increase in cAMP accumulation in rat (INS‐1 832/3) and human (EndoC‐βH1) pancreatic β‐cell lines as well as rat, mouse and human hepatocytes (Table 1). In addition, MEDI0382 potentiated glucose‐stimulated insulin secretion in the rat (INS‐1 832/3) pancreatic β‐cell line and increased glucose output in rat hepatocytes (Figure 2 and Table 1). MEDI0382 showed >10,000‐fold selectivity for GLP‐1 and glucagon receptor activation over related Class B G protein‐coupled receptors such as those for human GIP, GLP‐2 and secretin (Figure S1, Supporting Information).
Figure 2.
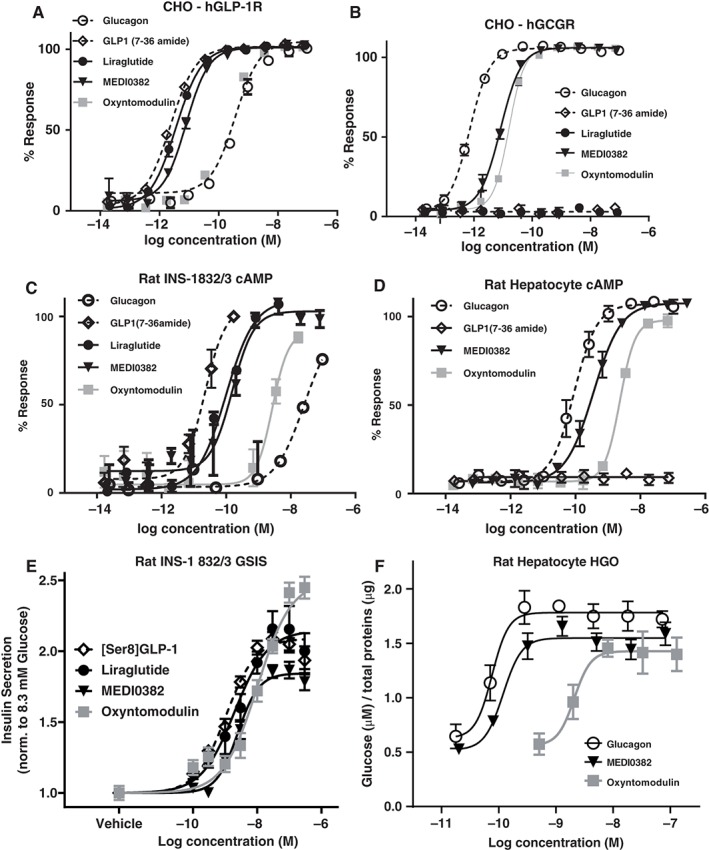
Potency of MEDI0382 in transfected and physiologically relevant endogenous receptor systems. Representative concentration‐response curves for MEDI0382, GLP‐1, glucagon (GCG), oxyntomodulin (OXM) and liraglutide in cAMP accumulation assays in CHO cell lines expressing human GLP‐1 receptors (A), human GCG receptors (B), rat INS‐1 832/3 β–cell line (C) and rat hepatocytes (D) all in the presence of 0.1% BSA. Concentration‐response curve for MEDI0382, GLP‐1, liraglutide and OXM in rat INS‐1 832/3 glucose‐stimulated insulin secretion assay (GSIS) in the presence of 0.2% BSA (E). Concentration‐response curve for MEDI0382, GCG and OXM in rat hepatocyte glucose output (HGO) assay in the presence of 0.1% BSA (F). Values are mean (±SEM) from duplicate analysis fitted with 4‐parameter logistic fit to determine EC50. Data shown representative of n ≥ 3 experiments.
Table 1.
Potency (EC50 values) for MEDI0382, GLP‐1, glucagon, oxyntomodulin and liraglutide for transfected receptors expressed in CHO cells and at endogenous receptor populations in the rat INS‐1 832/3 β–cell line, human EndoC‐βH1 β–cell line, and primary human, mouse and rat hepatocytes for both second messenger and functional readouts
| Agonist | CHO‐human GLP‐1R cAMP | CHO‐human GCGR cAMP | Rat INS‐1 832/3 β‐cell cAMP | Human EndoC β‐cell cAMP | Rat hepatocyte cAMP | Mouse hepatocyte cAMP | Pooled human hepatocyte cAMP | Rat INS‐1 832/3 β‐cell GSIS | Rat hepatocyte HGO |
|---|---|---|---|---|---|---|---|---|---|
| GLP‐1 1 | 2.0 ± 0.1 | Inactive | 32 ± 5 | 219 ± 75 | Inactive | Inactive | Inactive | 1068 ± 2732 | Inactive |
| Glucagon | 264 ± 101 | 1.3 ± 0.08 | 10708 ± 6737 | 22306 ± 11141 | 75 ± 8 | 58 ± 11 | 665 ± 141 | Not tested | 68 ± 24 |
| MEDI0382 | 6.9 ± 0.6 | 10.2 ± 1.5 | 226 ± 66 | 1051 ± 316 | 462 ± 57 | 840 ± 80 | 2447 ± 151 | 1506 ± 447 | 83 ± 28 |
| Oxyntomodulin | 102 ± 26 | 18.1 ± 1.2 | 2286 ± 297 | 4253 ± 651 | 2277 ± 755 | 1395 ± 190 | 9963 ± 2310 | 33043 ± 5025 | 1954 ± 1139 |
| Liraglutide | 3.2 ± 0.7 | Inactive | 81 ± 10 | 285 ± 69 | Not tested | Not tested | Not tested | 1430 ± 372 | Not tested |
Values are geometric mean (± SEM) EC50 (pM) from n ≥ 3 independent experiments in the presence of 0.1% or 0.2% (GSIS) BSA.
GLP‐1 = GLP‐1 (7‐36)NH 2.
GLP‐1 = ser8 GLP‐1.
Table 2.
Potency (EC50 values) and relative potency of MEDI0382 at human, cynomolgus monkey, mouse and rat transfected GLP‐1 (GLP‐1R) or glucagon (GCGR) receptors expressed in CHO cells measured by cAMP accumulation in the presence of physiological plasma albumin levels
| Species | GLP‐1R EC50 (pM) | GCGR EC50 (pM) | GLP‐1R activity relative to GLP1 (%) 1 | GCGR activity relative to glucagon (%) 2 | GCGR/GLP‐1R ratio 3 |
|---|---|---|---|---|---|
| Human | 188 ± 43 | 682 ± 154 | 1.02 | 0.19 | 0.19 |
| Monkey | 5.2 ± 0.78 | 318 ± 35 | 8.59 | 1.32 | 0.15 |
| Mouse | 74 ± 12 | 614 ± 141 | 2.88 | 0.71 | 0.25 |
| Rat | 50.6 ± 8.7 | 24173 ± 4240 | 1.13 | 0.13 | 0.12 |
Values are geometric mean (± SEM) EC50 (pM) from n = 4 independent experiments in the presence of 4.4% human albumin, 4.2% monkey albumin, 3.2% mouse albumin or 3.2% rat albumin.
GLP‐1R activity relative to GLP‐1% = (geometric mean EC 50 GLP1/geometric mean EC 50 MEDI0382) × 100.
GlucR activity relative to glucagon % = (geometric mean EC 50 glucagon/geometric mean EC 50 MEDI0382) × 100.
GlucR/GLP‐1R ratio = GlucR activity relative to glucagon %/GLP‐1R activity relative to GLP‐1%.
3.2. Acute administration of MEDI0382 reduces food intake in mice
MEDI0382 (10 nmol/kg, s.c.) robustly suppressed food intake in DIO mice relative to vehicle‐treated controls during 0‐12 hours after administration (Figures 3A and S2, Supporting Information). Mean food intake was reduced during this time interval to approximately 31% of control mice treated with vehicle (p < .001). The effect of MEDI0382 was evident early (0‐2 hours postdose), at which time oxyntomodulin (1000 nmol/kg, s.c.) also significantly suppressed food intake (Figure S2A, Supporting Information). Oxyntomodulin was ineffective during the 0‐12 hours interval (Figure S2B, Supporting Information), as expected from its short plasma half‐life.15, 25 The terminal plasma half‐life of MEDI0382 in mice after subcutaneous administration was measured in a separate study and was approximately 5 hours (data not shown) which is similar to liraglutide (c. 7 hours).26 Liraglutide (10 nmol/kg, s.c.) reduced food intake to approximately 46% relative to vehicle controls at 0‐12 hours (p < .001), but not during 0‐2 hours (Figure S2A and B, Supporting Information). None of the drugs administered had a significant effect during the 12‐24 hours time interval after a single administration (Figure S2C, Supporting Information).
Figure 3.
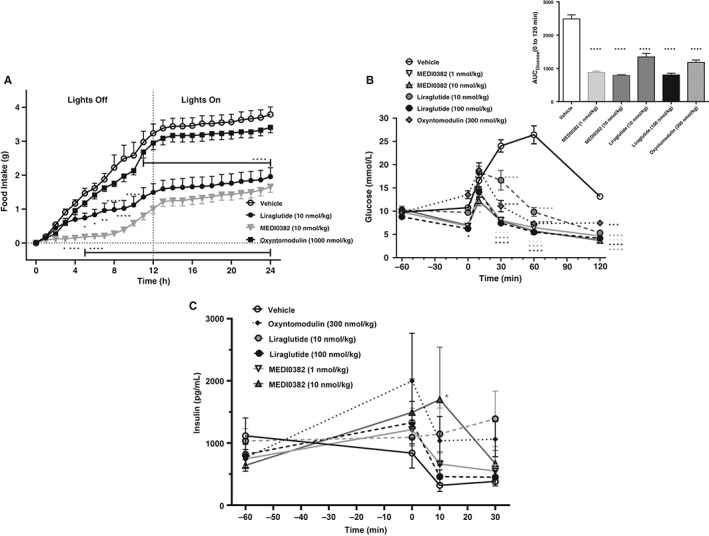
Acute effects of MEDI0382, liragutide or oxyntomodulin on food intake and glucose tolerance in male DIO mice. Cumulative food intake during 0‐24 hours after s.c. administration of vehicle, MEDI0382 (10 nmol/kg), liraglutide (10 nmol/kg) or oxyntomodulin (1000 nmol/kg) in DIO mice (A; n = 8 mice/group). Concentrations of blood glucose (B) and plasma insulin (C) after single s.c. administration of vehicle, MEDI0382 (1 and 10 nmol/kg), liraglutide (10 and 100 nmol/kg) or oxyntomodulin (300 nmol/kg) prior to administration of glucose (1.5 g/kg) by i.p. administration (n = 4‐7 mice/group). Time 0 is immediately prior to glucose challenge. Values are presented as mean (± SEM). *p < .05; **p < .01; ***p < .001; ****p < .0001 compared to vehicle.
The effect of MEDI0382‐treatment (30 or 100 nmol/kg s.c.) was also compared to treatment with liraglutide (30 nmol/kg s.c.) and the selective glucagon receptor agonist IUB288 (30 or 100 nmol/kg s.c.) in lean male GLP‐1 receptor KO mice and their wildtype littermates (Supplementary Figure S3A and B, Supporting Information). Neither MEDI0382 nor liraglutide had an effect on food intake in GLP‐1 receptor KO mice, but both peptides robustly reduced food consumption in wildtype mice. IUB288 failed to affect food intake in both GLP‐1 receptor KO and wildtype mice.
3.3. Acute administration of MEDI0382 improves glucose control in mice
Following single s.c. administration of vehicle, MEDI0382, oxyntomodulin or liraglutide to DIO mice, blood glucose levels measured prior to an intraperitoneal glucose tolerance test (IPGTT; t = 0) decreased significantly in mice dosed with liraglutide at 100 nmol/kg (Figure 3B; p < .05). After the glucose challenge, only 10 nmol/kg MEDI0382 reduced blood glucose levels (p < .05) at the 15 minutes time point compared with vehicle‐treated mice. However, at the 30, 60 and 120 minutes time points post‐challenge, MEDI0382, oxyntomodulin and liraglutide reduced glucose excursion at all dose levels (p < .001) compared with vehicle control (Figure 3B). Glucose tolerance was significantly improved in all treatment groups (p < .0001) with glucose AUC's of 2487, 877, 785, 1345, 801 and 1182 mmol/L*min for vehicle, 1 nmol/kg MEDI0382, 10 nmol/kg MEDI0382, 10 nmol/kg liraglutide, 100 nmol/kg liraglutide and 300 nmol/kg oxyntomodulin, respectively (Figure 3B inset).
Plasma insulin levels measured at the −60, 0, 10 and 30 min time points were variable and only the high dose of MEDI0382 (10 nmol/kg) showed a significant increase in insulin (p < .05) at the 10 minutes time‐point compared to vehicle‐treated mice (Figure 3C).
The acute effect of MEDI0382 on glucose control was also determined in lean male GLP‐1 receptor KO mice and their wildtype C57Bl/6 littermates (Figure S3C–F, Supporting Information). Treatment with MEDI0382 or liraglutide (both at 3 nmol/kg, s.c.) had no effect on glucose excursion compared to vehicle treatment in GLP‐1 receptor KO mice following an IPGTT (Figure S3C and D, Supporting Information). In contrast, treatment with MEDI0382 or liraglutide robustly lowered glucose excursion in wildtype mice compared to vehicle treatment. MEDI0382 had a greater effect on reducing glucose excursion than liraglutide at an equimolar dose level. In lean male GLP‐1 receptor KO mice which had been fasted for 2 hours and treated with somatostatin (10 mg/kg, s.c.) to prevent pancreatic release of glucagon and insulin, treatment with MEDI0382 (3, 10 or 30 nmol/kg, s.c.) robustly elevated glucose levels compared to vehicle‐treated mice, and had an effect similar to that elicited by the glucagon receptor agonist IUB 288 (30 nmol/kg, s.c.; Figure S3E, Supporting Information). Maximal effect on glucose elevation was evident at the lowest dose of MEDI0382 (3 nmol/kg) tested. In contrast, MEDI0382‐treatment reduced fasted blood glucose levels in wildtype mice (Figure S3F, Supporting Information). IUB288 substantially, but transiently, elevated blood glucose level following s.c. dosing in wildtype mice treated with somatostatin, but glucose then declined to a level lower than vehicle‐treated mice at 2 and 3 hours post‐dose.
3.4. Repeated administration of MEDI0382 reduces body weight and food intake and improves glucose tolerance in DIO mice
Repeated once‐daily s.c. administration of MEDI0382 or liraglutide to DIO mice significantly (p < .05) reduced body weight (Figure 4A). The mean body weight of vehicle‐treated animals increased by 2.5% of starting body weight over the course of the 4‐week study, whereas the mean body weight loss was 21%, 30% and 21% of starting body weight at dose levels of 10 nmol/kg MEDI0382, 30 nmol/kg MEDI0382 and 40 nmol/kg liraglutide, respectively. The average steady state plasma exposure (Css, average) at the end of the study was 12.3, 38.3 and 93.0 nmol/L at dose levels of 10 nmol/kg MEDI0382, 30 nmol/kg MEDI0382 and 40 nmol/kg liraglutide, respectively.
Figure 4.
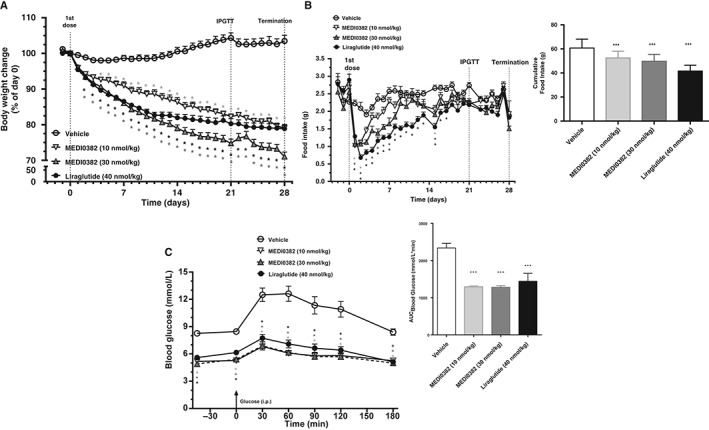
Effects of MEDI0382 on body weight (A), food intake (B) and glucose tolerance (C) in male DIO mice after repeated daily s.c. administration of vehicle, MEDI0382 (10 or 30 nmol/kg) or liraglutide (40 nmol/kg) for 28 days. Glucose tolerance (IPGTT) was conducted by i.p. administration of glucose (1 g/kg) on day 21. Time 0 is immediately prior to glucose challenge. Insets show overall area under the curve (AUC) values for food intake (B) and blood glucose (C). Values are presented as mean (± SEM); n = 11‐12 mice/group. *p < .05; ***p < .001 compared to vehicle.
The marked reductions in body weight upon repeated treatment with MEDI0382 and liraglutide were reflected in an immediate suppression of food intake compared to vehicle‐treated mice (p < .05), with the hypophagic effect reaching its maximum on Day 1‐2 (Figure 4B). Food intake gradually returned to baseline levels and normalized from day 9 for MEDI0382 and from day 18 for liraglutide. Cumulative total food intake was significantly reduced (p < .001) by both MEDI0382 (10 and 30 nmol/kg doses) and liraglutide (40 nmol/kg) when compared to vehicle treatment (Figure 4B inset). The impact of liraglutide on food intake was greater than that produced by the high dose of MEDI0382 (p < .05). The mean cumulative food intake at the end of study was 61, 53, 50 and 42 g for vehicle, 10 nmol/kg MEDI0382, 30 nmol/kg MEDI0382 and 40 nmol/kg liraglutide‐treated groups, respectively.
In order to assess the metabolic impact of MEDI0382, an IPGTT was performed on day 21 (Figure 4C). Glucose tolerance was significantly improved (p < .001) in all treatment groups with glucose AUC's of 2339, 1294, 1281, and 1442 mmol/L*min for vehicle, 10 nmol/kg MEDI0382, 30 nmol/kg MEDI0382 and 40 nmol/kg liraglutide, respectively. Furthermore, blood glucose levels measured prior to the glucose challenge (t = 0 minute) were also reduced in all treatment groups (p < .05). Improvements in several metabolic parameters measured at the end of the study, e.g. fat mass, liver triglycerides and plasma biomarkers such as fasting insulin, glucose and alanine aminotransferase (ALT), were noted following repeated treatment with MEDI0382 or liraglutide (Figures S4–S6, Supporting Information).
3.5. Energy expenditure in DIO mice
DIO mice were dosed daily for 3 weeks with either vehicle or MEDI0382 (10 nmol/kg) and an additional group of vehicle‐treated mice were pair‐fed with the same amount of food consumed by the MEDI0382‐treated group in order to determine the impact of MEDI0382 administration on metabolic rate by indirect calorimetry assessment during the last 4 days of treatment (Figure 5). Food intake of MEDI0382‐treated mice on day 0‐16 was significantly reduced by 19% relative to vehicle controls and was matched in the pair‐fed group (17% lower compared to vehicle, both p < .001 compared to vehicle; data not shown). Body weight was equally reduced by MEDI0382 and pair‐feeding relative to vehicle controls through the first 10 days of treatment, whereupon the body weights of pair‐fed mice stabilized while MEDI0382‐treated mice continued to lose weight to a greater extent compared to pair‐fed mice (Figure 5A). MEDI0382 administration was associated with significantly reduced fat mass relative to vehicle and pair‐fed groups (p < .05), whereas fat‐free mass was not different between groups (data not shown). Indirect calorimetry revealed that VO2 (rate of oxygen consumption, a proxy for metabolic rate) was elevated in mice dosed with MEDI0382 (Figure 5B). Mean 24 hours VO2 was significantly higher (by 22%, p < .05) relative to vehicle controls, but did not quite reach statistical significance compared to pair‐fed controls (15% increase, p = .077, Figure 5C). The respiratory exchange ratio (RER) was low overall (Figure 5D), as expected for mice on high‐fat diet when fat utilization is forced by the diet. Nevertheless, MEDI0382‐treated mice exhibited significantly lower mean 24 hours RER relative to vehicle controls (p < .05), with a strong trend apparent when compared to pair‐fed mice (p = .064; Figure 5E). Analysis of ambulatory activity showed no significant difference in total distance travelled between groups (data not shown).
Figure 5.
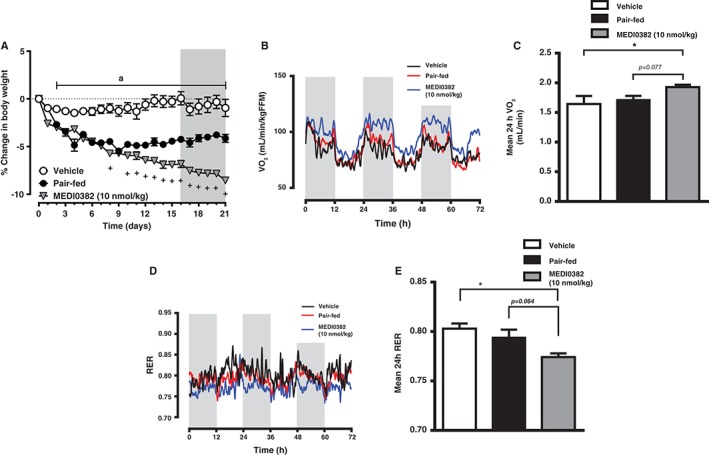
Body weight and energy expenditure parameters in male DIO mice after repeated daily s.c. administration of vehicle (ad libitum or pair‐fed) or MEDI0382 (10 nmol/kg). Change in body weight of mice expressed as percent change from baseline (A). Shaded area from day 16‐21 indicates period when mice were subjected to indirect calorimetry measurement. Diurnal (B) and 24 hours (C) mean rate of oxygen consumption (VO2) normalized to fat‐free mass over 3‐day recording period. Diurnal (D) and mean 24 hours (E) respiratory exchange ratio (RER). Data in (B) and (D) are smoothed to more clearly show circadian patterns. Shaded areas in (B) and (D) represent lights off period. ap < .05 for vehicle compared to both pair‐fed and MEDI0382 groups, +p < .05 MEDI0382 compared to pair‐fed, *p < .05 compared to vehicle.
3.6. Repeated administration of MEDI0382 reduces body weight in cynomolgus monkeys
The terminal plasma half‐life of MEDI0382 in cynomolgus monkeys after s.c. administration was measured in a separate single‐dose study and ranged from 5 to 8 hours (data not shown). The body weights of healthy cynomolgus monkeys following daily s.c. administration of MEDI0382 at dose levels of 8, 16 or 27 nmol/kg/d for 8 weeks reduced in a dose‐dependent fashion (Figure 6). The mean body weights of control animals increased by 9% (males) and 10% (females) of starting body weight over the course of the 8‐week study, whereas the mean body weight loss (with range for individual animals in parentheses) on the last day of dosing (Day 57) was 5% (7% to 4%), 7% (12% to 2%) and 11% (16% to 8%) of starting body weight for males and 5% (9% to 2%), 11% (22% to 4%) and 13% (20% to 6%) of starting body weight for females at dose levels of 8, 16 or 27 nmol/kg/d, respectively. Significant body weight loss compared to vehicle‐treated animals occurred mainly during the first 2 weeks (p < .05—p < .001) and thereafter stabilized for the remainder of the treatment period. Bodyweight loss was not associated with any adverse clinical signs or vomiting in monkeys except for one female animal dosed at 27 nmol/kg/d in the 8‐week study which was killed prematurely on Day 36 because of poor clinical condition (subdued, thin and dehydrated with multiple skin lesions) which was considered to be related to significant weight loss in this individual (up to 20% of starting bodyweight).
Figure 6.
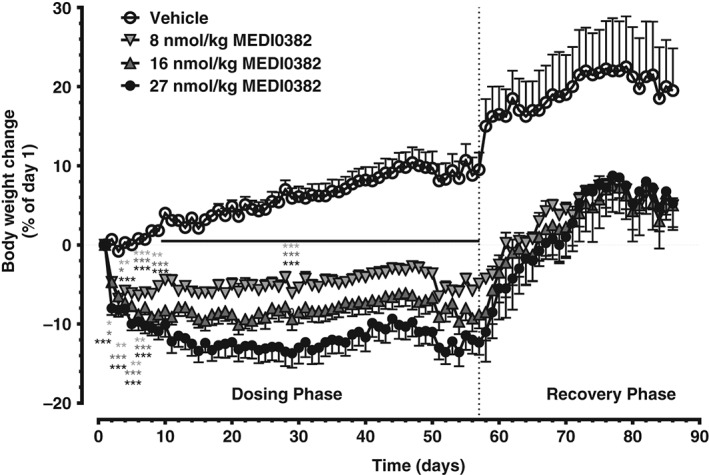
Effect of MEDI0382 on body weight in cynomolgus monkeys after repeated daily administration of vehicle or MEDI0382 (8, 16 or 27 nmol/kg/d; 30, 60 or 100 µg/kg/d, s.c.) for 57 days. Values are mean (± SD); n = 5 male and 5 female animals/group during dosing phase; n = 2 male and 2 female animals/group during recovery phase. *p < .05; **p < .01; ***p < .001 compared to vehicle.
Body weights generally returned to pretreatment values within approximately 2 weeks of cessation of treatment in all groups, but it is interesting to note that the body weights in all treated groups stabilized at a lower level than that in controls during the last 2 weeks of the treatment‐free period. The changes in bodyweight were associated with concomitant reductions in food consumption (Figure S7E, Supporting Information).
Fasting blood glucose and plasma insulin were measured in monkeys in a separate study following daily s.c. administration of MEDI0382 at dose levels of 0, 4, 8 or 27 nmol/kg/d for 29 days, but there were no notable changes in either parameter compared with baseline throughout the course of the study (Figure S7A–D, Supporting Information).
4. DISCUSSION
Approximately 80%‐90% of individuals with type 2 diabetes (T2D) are overweight or obese27 and weight reduction by diet or drug intervention is associated with reduced blood glucose levels and reduced cardiovascular risk factors.28, 29, 30 GLP‐1 analogues have been shown to improve glycaemic control and lead to reduction in insulin dose in T2D subjects.31, 32, 33 GLP‐1 agonists also reduce body weight in animal models of obesity and induce modest weight loss in humans.4, 34, 35, 36 The GLP‐1 analogue Saxenda (liraglutide 3 mg), which was approved in 2014 for treatment of obese adults with weight‐related health conditions, has been shown in clinical trials to reduce body weight by 5%–6% versus placebo after 56 weeks.3, 5 MEDI0382 was designed to achieve glucose control equivalent to that of liraglutide by virtue of its GLP‐1 receptor agonist activity, and shows broadly similar potency to liraglutide in human and rat pancreatic β‐cell lines and in recombinant GLP‐1 receptor cell lines (Table 1). The GLP‐1 activity was complemented with glucagon receptor agonist activity in a single peptide to harness the additional effects of glucagon on energy intake, metabolic rate and energy expenditure and hence further reduce bodyweight.37, 38, 39, 40
This is the first report of the use of more physiologically relevant endogenous receptor systems, namely human and rodent pancreatic β‐cells and hepatocytes, utilizing both second messenger (cAMP) and functional (GSIS and HGO) endpoints for characterization of the potency profile of a dual GLP‐1/glucagon receptor agonist. The potencies in cAMP and biological effect assays at endogenous receptor systems are right shifted in comparison to transfected cell lines for all molecules tested, presumably because of increased receptor reserve resulting from receptor overexpression in transfected receptor systems (Table 1). The relative potency of MEDI0382 in the human and rat pancreatic β‐cell lines and hepatocytes when compared to GLP‐1, glucagon, oxyntomodulin and liraglutide was, however, similar to that observed in transfected cell lines (Table 1). The balance of activity at the GLP‐1 and glucagon receptors in humans, determined using transfected cell lines, was confirmed to be biased approximately fivefold towards GLP‐1 receptor activation versus glucagon receptor activation (Table 2) and is predicted to provide significant weight loss and glucose control in humans by systems biology modelling of clinical glucagon and GLP‐1 data (data not shown). Furthermore, the balance of agonistic activity at each receptor is preserved across species (Table 2), thus allowing its pharmacology to be evaluated in different preclinical models.
MEDI0382 elicited profound bodyweight loss in DIO mice and healthy cynomolgus monkeys, and this is the first time that bodyweight reductions have been shown to translate from rodents to non‐human primates for a dual GLP‐1/glucagon receptor agonist. In monkeys, significant and sustained dose‐dependent body weight loss (of up to c. 20% compared to starting body weight in individual animals dosed s.c. at 27 nmol/kg for 8 weeks) was observed, which is remarkable considering that these animals were young, lean and still growing. Interestingly, bodyweights of monkeys treated with MEDI0382 appeared to stabilize at a lower level than did control animals during the last 2 weeks of the treatment‐free period in all treated groups (Figure 6), which suggests that treated animals do not attain the same bodyweights as control animals, even after cessation of treatment, and opens up the intriguing possibility that the threshold at which bodyweight is maintained has been re‐set.
In DIO mice, mean body weights decreased by up to 30% compared with starting bodyweight after daily s.c. administration of MEDI0382 for 4 weeks at a dose level of 30 nmol/kg. Liraglutide was used as a comparator to facilitate translation to the clinic, since it is currently the only incretin‐based therapy to have been approved for treatment of obesity. At the end of the treatment period, mean body weight loss in mice treated at the lowest dose level of MEDI0382 (10 nmol/kg) was similar to that in animals dosed with liraglutide at 40 nmol/kg (21% reduction compared to starting bodyweight in both cases). This facilitates a direct comparison of the two interventions without having to account for any differential effects of weight loss on metabolic parameters (Figures S4–S6, Supporting Information). The dose of liraglutide (40 nmol/kg) was chosen to mimic steady state unbound plasma drug exposure in humans at the approved dose level of 3 mg 26, 41, which is also tolerable in terms of nausea and vomiting in humans. It was not possible to compare higher doses of liraglutide and MEDI0382 since the bodyweight loss caused by higher doses of MEDI0382 would have exceeded the ethical limits set by the project license under which the studies were conducted. It is interesting to note that the average steady state plasma exposure (Css, average) at which similar bodyweight loss was achieved was notably lower for MEDI0382 at a dose level of 10 nmol/kg (12.3 nM) than for liraglutide (93 nM) and this provides indirect support for the contribution of glucagon receptor agonism to the weight loss effects observed with MEDI0382. The MEDI0382 exposures are >10‐fold greater than the EC50 determined in the mouse hepatocyte cAMP potency assay in vitro. Furthermore, the rate of decline in bodyweight of liraglutide‐treated animals decreased after approximately 14 days, whereas MEDI0382‐treated animals continued to lose weight throughout the 28‐day period (Figure 4A). Weight loss was accompanied by a significant reduction in body fat in the subcutaneous and visceral fat compartments (Figure S4, Supporting Information). Plasma parameters measured at the end of treatment in DIO mice showed an increase in β‐hydroxybutyrate and kisspeptin levels (Figure S6, Supporting Information), suggesting a glucagon receptor activation‐driven increase in fatty acid oxidation.42, 43 The reduction in bodyweight also corresponded with a reduction in food intake by mice and monkeys. Although weight loss in DIO mice at a dose level of MEDI0382 of 10 nmol/kg was similar to that in animals dosed with liraglutide (40 nmol/kg), the reduction in food intake was less for MEDI0382 (Figure 4B), which suggests that the additional weight loss induced by MEDI0382 was unlikely to be related to increased nausea or taste aversion. Moreover, there were no differences in locomotor activity after repeated dosing of DIO mice with MEDI0382 compared to vehicle‐treated animals in the energy expenditure study. Since the additional weight loss evoked by MEDI0382 was not just a consequence of reduced food intake, other glucagon receptor‐driven mechanisms, such as increased energy expenditure,40 were considered likely to contribute to this effect.
This proposal is supported by evidence from a separate indirect calorimetry study conducted in DIO mice, illustrating that MEDI0382‐treated mice exhibit greater bodyweight loss compared with both vehicle‐treated and pair‐fed control mice treated with vehicle. This result suggests that the effects of MEDI0382 on bodyweight are not simply a reflection of reduced energy intake, but are also due to increased energy expenditure (Figure 5). MEDI0382‐treated mice exhibited higher oxygen consumption (VO2), suggesting enhanced metabolic rate, and a lower respiratory exchange ratio (RER), indicative of preferential fat oxidation, compared with vehicle and pair‐fed controls, which may be responsible for the greater bodyweight loss produced by MEDI0382 compared to pair‐fed controls. In this context, oxyntomodulin and its analogues, but not GLP‐1 analogues, have been reported to increase energy expenditure in rodents and overweight humans, and these activities contribute to the weight loss effects.11, 16, 44, 45, 46, 47, 48 Further support for the role of glucagon receptor activation by MEDI0382, in energy expenditure as compared with energy intake, comes from studies measuring the acute effects of MEDI0382, liraglutide and IUB288 (a selective glucagon agonist) in GLP‐1R KO mice and wild‐type C57Bl/6 littermates (Figure S3, Supporting Information). MEDI0382 and liraglutide robustly reduced food intake and improved glucose tolerance in wild type mice (Figure S3B and D, Supporting Information), but these effects were abolished in GLP‐1R KO mice (Figure S3A and C, Supporting Information). IUB288 also failed to reduce food intake in both GLP‐1R KO mice and wild type littermates (Figure S3A and B, Supporting Information). Similarly, in 2‐hours fasted wildtype mice dosed with somatostatin to prevent release of pancreatic glucagon and insulin, glucose control was also driven by GLP‐1 receptor activation (Figure S3F, Supporting Information). In contrast, in GLP‐1 receptor KO mice dosed with somatostatin, MEDI0382 elicited a robust maximal elevation in glucose levels at the lowest dose tested (3 nmol/kg; Figure S3E, Supporting Information). This was the same dose that elicited robust glucose tolerance in an IPGTT in wildtype mice. Thus, the energy intake and glucose control effects of MEDI0382 are driven mainly by GLP‐1 receptor activation, whereas blood glucose elevation (in the absence of GLP‐1 receptors) and energy expenditure appear to be mediated by glucagon receptor activation. Taken together, our results indicate that MEDI0382 possesses a strong glucagon receptor tone that is counterbalanced by the GLP‐1 activity of the peptide.
It is interesting to note that the reduction in food intake appeared to parallel body weight loss in cynomolgus monkeys and both stabilized at a lower level after approximately 2 weeks of treatment (Figures 6 and S7E, Supporting Information) whereas in DIO mice, food intake normalized after approximately 9 days although bodyweights continued to decline (Figure 4A and B). It is possible that an increase in food intake in monkeys was obscured by an increased intake of supplementary food (bread, biscuits and/or malt loaf with jam and peanut butter) which was provided to help minimize weight loss because this was more palatable than pelleted diet and the amount of supplementary food eaten was not recorded. It remains to be seen whether the food‐intake and weight‐loss profile of MEDI0382 in humans translates best to DIO rodents or lean monkeys.
MEDI0382 suppressed the acute rise in plasma glucose after glucose challenge in DIO mice after a single dose, or after repeated daily dosing for 21 days (Figures 3B and 4C). The effect of MEDI0382 was similar to that of liraglutide (40 nmol/kg) at both dose levels (10 and 30 nmol/kg) after repeated dosing. There was a significant reduction in plasma insulin after repeated dosing with MEDI0382 or liraglutide for 21 days (Figure S6A, Supporting Information) which is probably a consequence of improved insulin sensitivity induced by weight loss. A significant reduction in fat mass, accompanied by a profound improvement in liver health, characterized by decreased liver triglyceride content and plasma ALT levels (Figures S4 and S5, Supporting Information), was observed after repeated dosing and this is likely to contribute to increased insulin‐mediated glucose disposal in fat and liver. The effects of MEDI0382 on glycaemic control after repeated dosing are therefore in line with those of liraglutide and are consistent with the similar GLP‐1 receptor potency of the two molecules. The fact that there were no notable changes in plasma glucose or insulin in healthy monkeys suggests that the balance of activities at GLP‐1 and glucagon receptors is optimal for both glycaemic control and safety.
In summary, MEDI0382 is a novel dual GLP‐1/glucagon receptor peptide agonist with a balance of agonism at GLP‐1 and glucagon receptors which provides glycaemic control following glucose challenge in obese insulin‐resistant mice and in lean mice, without having an adverse effect on resting glucose levels. The key differentiator from GLP‐1 agonists such as liraglutide is the enhanced weight loss which is a consequence of the glucagon component and has been demonstrated in DIO mice and, for the first time, in non‐human primates. MEDI0382 has recently entered clinical development to evaluate its potential as a treatment for overweight or obese individuals with T2D.19
Supporting information
Figure S1‐S7. Supporting Information ‐ Figures
ACKNOWLEDGMENTS
We would like to thank Caroline Brennan (Envigo, UK), David Fairman (formerly of MedImmune, now at GSK) and Lolke de Haan (MedImmune) for assistance with the design and conduct of in vivo studies, Jacqueline Metcalfe and Nicole Burmeister (MedImmune) for assistance with peptide synthesis and Andie Collinson (MedImmune) for conduct of in vitro screening assays. We also wish to thank Louise Lantier and Staci Bordash (Mouse Metabolic Phenotypic Center, Vanderbilt University) for assistance in performing the indirect calorimetry study and Lambertus Benthem (formerly of AstraZeneca) for input into the pharmacology package used to help select MEDI0382 for further development.
Financial Disclosure
There was no external funding source for this study.
Author contributions
M.A.B designed MEDI0382. R.J., M.A.B, D.C.H. and R.J.‐L defined the optimal in vitro properties for the peptide. D.C.H., A.K., N.B., H.S., A.R. and J.N. were responsible for the design and conduct of in vitro potency experiments. G.H., M.F. and T.K. helped design, conduct and analyse the DIO mouse study. S.W. and S.O. helped design, conduct and analyse acute studies in GLP‐1R KO mice. A.K., J.L.T., M.F.‐F., R.J.‐L., L.J., J.G., S.J.H., and M.P.C. were responsible for design, data analysis and/or interpretation of in vivo studies. S.J.H., A.K., D.C.H., J.L.T., M.F.‐F., R.J.‐L., R.J., L.J., C.R. and M.P.C. contributed to and reviewed the manuscript.
Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, Jansson‐Löfmark R, Naylor J, Rossi A, Bednarek MA, Bhagroo N, Salari H, Will S, Oldham S, Hansen G, Feigh M, Klein T, Grimsby J, Maguire S, Jermutus L, Rondinone CM and Coghlan MP. Robust anti‐obesity and metabolic effects of a dual GLP‐1/glucagon receptor peptide agonist in rodents and non‐human primates, Diabetes Obes Metab 2016, 18, 1176–1190. DOI:10.1111/dom.12735
Conflict of Interest: All authors are employees of MedImmune, AstraZeneca or Gubra.
REFERENCES
- 1. Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819–837. [DOI] [PubMed] [Google Scholar]
- 2. Uccellatore A, Genovese S, Dicembrini I, Mannucci E, Ceriello A. Comparison review of short‐acting and long‐acting glucagon‐like peptide‐1 receptor agonists. Diabetes Ther. 2015;6:239–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wadden TA, Hollander P, Klein S, et al. Weight maintenance and additional weight loss with liraglutide after low‐calorie‐diet‐induced weight loss: the SCALE maintenance randomized study. Int J Obes. 2013;37:1443–1451. [DOI] [PubMed] [Google Scholar]
- 4. Potts JE, Gray LJ, Brady EM, Khunti K, Davies MJ, Bodicoat DH. The effect of glucagon‐like peptide 1 receptor agonists on weight loss in type 2 diabetes: a systematic review and mixed treatment comparison meta‐analysis. PLoS One. 2015;10:e0126769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pi‐Sunyer X, Astrup A, Fujioka K, et al. SCALE obesity and prediabetes NN8022‐1839 study group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11–22. [DOI] [PubMed] [Google Scholar]
- 6. de Mello AH, Prá M, Cardoso LC, de Bona Schraiber R, Rezin GT. Incretin‐based therapies for obesity treatment. Metabolism. 2015;64:967–981. [DOI] [PubMed] [Google Scholar]
- 7. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effects of glucagon on food intake and body weight in man. J Appl Physiol. 1957;11:419–421. [DOI] [PubMed] [Google Scholar]
- 9. Geary N, Le Sauter J, Noh U. Glucagon acts in the liver to control spontaneous meal size in rats. Am J Physiol. 1993;264:R116–R122. [DOI] [PubMed] [Google Scholar]
- 10. Cohen MA, Ellis SM, Le Roux CW, et al. Oxyntomodulin suppresses appetite and reduces food intake in humans. J Clin Endocrinol Metab. 2003;88:4696–4701. [DOI] [PubMed] [Google Scholar]
- 11. Wynne K, Park AJ, Small CJ, et al. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double‐blind, randomized, controlled trial. Diabetes. 2005;54:2390–2395. [DOI] [PubMed] [Google Scholar]
- 12. Wynne K, Park AJ, Small CJ, et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes. 2006;30:1729–1736. [DOI] [PubMed] [Google Scholar]
- 13. Pocai A. Unraveling oxyntomodulin, GLP1's enigmatic brother. J Endocrinol. 2012;215:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsbøll T. Effect of oxyntomodulin, glucagon, GLP‐1 and combined glucagon + GLP‐1 infusion on food intake, appetite and resting energy expenditure. J Clin Endocrinol Metab. 2015;100:4541–4552. [DOI] [PubMed] [Google Scholar]
- 15. Schjoldager BT, Baldissera FG, Mortensen PE, Holst JJ, Christiansen J. Oxyntomodulin: a potential hormone from the distal gut. Pharmacokinetics and effects on gastric acid and insulin secretion in man. Eur J Clin Invest. 1988;18:499–503. [DOI] [PubMed] [Google Scholar]
- 16. Day JW, Ottaway N, Patterson JT, et al. A new glucagon and GLP‐1 co‐agonist eliminates obesity in rodent. Nat Chem Biol. 2009;5:749–757. [DOI] [PubMed] [Google Scholar]
- 17. Pocai A, Carrington PE, Adams JR, et al. Glucagon‐like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Day JW, Gelfanov V, Smiley DO, et al. Optimization of co‐agonism at GLP‐1 and glucagon receptors to safely maximize weight reduction in DIO‐rodents. Biopolymers. 2012;98:443–450. [DOI] [PubMed] [Google Scholar]
- 19. A multiple‐ascending‐dose study to evaluate the efficacy, safety, and pharmacokinetics (PK) of MEDI0382 in overweight and obese subjects with type 2 diabetes. NCT02548585. https://clinicaltrials.gov/ct2/show/NCT02548585. Accessed July 19, 2016.
- 20. Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon‐like peptide‐1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–1669. [DOI] [PubMed] [Google Scholar]
- 21. Havelund S, Plum A, Ribel U, et al. The mechanism of protraction of insulin detemir, a long‐acting, acylated analog of human insulin. Pharm Res. 2004;21:1498–1504. [DOI] [PubMed] [Google Scholar]
- 22. Butler R, Hornigold DC, Huang L, et al. Use of the site specific retargeting Jump‐In™ platform cell line to support biologic drug discovery. J Biomol Screen. 2015;20:528–535. [DOI] [PubMed] [Google Scholar]
- 23. Naylor J, Rossi A, Hornigold DC. Acoustic dispensing preserves the potency of therapeutic peptides throughout the entire drug discovery workflow. J Lab Autom. 2016;21:90–96. [DOI] [PubMed] [Google Scholar]
- 24. Habegger KM, Stemmer K, Cheng C, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62:1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baldissera FG, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV. Oxyntomodulin (glicentin‐(33‐69)): pharmacokinetics, binding to liver cell membranes, effects on isolated perfused pig pancreas, and secretion from isolated perfused lower small intestine of pigs. Regul Pept. 1988;21:151–166. [DOI] [PubMed] [Google Scholar]
- 26. Center for Drug Evaluation and Research, United States Food and Drug Administration . Victoza pharmacology review. 2009; NDA 22–341.
- 27. Eberhardt MS, Ogden C, Engelgau M, Cadwell B, Hedley AA, Saydah SH. Prevalence of overweight and obesity among adults with diagnosed diabetes‐‐United States, 1988‐1994 and 1999‐2002, Centers for Disease Control and Prevention (CDC). MMWR Morb Mortal Wkly Rep. 2004;53:1066–1068. [PubMed] [Google Scholar]
- 28. Elhayany A, Lustman A, Abel R, Attal‐Singer J, Vinker S. A low carbohydrate Mediterranean diet improves cardiovascular risk factors and diabetes control among overweight patients with type 2 diabetes mellitus: a 1‐year prospective randomized intervention study. Diabetes Obes Metab. 2009;12:204–209. [DOI] [PubMed] [Google Scholar]
- 29. Blonde L, Pencek R, MacConell L. Association among weight change, glycemic control, and markers of cardiovascular risk with exenatide once weekly: a pooled analysis of patients with type 2 diabetes. Cardiovasc Diabetol. 2015;14:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorber D. GLP‐1 receptor agonists: effects on cardiovascular risk reduction. Cardiovasc Ther. 2013;31:238–249. [DOI] [PubMed] [Google Scholar]
- 31. Diamant M, Nauck MA, Shaginian R, et al. Glucagon‐like peptide‐1 receptor agonist or bolus insulin with optimized basal insulin in diabetes. Diabetes Care. 2014;37:2763–2773. [DOI] [PubMed] [Google Scholar]
- 32. van Velsen EF, Lamers J, Blok V, van Leendert RJ, Kiewiet‐Kemper RM. A prospective study of concomitant GLP‐1 analogue and insulin use in type 2 diabetes in clinical practice. Neth J Med. 2014;72:523–527. [PubMed] [Google Scholar]
- 33. de Wit HM, Vervoort GMM, Jansen HJ, de Grauw WJC, de Galan BE, Tack CJ. Liraglutide reverses pronounced insulin‐associated weight gain, improves glycaemic control and decreases insulin dose in patients with type 2 diabetes: a 26 week, randomised clinical trial (ELEGANT). Diabetologia. 2014;57:1812–1819. [DOI] [PubMed] [Google Scholar]
- 34. Young AA, Gedulin BR, Bhavsar S, et al. Glucose‐lowering and insulin‐sensitizing actions of exendin‐4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes. 1999;48:1026–1034. [DOI] [PubMed] [Google Scholar]
- 35. Davies M, Heller S, Sreenan S, et al. Once‐weekly exenatide versus once‐ or twice‐daily insulin detemir: randomized, open‐label, clinical trial of efficacy and safety in patients with type 2 diabetes treated with metformin alone or in combination with sulfonylureas. Diabetes Care. 2013;36:1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon‐like peptide‐1 receptor agonists on weight loss: systematic review and meta‐analyses of randomised controlled trials. BMJ. 2012;344:d7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nair KS. Hyperglucagonemia increases resting metabolic rate in man during insulin deficiency. J Clin Endocrinol Metab. 1987;64:896–901. [DOI] [PubMed] [Google Scholar]
- 38. Tan TM, Field BC, McCullough KA, et al. Coadministration of glucagon‐like peptide‐1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62:1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cegla J, Troke RC, Jones B, et al. Coinfusion of low‐dose GLP‐1 and glucagon in man results in a reduction in food intake. Diabetes. 2014;63:3711–3720. [DOI] [PubMed] [Google Scholar]
- 40. Salem V, Izzi‐Engbeaya C, Coello C, et al. Glucagon increases energy expenditure independently of brown adipose tissue activation in humans. Diabetes Obes Metab. 2016;18:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Center for Drug Evaluation and Research, United States Food and Drug Administration . Liraglutide injection, 3 mg briefing document. 2014; NDA 206321.
- 42. Liu W, Beck BH, Vaidya KS, et al. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 2014;74:954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song WJ, Mondal P, Wolfe A, et al. Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. 2014;19:667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dakin CL, Small CJ, Batterham RL, et al. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology. 2004;145:2687–2695. [DOI] [PubMed] [Google Scholar]
- 45. Druce MR, Bloom SR. Oxyntomodulin: a novel potential treatment for obesity. Treat Endocrinol. 2006;5:265–272. [DOI] [PubMed] [Google Scholar]
- 46. Harder H, Nielsen L, Tu DT, Astrup A. The effect of liraglutide, a long‐acting glucagon‐like peptide 1 derivative, on glycemic control, body composition, and 24‐h energy expenditure in patients with type 2 diabetes. Diabetes Care. 2004;27:1915–1921. [DOI] [PubMed] [Google Scholar]
- 47. van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WH. Effects of the once‐daily GLP‐1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non‐diabetic adults. Int J Obes. 2014;38:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long‐acting glucagon‐like peptide‐1 analog, reduces body weight and food intake in obese candy‐fed rats, whereas a dipeptidyl peptidase‐IV inhibitor, vildagliptin, does not. Diabetes. 2007;56:8–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S7. Supporting Information ‐ Figures