

ABSTRACT
As whole genome sequencing becomes cheaper and faster, it will progressively substitute targeted next‐generation sequencing as standard practice in research and diagnostics. However, computing cost–performance ratio is not advancing at an equivalent rate. Therefore, it is essential to evaluate the robustness of the variant detection process taking into account the computing resources required. We have benchmarked six combinations of state‐of‐the‐art read aligners (BWA‐MEM and GEM3) and variant callers (FreeBayes, GATK HaplotypeCaller, SAMtools) on whole genome and whole exome sequencing data from the NA12878 human sample. Results have been compared between them and against the NIST Genome in a Bottle (GIAB) variants reference dataset. We report differences in speed of up to 20 times in some steps of the process and have observed that SNV, and to a lesser extent InDel, detection is highly consistent in 70% of the genome. SNV, and especially InDel, detection is less reliable in 20% of the genome, and almost unfeasible in the remaining 10%. These findings will aid in choosing the appropriate tools bearing in mind objectives, workload, and computing infrastructure available.
Keywords: whole genome sequencing, whole exome sequencing, NGS, NA12878, alignment, variant calling, bioinformatics, computing speed, benchmark
Introduction
As the price of whole exome and whole genome sequencing (WES and WGS) has dropped steeply in recent years, there has been a move within the clinical genetics community toward use of this technology in aiding, and confirming, diagnosis, particularly with regards to rare disease cases. Indeed, WES in particular has been shown to be effective in solving rare disease cases where initial screening of panels of candidate genes proved fruitless, with a typical success rate reported of ∼25% [Yang et al., 2014; Sawyer et al., 2015; Wright et al., 2015]. Given that this number is only representative of hard‐to‐solve cases, it is likely that the real figure, were WES to be used ab initio for genetic analysis, would be ∼50%–60%, since WES will identify nearly everything ascertained using a custom panel.
However, there are still some barriers preventing the wide‐scale adoption of this technology in a clinical diagnostic setting [Biesecker and Green, 2014; Dewey et al., 2014; Goldfeder et al., 2016]. In particular, WGS is still relatively expensive to sequence and analyze, though it will progressively substitute targeted sequencing in research and clinical diagnostics over time since it has the added advantage of allowing better identification of larger chromosomal events such as structural variants. Sequencing costs have dropped dramatically in recent years and systems such as the Illumina HiSeq X‐Ten can sequence up to 18,000 human genomes per year at an advertised cost of $1,000 per genome. However, the computing cost‐performance ratio is not dropping at the same rate, with computing performance only doubling approximately every 2 years according to Moore's law. As few labs have the capability to sequence, process, store, and interpret these large volumes of data, the optimal choice of tools and pipelines will impact strongly upon the computing resources required, even for smaller setups. Furthermore, there are still many parts of the genome that remain difficult to interrogate correctly using short‐read (100–150nt) sequencing because of the ubiquity of repetitive regions throughout the human genome. WES on the other hand, although more affordable and requiring less computing resources, suffers somewhat from uneven depth of coverage of targeted regions due to the target capture and PCR amplification steps. This means that successful variant detection in such regions is less assured.
Massively parallel short‐read sequencing on Illumina platforms typically results in the production of ∼40–400 million reads per exome or genome, respectively. However, this is just the first step toward obtaining biologically or medically meaningful results. This large volume of reads needs to undergo quality control, before being aligned to a reference genome. These alignments can then be interrogated for variant events, that is, positions where the sample sequenced differs from the reference genome sequence, using a range of bioinformatics tools, depending on the nature of the event of interest. As with traditional biology experiments, use of different tools and parameters at any step may result in different outcomes in terms of variant events identified. Thus, it is important to be aware of the qualities, limitations, and any inherent biases of the tools applied. Furthermore, in the context of large‐scale sequencing, the associated computational costs of analyzing such vast amounts of raw data must be taken into account.
As a result, there have been initiatives in recent years to assess the reliability of WGS and WES pipelines [Warden et al., 2014; Zook et al., 2014; Cornish and Guda, 2015; Highnam et al., 2015; Hwang et al., 2015], though the majority of benchmarking comparisons have focused solely on WES data. As tool development and improvement remains a very active area of research in this field, it is essential to keep abreast of the latest developments. Here, we detail a benchmark of all combinations of two alignment tools, BWA‐MEM [Li, 2013], and GEM3 (Marco‐Sola et al., manuscript in preparation), and three popular variant‐calling algorithms, FreeBayes [Garrison and Marth, 2012], GATK HaplotypeCaller [DePristo et al., 2011], and SAMtools [Li, 2011]. We have used publicly available WGS data (∼50× mean coverage) for the HapMap sample NA12878, and performed WES (∼90× mean coverage) on DNA from a reference immortalized cell line from NA12878. All steps of each benchmark have been run in parallel, using built‐in multi‐threading options when supported by the relevant tool, or through running multiple instances of each tool in parallel on smaller chunks of the input data when threading is not supported. We provide details of the computational costs incurred in each step, and compare the accuracy of the resulting variant calls with a high‐confidence whole genome reference call set for NA12878 compiled by the US National Institute for Science and Technology (NIST) and the Genome in a bottle (GIAB) consortium [Zook et al., 2014]. We find substantial differences in terms of computational costs between tools that have been designed to perform similar tasks, and some differences in the quality of the end results in terms of accuracy of variant detection. Furthermore, we have explored the relationship between the regions defined as reliably callable by Zook et al. [2014], the regions of the genome which are potentially uniquely mappable, and the actual coverage obtained in these regions using BWA‐MEM and GEM3. Finally, we have also studied the concordance of the variants identified by the three callers in the reliably callable and non‐reliably callable regions of the genome and the exome.
Material and Methods
Source of Raw WGS Reads
Reads corresponding to approximately 50× coverage for the HapMap sample NA12878 were downloaded from the European Nucleotide archive (www.ebi.ac.uk/ena/data/view/ERP001229). These FASTQ files, consisting of 101nt read pairs were generated by Illumina Inc. (Cambridge, UK) on four separate flow cells using a HiSeq2000 instrument, and released as part of the Platinum Genomes (http://www.illumina.com/platinumgenomes/).
WES Library Preparation and Sequencing
WES was performed at the Centro Nacional de Análisis Genómico (CNAG‐CRG, Barcelona, Spain) using an immortalized cell‐line sample from NA12878, obtained from the collection at NIGMS at Coriell Institute for Medical Research. Whole exome enrichment was undertaken with the SeqCap EZ MedExome Target Enrichment Kit (Roche NimbleGen, Madison, WI, US) following the manufacturer's protocol (version 5.1). Pre‐capture multiplexing was applied. Briefly, 100 ng of genomic DNA was fragmented with Covaris™ E210 and used for ligation of adapters containing Illumina‐specific indices with a KAPA DNA Library Preparation kit (Kapa Biosystems). Adapter ligated DNA fragments were enriched through nine cycles of pre‐capture PCR using KAPA HiFi HotStart ReadyMix (2×) (Kapa Biosystems, Wilmington, MA, US) and analyzed on an Agilent 2100 Bioanalyzer with the DNA 7500 assay. Sample NA12878 was pre‐capture pooled with two other libraries with a combined mass of 1,250 ng for the bait hybridization step (47ºC, 16 hr). After washes, the multiplexed captured library was recovered with capture beads and amplified with 14 cycles of post‐capture PCR using KAPA HiFi HotStart ReadyMix (2×). Size, concentration, and quality of the captured material were determined using an Agilent DNA 7500 chip. The success of the enrichment was measured by a qPCR SYBR Green assay on a Roche LightCycler® 480 Instrument evaluating one genomic locus with pre‐ and post‐captured material. The three library pool was sequenced on an Illumina HiSeq 2000 instrument in one sequencing lane following the manufacturer's protocol, with paired runs of 2 × 101 bp and 2 × 126 bp, to reach a median coverage of 90× for the ∼46.6 MB target region. Image analysis, base calling, and quality scoring of the run were performed using Illumina's Real Time Analysis software (RTA 1.13.48) and generation of FASTQ files performed by CASAVA.
Alignment and Variant Calling
Raw reads for both WGS and WES were mapped to a version of the human reference genome, GRCh37, which includes decoy sequence to improve the efficiency of read‐mapping (hs37d5), as used in the secondary phase of the 1000 genomes project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/). For the purposes of benchmarking alignment algorithm speed, we used a computing node equipped with 32 hardware threads, whereas for the purpose of benchmarking variant calling speed, we used a node equipped with 16 hardware threads (Supp. Table S1). We mapped all reads from each experiment with GEM3, an improved version of the GEM mapping algorithm [Marco‐Sola et al., 2012], and BWA‐MEM version 0.7.8 [Li, 2013]. Following alignment, we sorted the resulting BAM files, removed duplicate reads using PICARD (version 1.110; http://broadinstitute.github.io/picard), and performed indel realignment with GATK (version 3.3) to produce a master BAM for each experiment‐aligner combination (n = 4).
Each master BAM was subsequently used as input to recent versions of each of three variant calling algorithms, FreeBayes [Garrison and Marth, 2012] version 0.9.20, GATK [DePristo et al., 2011] HaplotypeCaller version 3.3, and SAMtools [Li, 2011] version 1.2. In the case of SAMtools, we tested two pairs of settings for variant calling, which we refer to here as normal and fast. The normal setting includes a probabilistic realignment step for the computation of base alignment quality, which is disabled in the fast mode. This step is expected to reduce false positive single nucleotide variant (SNV) calling due to misalignment. For FreeBayes, we used default settings for running parallel threads, and for HaplotypeCaller, we followed the up‐to‐date version of GATK best practices pipeline [Van der Auwera et al., 2013], but omitted their final variant quality score recalibration step (VQSR) for consistency, since a single WES experiment does not generate enough data for VQSR to be applicable. For the WES alignments, calling was performed as a single process since the quantity of input data is relatively small, whereas for the WGS samples, calling was separated by chromosome, in order to reduce the elapsed time required for variant calling.
Raw VCFs output by each of the variant calling pipelines contain large numbers of false‐positive variant calls. Thus, we discarded any variant positions for which the variant quality score (QUAL) reported was less than 30 to generate 16 final test call sets—eight for WGS and eight for WES. By definition, all positions that have a QUAL score in excess of 30 should have a greater than 99.9% probability of being a bona fide variant. We recorded elapsed time and CPU time required for each process to complete, and where a process was split into chunks, we report the sum of the individual chunks and the elapsed time required for the largest chunk to complete, since this will be rate‐determining. The exact commands used for all tools for alignment and variant calling are provided in Supp. Table S2.
Comparison with NIST Reference Variant Call Set
In order to validate the accuracy of SNV and short insertion and deletion variant (InDel) detection, we downloaded a publicly available set of reference calls for NA12878, generated by NIST/GIAB through the integration of call sets from a variety of pipelines [Zook et al., 2014]. To calculate specificity and sensitivity for each call set, we first compared calls within the ∼2.2 GB of the NA12878 genome which Zook et al. [2014] classified as being reliably callable, that is, excluding genomic regions containing known copy‐number and structural variants and simple repeats in this sample, which are problematic when mapping short reads (Supp. Table S3). Subsequently, we calculated mappability statistics for the whole genome based on paired‐end reads with an insert size of 300nt, allowing for two mismatches with respect to the reference, using the tool developed by Derrien et al. [2012], and further investigated the concordance of variant calls made by the three calling algorithms in the remaining ∼900 MB of the genome not included in the NIST reliably callable region. For WES variant call sets, we restricted the comparison to calls within the intersection of the NIST reliably callable regions and the target regions of the NimbleGen exome capture kit, representing ∼34.7 MB of genomic sequence. As coverage clearly impacts on the ability to correctly identify variants, we also investigated coverage in each of these regions using GATK's DepthOfCoverage tool, for a variety of minimum depths of coverage, while requiring a minimum mapping quality of 20 (Supp. Table S2). In order to normalize VCFs as far as possible, we ran the vcfallelicprimitives script from the VCFlib suite (https://github.com/vcflib/vcflib) as recommended by Zook et al. [2014], followed by left‐aligning and trimming variants, with GATK's LeftAlignAndTrimVariants tool (https://www.broadinstitute.org/gatk/guide/tooldocs/org_broadinstitute_gatk_tools_walkers_variantutils_LeftAlignAndTrimVariants.php), and used a custom perl script to normalize all genotype calls to either 0/1, 1/1, or 1/2. As the number of positions that are multi‐allelic for the alternative allele is insignificant (0.15% in NIST high‐confidence set) and full normalization of such positions is not trivial, we ignored them for this comparison. For each call set, we identified true positives as positions that were identical to the NIST reference data set at the level of chromosome, position, alternative allele observed, and genotype called, false positives as any call in our variant call sets that was absent from the NIST set, and false negatives as any call in the NIST set absent from our call set. Intersecting of datasets was performed using the BEDtools package [Quinlan, 2014] and Venn diagrams produced using the VennDiagram R package [Chen & Boutros, 2011].
Results and Discussion
Alignments, Mappability, Coverage, and Callability
Mapping metrics from the alignments generated by BWA‐MEM and GEM3 indicate that they perform quite similarly (Supp. Table S4). Both tools were able to align over 99% of the reads, independent of quality, though BWA‐MEM mapped a higher proportion of reads with high quality (i.e., most likely uniquely mapping reads) than did GEM3 for the WGS but not for the WES sample. This may explain the higher mismatch rate identified in the BWA‐MEM mappings in only the WGS data. As we would expect, the calculated insert size (distance between read1 and read2 including the length of the reads) was also very similar being slightly over 300 bp.
To estimate in which regions of the genome (or exome) it should be possible to reliably map reads, and confidently identify variants, we assessed the overlaps between the mappable and reliably callable regions of the genome and the exome and computed the actual coverage obtained by BWA‐MEM and GEM3 on these regions. Although according to our mappability metrics, ∼2.8 GB (90.04%) of the genome should be mappable, the NIST v2.18 dataset defined only ∼2.2 GB (70.91%) of the genome to be reliably callable (Supp. Table S5). Unsurprisingly, 99.91% of the reliably callable genome is mappable but, remarkably, 65.98% of the non‐reliably callable genome is also mappable. We identified similar trends in the exome, although the total length of the regions concerned are much shorter (Supp. Table S6).
Next, we computed the actual coverage on the full genome and on all combinations of mappable and reliably callable regions (Supp. Table S7). The overall mean coverage was 49.94 for BWA‐MEM and 49.22 for GEM3. In both cases, the mean coverage was around 5% points higher if computed only on the mappable, or only the reliably callable, regions of the genome. This figure only increased an additional ∼0.1 if regions which are both mappable and reliably callable are considered. The non‐reliably callable but mappable regions of the genome had above 50× coverage, whereas the non‐mappable regions of the genome only had a mean coverage of approximately 4×, and the value for the non‐reliably callable and non‐mappable regions was a little lower still. Although these metrics were also computed for the exome, results were not so conclusive because of the short length of the non‐mappable region (Supp. Table S8). These findings indicate that mappability provides a good approximation of the regions of the genome which will actually be well covered in the experiment, and also highlights that the definition of reliably callable used by NIST is particularly stringent.
We also assessed coverage and callability in regions of the medically interpretable genome (MIG), as defined by Patwardhan et al. [2015], representing a total of ∼11.7 MB of genome, of which 98.6% is targeted by the MedExome kit. Interestingly, only 78% of the MIG region is reliably callable according to NIST, suggesting that there are many medically relevant genes for which short‐read technology may not be sufficient for accurate variant identification. Nevertheless, our results clearly indicate that the MedExome kit does a good job of increasing coverage in these specific regions of interest, as the mean coverage increased to approximately ∼102× versus ∼84–88× for the non‐MIG regions of the MedExome (Supp. Table S8), indicating that this kit may be a good option for clinical applications. Comprehensive coverage and variant calling results on several samples captured with the MedExome kit will be published elsewhere.
Accuracy of Variant Calling
Variant calling results for WGS, split by event type, are shown in Table 1. Taking the NIST reference set as the ground truth, the numbers indicate there is no obvious “best” variant calling pipeline. In fact all pipelines tested here perform very well, particularly for SNVs where sensitivity ranged from 99.69% to 99.92% and specificity from 99.53% to 99.87% across pipelines, with 99% of SNVs being called by all three callers (Figs. 1 and 2). InDel events are known to be more problematic to align and call consistently [O'Rawe et al., 2013; Fang et al., 2014, Hasan et al., 2015], and we observe sensitivity ranging from 79.80% to 98.69%, specificity from 77.52% to 99.07%, and concordance across all three callers of only 62%–66%. The results on the exome showed similar trends, with very high sensitivity and specificity for SNVs and lower for InDels (Supp. Table S9).
Table 1.
Summary of Variant Calling for Eight Pipelines for the WGS Sample
| Dataset | Total calls | TP | FP | FN | Specificity | Sensitivity | F1 score |
|---|---|---|---|---|---|---|---|
| Whole genome SNVs | |||||||
| NIST v2.18 Gold Standard | 2,740,732 | ||||||
| BWA‐MEM‐MEM + FreeBayes | 2,744,545 | 2,738,200 | 6,345 | 2,532 | 0.99769 | 0.99908 | 0.99838 |
| BWA‐MEM + HaplotypeCaller | 2,748,582 | 2,738,426 | 10,156 | 2,306 | 0.99631 | 0.99916 | 0.99773 |
| BWA‐MEM + SAMtools fast | 2,748,866 | 2,738,489 | 10,377 | 2,243 | 0.99622 | 0.99918 | 0.99770 |
| BWA‐MEM + SAMtools normal | 2,736,410 | 2,732,882 | 3,528 | 7,850 | 0.99871 | 0.99714 | 0.99792 |
| GEM3 + FreeBayes | 2,742,937 | 2,735,581 | 7,356 | 5,151 | 0.99732 | 0.99812 | 0.99772 |
| GEM3 + HaplotypeCaller | 2,745,423 | 2,738,414 | 7,009 | 2,318 | 0.99745 | 0.99915 | 0.99830 |
| GEM3 + SAMtools fast | 2,749,554 | 2,736,718 | 12,836 | 4,014 | 0.99533 | 0.99854 | 0.99693 |
| GEM3 + SAMtools normal | 2,736,871 | 2,732,313 | 4,558 | 8,419 | 0.99833 | 0.99693 | 0.99763 |
| Whole genome deletions | |||||||
| NIST v2.18 Gold Standard | 85,958 | ||||||
| BWA‐MEM + FreeBayes | 82,263 | 75,674 | 6,589 | 10,284 | 0.91990 | 0.88036 | 0.89970 |
| BWA‐MEM + HaplotypeCaller | 86,323 | 84,789 | 1,534 | 1,169 | 0.98223 | 0.98640 | 0.98431 |
| BWA‐MEM + SAMtools fast | 77,671 | 68,591 | 9,080 | 17,367 | 0.88310 | 0.79796 | 0.83837 |
| BWA‐MEM + SAMtools normal | 77,712 | 68,615 | 9,097 | 17,343 | 0.88294 | 0.79824 | 0.83846 |
| GEM3 + FreeBayes | 81,602 | 76,002 | 5,600 | 9,956 | 0.93137 | 0.88418 | 0.90716 |
| GEM3 + HaplotypeCaller | 86,132 | 84,783 | 1,349 | 1,175 | 0.98434 | 0.98633 | 0.98533 |
| GEM3 + SAMtools fast | 80,905 | 69,096 | 11,809 | 16,862 | 0.85404 | 0.80383 | 0.82818 |
| GEM3 + SAMtools normal | 80,955 | 69,124 | 11,831 | 16,834 | 0.85386 | 0.80416 | 0.82826 |
| Whole genome insertions | |||||||
| NIST v2.18 Gold Standard | 84,583 | ||||||
| BWA‐MEM + FreeBayes | 78,592 | 73,890 | 4,702 | 10,693 | 0.94017 | 0.87358 | 0.90565 |
| BWA‐MEM + HaplotypeCaller | 84,521 | 83,473 | 1,048 | 1,110 | 0.98760 | 0.98688 | 0.98724 |
| BWA‐MEM + SAMtools fast | 79,762 | 69,389 | 10,373 | 15,194 | 0.86995 | 0.82037 | 0.84443 |
| BWA‐MEM + SAMtools normal | 79,762 | 69,396 | 10,366 | 15,187 | 0.87004 | 0.82045 | 0.84452 |
| GEM3 + FreeBayes | 78,417 | 73,154 | 5,263 | 11,429 | 0.93288 | 0.86488 | 0.89760 |
| GEM3 + HaplotypeCaller | 83,973 | 83,189 | 784 | 1,394 | 0.99066 | 0.98352 | 0.98708 |
| GEM3 + SAMtools fast | 92,775 | 71,928 | 20,847 | 12,655 | 0.77530 | 0.85038 | 0.81111 |
| GEM3 + SAMtools normal | 92,795 | 71,938 | 20,857 | 12,645 | 0.77524 | 0.85050 | 0.81113 |
TP, true positives; FP, false positives; FN, false negatives; specificity, number of TP calls as a proportion of total calls; sensitivity, number of TP calls as a proportion of the number of NIST reference set calls; F1‐score, measure of overall accuracy calculated as (2 × TP)/((2 × TP) + FP + FN).
Figure 1.
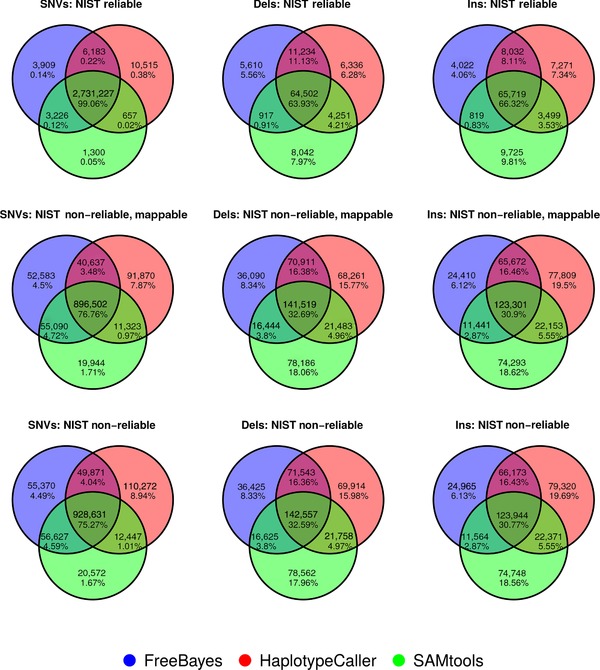
Venn diagrams illustrating concordance of variant identification for BWA‐MEM alignments. Separate Venn diagrams show the number and percentage of concordant calls for a particular variant type by pipeline for the NIST reliably callable regions, the NIST non‐reliably callable but mappable regions, and the NIST non‐reliably callable regions. SNVs, single nucleotide variants; Dels, deletions; Ins, insertions.
Figure 2.

Venn diagrams illustrating concordance of variant identification for GEM3 alignments. Separate Venn diagrams show the number and percentage of concordant calls for a particular variant type by pipeline for the NIST reliably callable regions, the NIST non‐reliably callable but mappable regions, and the NIST non‐reliably callable regions. SNVs, single nucleotide variants; Dels, deletions; Ins, insertions.
Our InDel results are somewhat surprising given that we performed indel realignment equivalently on the aligned BAMs prior to variant calling, indicating that there are substantial differences in the manner in which the callers identify InDels. Furthermore, the difference in total InDel variant calls differs substantially, but not consistently in direction, depending on the aligner that was used, suggesting that the underlying representation of InDels in the raw post‐alignment BAMs impacts on variant identification, even when indel realignment is performed prior to variant calling. In particular, there is a large difference in the number of InDels identified by SAMtools for the two WGS alignment datasets, with 16% more insertions, and 4% more deletions being called in the GEM3 datasets, whereas the difference is less than 1% for FreeBayes and HaplotypeCaller in each case (Table 1). Notably, the HaplotypeCaller combinations appear to perform substantially better than those of the other variant callers in terms of accuracy of InDel detection. However, this may reflect a bias in the high‐confidence calls in the NIST reference set, which is an integration of 14 different sequencing platform/aligner/variant caller workflows, but is heavily weighted toward BWA as an aligner (50% of workflows), and utilized either GATK HaplotypeCaller or GATK UnifiedGenotyper exclusively for variant identification.
To investigate the InDel observations further, we relaxed the requirement for genotype equivalence when testing for concordance with the reference set, that is, treating a heterozygote variant call as equivalent to a homozygote variant call for the same position and alternative allele and vice versa (Supp. Table S10). The overall pattern of results does not change, but the non‐HaplotypeCaller combinations improve somewhat, in particular in the case of FreeBayes where there is a substantial reduction in the number of false‐positive and false‐negative InDel calls. This may suggest that FreeBayes' InDel calling could be improved slightly.
We observe negligible differences in terms of InDel calls between the two modes of SAMtools tested, but for SNVs the normal mode is more specific but less sensitive than the fast mode. This reduction is sensitivity can be explained in part by a reduction of ∼0.5% in the total number of SNVs called when using the normal mode, in accordance with the stated objective of reducing the number of false positive SNV calls when using the normal setting. If we relax the requirement for genotype concordance again, the most marked improvement is the degree of reduction in false negatives in the fast mode (Supp. Table S10). This indicates that SAMtools fast‐mode identifies many of the same SNV variant positions as the normal mode, but differs in the genotype it assigns.
Since one of the major advantages of performing WGS versus WES is the ability to detect copy‐number (CNV) and structural variants (SV), and as GEM3 has been recently released, we ran Control‐FREEC [Boeva et al, 2012] and DELLY2 [Rausch et al, 2012] on the GEM3 and BWA‐MEM alignments to evaluate CNV and SV detection, respectively. Using the recommended settings provided by the developers (http://bioinfo-out.curie.fr/projects/freec/src/config.txt), Control‐FREEC identified exactly the same three significantly likely CNV events (two losses, and one gain), in each alignment. Furthermore, when the alignments were processed as a pseudo tumor–normal pair, no significant events were observed, indicating that there is no significant bias in normalized coverage between the alignment datasets (Supp. Table S11). DELLY2 identified a large number of putative SVs, including large deletions, duplications, and inversions, but no large insertions events (Supp. Table S12). The concordance of events varied between 0.37 and 0.86 depending on the type of event with deletions showing the highest concordance, followed by inversions and duplications. Although more events of each type were identified for the BWA‐MEM alignments, in the absence of a reference data set for SVs in this sample, it is impossible to establish how reliable the calls are, and whether one aligner is outperforming the other in terms of sensitivity and/or specificity. The NIST/GIAB consortium are actively working on developing reference call sets for CNVs and SVs in this sample (Justin Zook, personal communication), and it will be interesting to compare our findings with their reference call sets when they become available.
Concordance of Variant Calling Outwith NIST Reliably Callable Regions
It is not possible to establish the accuracy of variant calls outside the regions defined as reliably callable in the NIST v2.18 dataset because there are no reference calls available and, even if there were, they could not be taken as the “truth.” Therefore, we assessed the agreement of the different callers by comparing the concordance of calls in the NIST reliably callable region, the non‐reliably callable region and in the non‐reliably callable but mappable region (Figs. 1 and 2; Supp. Table S7). The percentage of each of these genomic regions covered by at least 10 reads (C10 in Supp. Table S7) was approximately 100, 66, and 96, respectively. Virtually all of the reliably callable region is mappable but the reverse is not true. Surprisingly, the concordance of results in the non‐reliably callable region and in the non‐reliably callable but mappable regions is very similar, though slightly better in the latter. In both cases, the three callers are concordant for over 75% of the SNVs, but only 30% of the insertions and deletions identified. This illustrates that mappability and reliably callable are not directly interchangeable. As mappability is assessed on the reference genome, and reliably callable regions were identified in a specific sample, with totally independent methods, it also indicates that there might be some features of certain genomic regions that make variant calling difficult even when it is possible to map to them uniquely, and/or that there may be sample specific limitations. These analyses could be further stratified in the future with the aim of expanding the reliably callable region of the genome.
Comparison of WGS and WES Variant Calls
The NA12878 WGS and WES datasets were generated at least 3 years apart by two different labs from two different sample aliquots. Nevertheless, we investigated the concordance of calls between the WES and WGS data for the GEM3 pipelines, restricting the region of interest to the 34.7 MB of exome capture region which NIST defined as reliably callable (Fig. 3). This region has excellent coverage in both WES and WGS data (C10 of 99.43 and 99.99, respectively; Supp. Tables S7and S8) and we observe an extremely high agreement between SNV calls (98.03%–99.46%). However, there is less concordance in InDel calls (65.76%–84.85%), with more events being identified in the MedExome WES data in general (the exception being FreeBayes insertions). Fang et al. [2014] reported only 52% concordance in a similar analysis using an older NimbleGen capture kit, and found that only 57% of WES‐specific InDels could be validated, whereas 84% of the WGS‐specific events tested were validated using an Illumina MiSeq. Thus, it is possible that the excess in InDel calls obtained with the MedExome might be false‐positives, as we have observed a similar trend for this particular kit in an inter‐exome comparison benchmark (manuscript in preparation). In addition, we observed a particularly large fraction of WES specific InDel calls in the HaplotypeCaller call set, the explanation of which will require further investigation. Overall, and even with the lower InDel concordance, this WGS and WES comparison indicates that Illumina NGS technology and the bioinformatics pipelines tested here are currently very consistent in their identification of germline variants. This is in contrast to recent work by Alioto et al. [2015], which reported much lower concordance in somatic mutation detection between different laboratories. They found that large variation in the quality of library construction would affect downstream results, especially for protocols using PCR. They also advocate the use of a combination of somatic mutation callers, in order to identify somatic mutations, as this improved accuracy of variant detection.
Figure 3.
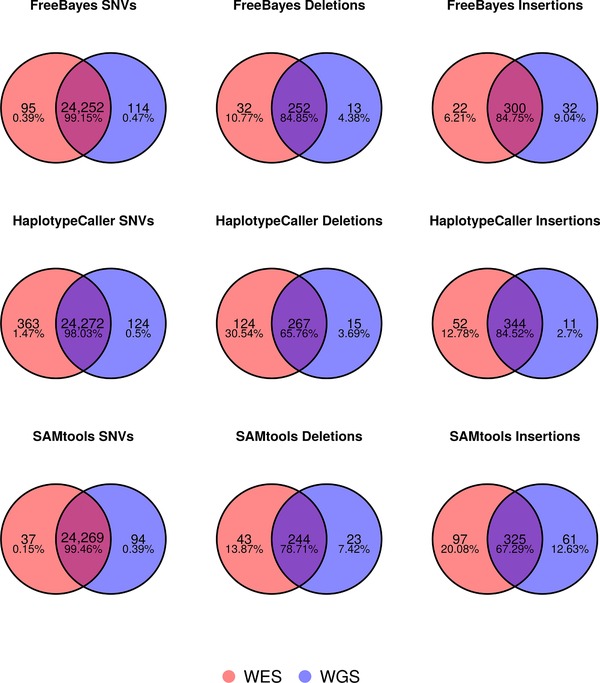
Venn diagrams illustrating concordance of WGS and WES variant identification. Separate Venn diagrams show the number and percentage of concordant calls for a particular variant type for the GEM3 pipelines for variants identified in the intersection of the NIST reliably callable region and exome capture regions (∼34.7 MB). SNVs, single nucleotide variants.
Computational Costs
Computational costs are an important consideration in NGS analyses, since many of the tools involved have significant resource requirements, or can work more efficiently when provided with access to more resources. Here, we considered primarily elapsed time (also known as wall‐clock time) in minutes and processing‐time (“CPU time”). As opposed to the actual time taken to complete a task (i.e., elapsed time), for tasks executed in parallel, CPU‐time is the sum of CPU times devoted to the task by each CPU running it. The difference between the two can be approximately illustrated by the following toy example. If a task (e.g., alignment of 100 million reads to a reference sequence) can be achieved in 60 min using a single processor, or 30 min using two processors, then the elapsed time has been reduced by half using an extra CPU. However, the CPU time is expected to remain the same (i.e., 60 min in total) in both cases. Note that in some cases, overheads due to the parallelization can be added to the CPU time of the parallel executions. Thus, if speed is of the essence, as may be the case in clinical diagnostics, then maximizing the number of processors available for a task may be the best strategy. On the other hand, if we have multiple tasks to perform, then assigning each task to a distinct CPU may be more efficient. It should also be noted that many programs used in genomics do not scale linearly with the number of CPU used, and will tend toward an asymptote due to input/output operations, inefficient parallelization, or RAM becoming limiting factors, and can even start to become less efficient when divided into too many small tasks.
Here, we observe that the GEM3 mapping algorithm is substantially faster than BWA‐MEM, requiring only 6 min to fully map 96.5 million WES reads, and 98 min for 1,708 million WGS reads, using 32 threads, representing just 40% and 20% of the time required by BWA‐MEM respectively, though exploiting twice as much RAM. We observe that both algorithms make good use of the available CPU threads, as indicated by the CPU‐time being between 26‐fold and 31‐fold the elapsed time, and thus the differences in CPU time are similar to those of elapsed time (Table 2). We have also tested a newly developed GPU implementation of GEM3, and found it to reduce mapping time by at least a further 20% for WGS, essentially becoming limited only by input/output operations (Marco‐Sola et al., manuscript in preparation).
Table 2.
Elapsed and CPU Times (Minutes), and Maximum RAM (GB) Requirements for Alignment Algorithms
| Exome | Genome | |||||||
|---|---|---|---|---|---|---|---|---|
| BWA‐MEM | GEM3 | BWA‐MEM | GEM3 | |||||
| Total | Largest Chunk | Total | Largest Chunk | Total | Largest Chunk | Total | Largest Chunk | |
| Elapsed time | 16 | 7 | 6 | 2.5 | 570 | 149 | 120 | 31 |
| CPU time | 468 | 192 | 162 | 66 | 17,706 | 4,633 | 3,458 | 904 |
| Max RAM | 7.6 | 15.4 | 8.3 | 15.7 | ||||
Alignment was performed on a compute node that allowed for up to 32 processing threads, supported by 256 GB of available RAM. Elapsed time is the real time required for the process to complete, whereas CPU time is the sum of the times that CPU threads were actively processing. Exome data came from 3 FASTQ files, whereas WGS came from 4 FASTQ files, the reads of which were mapped independently. The largest chunk time is the maximum time required for mapping of the largest individual chunk of a particular dataset.
Regarding variant calling, we observe that FreeBayes is much faster than the other algorithms, requiring 41 min to process the WES samples, and 155–165 min for the WGS samples (Table 3). SAMtools requires approximately 200 min for the WES samples and between 3,045 and 3,559 min for the WGS, whereas HaplotypeCaller requires between 269 and 362 min for the WES samples, and between 2,155 and 3,559 min for the WGS samples. FreeBayes achieves this significant reduction through efficient parallelization of the variant calling task, making better use of the available hardware threads as clearly shown by the fact that the CPU time for FreeBayes is 12‐fold greater than the elapsed time, whereas the equivalent value is between threefold and fivefold for GATK, and just onefold for SAMtools. As SAMtools and HaplotypeCaller are less able to exploit the availability of multiple CPUs per task, it is more efficient when using these algorithms to separate large tasks into smaller individual tasks. Thus, in order to reduce the elapsed time required for whole genome variant calling by SAMtools and HaplotypeCaller, we split calling into chromosome‐level of chunks. This resulted in a reduction in the elapsed time required by an order of magnitude, whereas the CPU time remains constant. Since chromosome 1 is the longest chromosome, it is typically that for which variant calling takes the longest, and thus provides an upper bound for time required to variant call a WGS sample, without applying a more complicated scatter‐gather approach. Nevertheless, with the exception of SAMtools fast mode, the time required to call variants on chromosome 1 was still longer than that required by FreeBayes to complete calling on the whole genome, and FreeBayes remained the most efficient caller in terms of CPU time without the need for splitting by chromosome.
Table 3.
Elapsed and CPU Times (Minutes) for Variant Calling
| BWA‐MEM Exome | GEM3 Exome | BWA‐MEM WGS | GEM3 WGS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total elapsed time | CPU time | Total elapsed time | CPU time | Total elapsed time | Largest chunk elapsed time | CPU time | Total elapsed time | Largest chunk elapsed time | CPU time | |
| FreeBayes | 45 | 558 | 41 | 497 | 155 | N/A | 2,104 | 165 | N/A | 2,156 |
| HaplotypeCaller | 362 | 1,119 | 269 | 1,284 | 2,155 | 241 | 7,930 | 2,681 | 238 | 8,231 |
| SAMtools normal | 199 | 201 | 211 | 213 | 3,045 | 250 | 3,025 | 3,559 | 334 | 3,537 |
| SAMtools fast | 103 | 104 | 114 | 116 | 1,328 | 117 | 1,309 | 1,610 | 139 | 1,591 |
Elapsed time is the real time required for the process to complete, whereas CPU time is the sum of the times that CPU threads were actively processing. For exome samples, variant calls were generated in a single process, whereas for WGS samples variant calling was performed on each chromosome independently, except in the case of FreeBayes which was sufficiently fast that separating into chunks was unnecessary.
It should be further noted that the timings for HaplotypeCaller do not include those of the base quality score recalibration (BQSR) step, which is currently recommended in GATK Best Practices and adds a large overhead of approximately an extra hour to the WES variant calling, and ∼12 hr for WGS variant calling (∼100 min for chromosome 1). However, Warden et al. [2014] found that BQSR had a relatively modest effect, only impacting on 2%–4% of variant calls, and it is possible that BQSR may be of less relevance when the raw sequencing data is of high quality. This may be worthy of further investigation, given the additional computational burden implied by this step. While variant calling timings described here were produced on a single node with 16 available CPUs, it has been shown that scalability of HaplotypeCaller performance decreases markedly after ∼4 threads are used [Kawalia et al., 2015; Intel Corporation, 2016], and as SAMtools does not support threading the availability of multiple CPUs has no effect on performance. Thus, a more resource‐efficient way of using HaplotypeCaller, when the computing resources are available, is to run by chromosome, requesting four threads apiece (a total of 100 threads, including mitochondrion), and for SAMtools to run by chromosome requesting a single thread for each (25 threads). In our experience, 5 GB of RAM per CPU is sufficient for each of these tools when run in this manner.
Concluding Remarks
With WGS progressively becoming standard practice for research and diagnostics, it will be essential to process large amounts of samples with very high accuracy, in the shortest turnaround time possible, using the minimum possible resources. Here, we have compared tools that are well‐established, and frequently used in the genomics field, but interesting new tools are constantly being developed [Kelly et al, 2015], and established tools may also be improved upon [Kathiresan et al, 2014], in this rapidly advancing field. The high concordance of results obtained between the WGS and WES data generated by different labs, in different years, from different aliquots of the NA12878 reference sample, indicates that Illumina NGS technology and protocols have reached an impressive mature state. Regardless of the differences observed in run times for the tools tested, our results show that germline SNV detection is a very reliable process in ∼70% of the genome. The InDel results on this 70% of the genome are less impressive, partly because of the technical difficulty of identifying these variants, and partly because of the likely bias toward a particular combination of aligner and caller in generation of the reference data set. Nevertheless, the pipeline combinations involving HaplotypeCaller and FreeBayes achieved accuracy in excess of 90% for InDels if we allow for genotype discordance. Furthermore, variants, especially SNVs, can still be quite reliably called in an additional 20% of the genome. Further studies and germline reference data sets will help to estimate the accuracy of the aligning and variant callings tools on these regions with the aim of increasing the reliably callable percentage of the genome. For the remaining ∼10% of the genome, it seems unfeasible to conduct variant calling exercises with the sequencing technology and tools assayed in this benchmark. The development of new technologies and tools, in particular long‐read sequencing and alignment, should help to shed some light on these remaining dark regions of the genome.
Disclosure statement: The authors declare no conflict of interest.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Text
Acknowledgments
We thank Raul Tonda for help with pipeline implementation and figure generation, and Nvidia for their donation of part of the systems used in this work. We thank two anonymous reviewers for their suggestions which helped improve the article.
Contract Grant Sponsors: Spanish Ministry of Economy and Competitiveness; Generalitat de Catalunya; European Regional Development Fund (ERDF); RD‐Connect Project (EC FP7/2007‐2013 #305444); ELIXIR‐EXCELERATE (EC H2020 #676559); MICINN (TIN2014‐53234‐ C2‐1‐R).
Ethical Compliance: The results described in this article have been generated with publicly available samples and data.
For the Next Generation Sequencing special issue
References
- Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, Heisler LE, Beck TA, Simpson JT, Tonon L, Sertier AS, Patch AM, et al. 2015. A comprehensive assessment of somatic mutation detection in cancer using whole‐genome sequencing. Nat Commun 6:10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Green RC. 2014. Diagnostic clinical genome and exome sequencing. N Engl J Med 370:2418–2425. [DOI] [PubMed] [Google Scholar]
- Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix‐Lerosey I, Delattre O, Barillot E. 2012. Control‐FREEC: a tool for assessing copy number and allelic content using next‐generation sequencing data. Bioinformatics 28:423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Boutros PC. 2011. VennDiagram: a package for the generation of highly‐customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish A, Guda C. 2015. A comparison of variant calling pipelines using genome in a bottle as a reference. BioMed Res Int 2015:456479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Bank, E , Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis A, del Angel G, Rivas MA, Hanna M, McKenna A, Fennel TJ, et al. 2011. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, J Estellé, Marco‐Sola S, Knowles DG, Raineri E, R Guigó, Ribeca P. 2012. Fast computation and applications of genome mappability. PLoS ONE 7:e30377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enn, GM , David SP, Pakdaman N, Ormond KE, et al. 2014. Clinical interpretation and implications of whole‐genome sequencing. JAMA 311:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Wu Y, Narzisi G, O'Rawe JA, Barrón LTJ, Rosenbaum J, Ronemus M, Iossifov I, Schatz MC, Lyon GJ. 2014. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med 6:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype‐based variant detection from short‐read sequencing. arXiv:1207.3907.
- Goldfeder RL, Priest JR, Zook JM, Grove ME, Waggot D, Wheeler MT, Salit M, Ashley EA. 2016. Medical implications of technical accuracy in genome sequencing. Genome Med 8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MS, Wu X, Zhang L. 2015. Performance evaluation of indel calling tools using real short‐read data. Human Genomics 9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highnam G, Wang JJ, Kusler D, Zook J, Vijayan V, Leibovich N, Mittelman D. 2015. An analytical framework for optimizing variant discovery from personal genomes. Nat Commun 6:6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Kim E, Lee I, Marcotte EM. 2015. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep 5:17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intel Corporation . 2016. Infrastructure for Deploying GATK Best Practices Pipeline. http://www.intel.com/content/www/us/en/healthcare-it/solutions/documents/deploying-gatk-best-practices-paper.html.
- Kathiresan N, Temanni MR, Al‐Ali R. 2014. Performance improvement of BWA MEM algorithm using data‐parallel with concurrent parallelization. In: 2014 International Conference on Parallel, Distributed and Grid Computing (PDGC), p 406–411.
- Kawalia A, Motameny S, Wonczak S, Thiele H, Nieroda L, Jabbari K, Borowski S, Sinha V, Gunia W, Lang U, Achter V, Nürnberg P. 2015. Leveraging the power of high performance computing for next generation sequencing data analysis: Tricks and twists from a high throughput exome workflow. PLoS ONE 10:e0126321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BJ, Fitch JR, Hu Y, Corsmeier DJ, Zhong H, Wetzel AN, Nordquist RD, Newsom DL, White P. 2015. Churchill: an ultra‐fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population‐scale genomics. Genome Biol 16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv:1303.3997.
- Marco‐Sola S, Sammeth M, Guigó R, Ribeca P. 2012. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods 9:1185–1188. [DOI] [PubMed] [Google Scholar]
- O'Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, Bodily P, Tian L, Hakonarson H, Johnson W, Wei Z, Wang K, et al. 2013. Low concordance of multiple variant‐calling pipelines: practical implications for exome and genome sequencing. Genome Med 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, AR . 2014. BEDTools: The Swiss‐Army tool for genome feature analysis. Curr Protoc Bioinformatics 11:12.1–12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan A, Harris J, Leng N, Bartha G, Church DM, Luo S, Haudenschild C, Pratt M, Zook J, Salit M, Tirch J, Morra M, et al. 2015. Achieving high‐sensitivity for clinical applications using augmented exome sequencing. Genome Med 7:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired‐end and split‐read analysis. Bioinformatics 28:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentrube, J , Smith A, Bedford HM, Bernard G, Bernie, FP , Brais B, Bulman DE, Warman Chardon J, et al. 2015. Utility of whole‐exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet 89:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hart, C , Poplin R, del Angel G, Levy‐Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, et al. 2013. From fastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden CD, Adamson AW, Neuhausen SL, Wu X. 2014. Detailed comparison of two popular variant calling packages for exome and targeted exon studies. Peer J 2:e600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, Van Kogelenberg M, King DA, Ambridge K, Barrett DM, Bayzetinova T, Bevan AP, Bragin E, et al. 2015. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome‐wide research data. Lancet 385:1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, et al. 2014. Molecular findings among patients referred for clinical whole‐exome sequencing. JAMA 312:1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M. 2014. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32:246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Text