

Summary
Naltrexone/bupropion extended release (NB) is indicated as an adjunct to a reduced‐calorie diet and increased physical activity for chronic weight management in adults with an initial body mass index of ≥30 or ≥27 kg m−2 and ≥1 weight‐related comorbidity (e.g. hypertension, type 2 diabetes and dyslipidaemia). In phase 3 clinical studies, nausea occurred in significantly higher proportions of subjects randomized to NB vs. placebo (PBO). In this pooled analysis of three phase 3, 56‐week, PBO‐controlled studies, we characterized nausea and weight loss in NB‐ and PBO‐treated subjects without diabetes. Subjects receiving NB (n = 1778) lost significantly more weight than those receiving PBO (n = 1160). Weight change was not significantly different between subjects reporting and not reporting nausea in either treatment arm. Severity of nausea was mild to moderate in ≥95% of all cases. In the NB arm, the highest incidence of nausea onset (9%) was reported during week 1. The median duration of mild, moderate and severe nausea in subjects receiving NB was 14, 9 and 13 days, respectively. Our results demonstrate that nausea associated with NB is rarely severe, primarily occurs early in treatment and is not a contributor to weight loss.
Keywords: Adverse events, naltrexone/bupropion extended release, nausea, obesity
What is already known about this subject?
Four phase 3, placebo (PBO)‐controlled studies evaluated the efficacy and safety of naltrexone/bupropion extended release (NB; Contrave® and Mysimba™) as an adjunct to a reduced‐calorie diet and increased physical activity in adults with an initial BMI of ≥30 or ≥27 kg m−2 in the presence of ≥1 weight‐related comorbidity (e.g. hypertension, type 2 diabetes and dyslipidaemia).
Subjects receiving daily doses totalling 32 mg naltrexone extended‐release (ER) and 360 mg bupropion ER lost significantly more weight (5.0–.3%) than those receiving PBO (1.2–5.1%). NB‐treated subjects were also significantly more likely than PBO‐treated subjects to achieve at least 5% weight loss from baseline to week 56 (45–51% vs. 16–19%). Nausea was reported in significantly higher proportions of subjects randomized to receive NB (29.2–42.3%) vs. PBO (5.3–10.5%) in these studies.
What this study adds?
In a pooled analysis of the three phase 3 studies enrolling subjects without diabetes, weight change was not significantly different between subjects reporting nausea and not reporting nausea in either treatment arm.
Severity of nausea was mild to moderate in 95 and 99% of cases in the NB and PBO arms, respectively.
The highest incidence of nausea onset occurred during week 1, with 9% of NB‐treated subjects reporting nausea during this time frame; the median duration of mild, moderate and severe nausea in subjects treated with NB was 14, 9 and 13 days, respectively.
Introduction
Recent estimates suggest that 35% of adults in the United States are obese 1. Excess body weight is a risk factor for numerous conditions, including hypertension, dyslipidaemia, type 2 diabetes, coronary artery disease, stroke, gallbladder disease, osteoarthritis, sleep apnoea and some cancers. Even modest weight loss (as little as 5%) in obese and overweight individuals has been shown to improve cardiometabolic risk profiles 2. Lifestyle modification has long been the cornerstone of weight control 3; however, many individuals are not able to achieve or maintain weight loss with diet and exercise alone. Thus, several pharmacotherapies have been developed as adjuncts to lifestyle modification for chronic weight management.
Naltrexone/bupropion extended release (NB) was developed based on pre‐clinical evidence that its components act in the hypothalamic melanocortin and reward systems to reduce food intake, resulting in weight loss 4. Pre‐clinical evidence suggests that bupropion stimulates hypothalamic pro‐opiomelanocortin (POMC) neurons, with downstream effects that modulate food intake and energy expenditure 5. Naltrexone blocks opioid receptor‐mediated hypothalamic pro‐opiomelanocortin POMC autoinhibition, which further augments POMC firing. In early studies, bupropion and naltrexone synergistically reduced food intake in obese mice when compared to naltrexone or bupropion monotherapy 6. A proof‐of‐concept study in obese people also showed that the combination produced a greater reduction in body weight than either agent as monotherapy 7.
The efficacy and safety of NB as an adjunct to a reduced‐calorie diet and increased physical activity in adults with an initial body mass index (BMI) of ≥30 (obese) or ≥27 kg m−2 (overweight) in the presence of ≥1 weight‐related comorbidity (e.g. hypertension, type 2 diabetes and dyslipidaemia) were evaluated in four phase 3 clinical studies 8, 9, 10, 11. After 56 weeks, subjects randomized to receive NB lost significantly more weight on average (5.0–9.3%) compared with subjects receiving the placebo (PBO) (1.2–5.1%) 8, 9, 10, 11. NB‐treated subjects were also significantly more likely than PBO‐treated subjects to achieve at least 5% weight loss from baseline to week 56 (45–51% vs. 16–19%). NB was approved by the US Food and Drug Administration in September 2014 (under the trade name of Contrave®). Marketing authorization for NB (marketed as Mysimba™) was granted in the European Union in March 2015.
In all four studies, nausea was reported in significantly higher proportions of subjects randomized to receive NB (29.2–42.3%) vs. PBO (5.3–10.5%) 8, 9, 10, 11. Here, we characterize the adverse event (AE) of nausea and evaluate its relationship to weight loss using a pooled analysis of the three Contrave Obesity Research (COR) studies that enrolled subjects without diabetes: COR‐I, COR‐II and COR‐behavior modification (BMOD).
Materials and methods
Study design
This was a pooled analysis of three phase 3, 56‐week, randomized, PBO‐controlled studies conducted in the United States 8, 9, 11; the designs of these studies are summarized in Table 1. All three studies enrolled adults aged 18–65 years without diabetes who had a BMI of 30–45 kg m−2 or a BMI of 27–45 kg m−2 with dyslipidaemia or hypertension. The co‐primary efficacy endpoints were percentage change in body weight from baseline and the proportion of participants who achieved a decrease in body weight of ≥5% at week 28 (COR‐II) or week 56 (COR‐I and COR‐BMOD). The COR‐Diabetes study 10 was not included in the analysis because nearly 80% of study subjects were concomitantly taking metformin, a medication for diabetes that is itself associated with nausea 12. Detailed descriptions of the studies and results have been published elsewhere 8, 9, 11.
Table 1.
Study designs of the COR‐I, COR‐II and COR‐BMOD studies
| Design parameter | COR‐I 9 | COR‐II 8 | COR‐BMOD 11 * |
|---|---|---|---|
| Treatments (randomized subjects) | NB32 (n = 583), NB16 (n = 578) or placebo (n = 581) | NB32† (n = 1001) or placebo (n = 495) | NB32 (n = 591) or placebo (n = 202) |
| Duration of dose escalation period | 4 weeks | 4–5 weeks | 4 weeks |
| Recommended caloric intake | Subjects were instructed to follow a hypocaloric diet (500 kcal d−1 deficit) | Subjects were instructed to follow a hypocaloric diet (500 kcal d−1 deficit) |
Individual goals for energy intake were based on initial body weight Subjects were instructed to consume ~15–20% of energy from protein, ≤30% energy from fat and the remainder from carbohydrate |
| Behavioural modification | Subjects were given advice on lifestyle modification (including instructions to increase physical activity) | Subjects were given advice on lifestyle modification (including instructions to increase physical activity) |
28 total 90‐min educational sessions delivered to groups of subjects by qualified professionals Subjects were instructed in measuring portion sizes, counting calories and keeping food diaries Subjects were encouraged to gradually increase to 360 min of physical activity per week and to keep daily records of their activity Written manuals included materials from the LEARN Program for Weight Management 18, the Diabetes Prevention Program 19 and other handouts used in prior studies |
Subjects who discontinued the study drug but continued the behavioural modification programme were included in the pooled analysis.
NB32 subjects with <5% weight loss at visits between weeks 28 and 44 inclusive were re‐randomized (double‐blind, 1:1 ratio) to continue receiving NB32 (n = 128) or escalate to NB48 (i.e. daily dose totalling 48 mg naltrexone ER and 360 mg bupropion ER; n = 123) for the remainder of the study.
ER, extended release; NB16, daily dose totalling 16 mg naltrexone ER and 360 mg bupropion ER; NB32, daily dose totalling 32 mg naltrexone ER and 360 mg bupropion ER.
Following screening, participants in the trial were randomized to receive 32 mg day−1 naltrexone SR and 360 mg day−1 bupropion SR (NB32) or a matching PBO, administered in divided doses, twice daily. In all trials, the study drug was escalated weekly over the first 4 weeks: one tablet in the morning during week 1, two tablets in week 2 (one morning and one evening), three tablets in week 3 (two morning and one evening) and the full dose in week 4 (two morning and two evening). In the pooled analysis, subjects from COR‐I and COR‐BMOD studies were randomized to NB32 or PBO for 56 weeks. Among subjects from COR‐II, 251 subjects receiving NB32 who failed to achieve or maintain at least ≥5% body weight loss from baseline were re‐randomized (beginning at weeks 28 and 44 inclusive) to continue NB32 (128 subjects) or receive a higher dose of naltrexone SR, NB48 (123 subjects).
Outcomes of interest for this analysis were baseline characteristics; subject disposition; percentage change in body weight from baseline (overall and by week); proportion of subjects meeting 5, 10 and 15% weight loss thresholds; nausea incidence and intensity (mild, moderate or severe) overall and by week, as collected from adverse reports; weight loss by report of nausea and nausea intensity; duration of nausea by nausea intensity; weight loss by duration of nausea; discontinuations due to nausea or other AEs (overall and by week); and incidence of the most common AEs. AEs, including nausea, were reported spontaneously and recorded at each study visit. Within each treatment arm, outcomes of interest were determined for total subjects, subjects reporting nausea and subjects not reporting nausea.
Statistical analyses
Analyses of disposition, baseline characteristics and safety‐related endpoints were based on the safety population, which consisted of subjects who received ≥1 dose of the study drug. Efficacy analyses were based on the modified intent‐to‐treat (mITT) population, which included all randomized participants with a baseline and ≥1 post‐baseline weight assessment on the study drug. Missing post‐baseline efficacy data were imputed using the last observation carried forward. Baseline characteristics, incidence and intensity of nausea, duration of nausea and discontinuations because of nausea or other AEs were summarized descriptively.
Group differences (i.e. nausea vs. no‐nausea subgroups within each treatment arm) for disposition were analysed using a Chi‐square test. Differences in percentage change from baseline in body weight were evaluated using an analysis of covariance model, with factors for study, nausea status (yes/no) and baseline body weight as the covariate; for percentages of subjects achieving each weight loss threshold, group differences were evaluated using a logistic regression model, with factors for study, nausea status (yes/no) and baseline body weight as a covariate. Statistical significance was defined by P‐values of <0.05.
Results
Subject demographics
The safety population included 2149 and 1261 subjects in the NB and PBO arms, respectively. In the NB arm, 30.7% of subjects reported nausea vs. 6.7% of subjects in the PBO arm. Demographic and baseline clinical characteristics were generally similar for subjects reporting nausea and not reporting nausea, although some imbalances in gender, weight and height were noted (Table 2). In both treatment arms, subjects reporting nausea were significantly more likely to be female than those not reporting nausea. Among the 660 subjects in the NB arm that reported nausea, 92.7% were female; among the 1489 subjects in the NB arm that did not report nausea, 82.9% were female (P < 0.001). Among the 85 subjects in the PBO arm that reported nausea, 94.1% were female; among the 1176 subjects that did not report nausea, 85.5% were female; P < 0.026).
Table 2.
Demographics and baseline clinical characteristics (safety population)
| NB | PBO | |||||||
|---|---|---|---|---|---|---|---|---|
| Total (n = 2149) | Nausea (n = 660) | No nausea (n = 1489) | P‐value* | Total (n = 1261) | Nausea (n = 85) | No nausea (n = 1176) | P‐Value* | |
| Age, years | 0.590 | 0.060 | ||||||
| Mean (SD) | 44.8 (10.96) | 44.6 (11.02) | 44.9 (10.94) | 44.3 (11.27) | 42.1 (11.44) | 44.4 (11.25) | ||
| Median (min, max) | 45.0 (18, 65) | 45.0 (18, 65) | 45.0 (18, 65) | 45.0 (18, 66) | 41.0 (19, 66) | 45.0 (18, 65) | ||
| Gender | <0.001 | 0.026 | ||||||
| Female, n (%) | 1847 (85.9) | 612 (92.7) | 1235 (82.9) | 1085 (86.0) | 80 (94.1) | 1005 (85.5) | ||
| Race, n (%) | 0.057 | 0.587 | ||||||
| White | 1664 (77.4) | 498 (75.5) | 1166 (78.3) | 991 (78.6) | 63 (74.1) | 928 (78.9) | ||
| Black or African | 377 (17.5) | 133 (20.2) | 244 (16.4) | 221 (17.5) | 20 (23.5) | 201 (17.1) | ||
| American Asian | 23 (1.1) | 8 (1.2) | 15 (1.0) | 10 (0.8) | 1 (1.2) | 9 (0.8) | ||
| Native Hawaiian or Pacific Islander | 9 (0.4) | 3 (0.5) | 6 (0.4) | 5 (0.4) | 0 | 5 (0.4) | ||
| American Indian or Alaska native | 35 (1.6) | 4 (0.6) | 31 (2.1) | 20 (1.6) | 1 (1.2) | 19 (1.6) | ||
| Other | 41 (1.9) | 14 (2.1) | 27 (1.8) | 14 (1.1) | 0 | 14 (1.2) | ||
| BMI, kg m−2 | 0.354 | 0.684 | ||||||
| Mean (SD) | 36.2 (4.35) | 36.1 (4.43) | 36.3 (4.31) | 36.3 (4.15) | 36.1 (3.93) | 36.3 (4.17) | ||
| Median (min, max) | 36.0 (27, 46) | 35.0 (27, 46) | 36.0 (27, 46) | 36.0 (27, 46) | 36.0 (29, 45) | 36.0 (27, 46) | ||
| Weight, kg | <0.001 | 0.621 | ||||||
| Mean (SD) | 100.1 (16.12) | 98.4 (15.91) | 100.9 (16.15) | 99.7 (15.07) | 98.9 (13.61) | 99.8 (15.17) | ||
| Median (min, max) | 98.0 (64, 168) | 97.0 (64, 154) | 99.0 (66, 168) | 98.0 (63, 162) | 98.0 (70, 134) | 98.0 (63, 162) | ||
| Height, cm | <0.001 | 0.715 | ||||||
| Mean (SD) | 166.0 (8.3) | 164.7 (7.6) | 166.5 (8.5) | 165.6 (8.1) | 165.3 (7.7) | 165.6 (8.1) | ||
| Median (min, max) | 165.0 (144, 208) | 164.0 (145, 191) | 165.0 (144, 208) | 165.0 (143, 196) | 165.0 (147, 189) | 165.0 (143, 196) | ||
| Hypertension, n (%) | 426 (19.8) | 125 (18.9) | 301 (20.2) | 0.494 | 254 (20.1) | 15 (17.6) | 239 (20.3) | 0.552 |
| Dyslipidaemia, n (%) | 1105 (51.4) | 316 (47.9) | 789 (53.0) | 0.029 | 625 (49.6) | 44 (51.8) | 581 (49.4) | 0.674 |
Based on a Chi‐square test for categorical variables and t‐test for continuous variables comparing the nausea and no‐nausea groups within each treatment arm.
BMI, body mass index; NB, naltrexone/bupropion extended release; PBO, placebo; SD, standard deviation.
Subject disposition
In the NB arm, fewer subjects reporting nausea completed the study compared with subjects not reporting nausea (48% vs. 58%; P < 0.001), whereas in the PBO arm, similar proportions of subjects reporting and not reporting nausea completed the study (60% vs. 53%; P = 0.215).
In both the NB and PBO arms, more subjects reporting nausea vs. not reporting nausea discontinued because of an AE (P < 0.001 for both comparisons). However, significantly more NB subjects not reporting nausea withdrew consent or were lost to follow‐up compared with those who did report nausea; significantly more PBO subjects not reporting nausea withdrew consent or withdrew for other reasons vs. those who did report nausea (Table 3).
Table 3.
Subject disposition (safety population)
| NB | PBO | |||||||
|---|---|---|---|---|---|---|---|---|
| Total (n = 2149) | Nausea (n = 660) | No nausea (n = 1489) | P‐value* | Total (n = 1261) | Nausea (n = 85) | No nausea (n = 1176) | P‐value* | |
| Completed treatment, n (%) | 1176 (54.7) | 316 (47.9) | 860 (57.8) | <0.001 | 675 (53.5) | 51 (60.0) | 624 (53.1) | 0.215 |
| Discontinued treatment, n (%) | ||||||||
| Adverse event | 503 (23.4) | 251 (38.0) | 252 (16.9) | <0.001 | 149 (11.8) | 20 (23.5) | 129 (11.0) | <0.001 |
| Withdrew consent | 178 (8.3) | 33 (5.0) | 145 (9.7) | <0.001 | 170 (13.5) | 3 (3.5) | 167 (14.2) | 0.005 |
| Lost to follow‐up | 164 (7.6) | 30 (4.5) | 134 (9.0) | <0.001 | 136 (10.8) | 8 (9.4) | 128 (10.9) | 0.673 |
| Other† | 128 (6.0) | 30 (4.5) | 98 (6.6) | 0.066 | 131 (10.4) | 3 (3.5) | 128 (10.9) | 0.032 |
Based on a Chi‐square test comparing the nausea and no‐nausea groups within each treatment arm.
Includes drug non‐compliance, insufficient weight loss, other primary reason not listed, protocol non‐compliance, subject became pregnant, enrolled but did not meet selection criteria or death.
NB, naltrexone/bupropion extended release; PBO, placebo.
In the NB arm, 5.7% of subjects discontinued because of nausea, and 17.7% discontinued because of other AEs compared with 0.2 and 11.6% of subjects, respectively, who discontinued in the PBO arm.
Weight loss
The mITT population, which was used for efficacy assessments, included 1778 and 1160 subjects in the NB and PBO arms, respectively. Beginning at the earliest post‐baseline assessment (week 4), subjects receiving NB lost more weight than subjects receiving PBO, irrespective of reported nausea (P < 0.001 at all assessments for comparisons between NB and PBO subjects not reporting nausea and between NB and PBO subjects reporting nausea; Fig. 1). Weight change was not significantly different within the NB treatment arm between subjects reporting and not reporting nausea at any treatment time point. In the NB arm, least squares (LS) mean (SE) percentage decrease in body weight was 7.3 (0.3) in subjects reporting nausea and 7.5 (0.2) in subjects not reporting nausea after 56 weeks (P = 0.562). The percentage decreases in body weight were similar between PBO subjects reporting and not reporting nausea at all time points; at week 56, LS mean (SE) percentage decreases were 3.0 (0.55) in subjects reporting nausea and 2.7 (0.2) in subjects not reporting nausea (P = 0.663).
Figure 1.
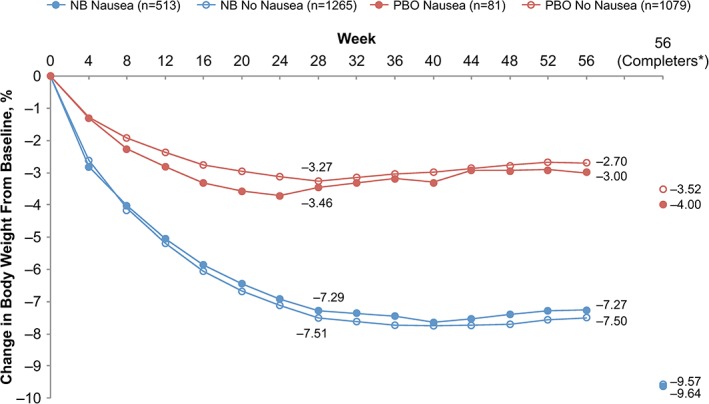
LS mean percentage change in body weight over time in subjects receiving NB and PBO who reported or did not report nausea (mITT population). LS, least squares; mITT, modified intent‐to‐treat; NB, naltrexone/bupropion extended release; PBO, placebo. *Subjects completing the 56‐week studies (NB nausea group, n = 296; NB no‐nausea group, n = 839; PBO nausea group, n = 50; PBO no‐nausea group, n = 613). Data point for NB no‐nausea group is obscured by NB nausea group.
In both the NB and the PBO arms, there were no significant differences between subjects who reported nausea and those who did not report nausea with respect to the proportion of subjects who achieved any of the weight loss thresholds (5, 10 or 15%; Fig. 2).
Figure 2.

Proportions of subjects achieving weight loss thresholds of ≥5, ≥10 and ≥15% at week 56 in subjects receiving NB and PBO who reported or did not report nausea (mITT population). mITT, modified intent‐to‐treat; NB, naltrexone/bupropion extended release; PBO, placebo.
Nausea intensity and onset
The severity of nausea was mild to moderate in 95% and 99% of cases in the NB and PBO arms, respectively. LS mean (SE) percentage reductions in body weight at week 56 were similar between subjects reporting mild vs. moderate nausea in both the NB [8.0 (0.4) vs. 6.6 (0.6); P = 0.062] and PBO [3.5 (0.7) vs. 2.7 (1.8); P = 0.515] arms. In the NB arm, 9% of subjects reported onset of nausea during week 1 (Fig. 3a). Beyond week 6, onset of nausea was reported in <1% of subjects receiving NB. Nausea was rarely reported in the PBO arm, but, similar to the NB arm, onset was most common during the early dose escalation period (Fig. 3b).
Figure 3.
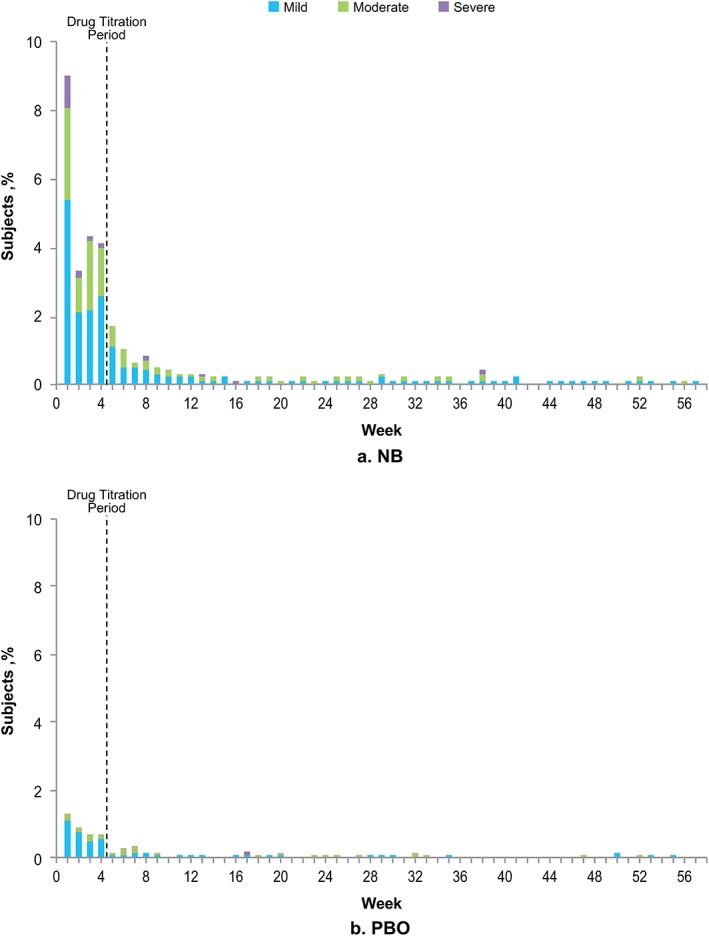
Onset of nausea by intensity in subjects receiving NB (graph a) and PBO (graph b) (safety population). NB, naltrexone/bupropion extended release; PBO, placebo.
Duration of nausea
In subjects receiving NB, the median duration of mild, moderate and severe nausea was 14, 9 and 13 days, respectively. Corresponding values for subjects in the PBO arm were 8, 4 and 33 days, respectively (one subject in the PBO arm reported severe nausea). LS mean (SE) percentage reductions in body weight at week 56 were similar between subjects with a duration of nausea <10 days vs. ≥10 days in both the NB [7.0 (0.6) vs. 7.6 (0.4); P = 0.367] and PBO [2.9 (0.9) vs. 3.4 (0.9); P = 0.674] arms. Only 10.3% of subjects treated with NB and 3.3% of subjects receiving PBO reported new or ongoing nausea beyond week 4.
Adverse events
In the NB and PBO arms, 87.2 and 74.1% of subjects reported an AE, respectively (Table 4). The AE profile of NB was consistent with previous reports 8, 9, 11. Nausea was the most frequently reported AE in the NB arm (30.7% vs. PBO, 6.7%). Subjects receiving both NB and PBO who reported nausea were more likely to report vomiting than those who did not report nausea (Table 4). In the NB arm, there were 124 events of vomiting in 660 subjects who reported nausea vs. 50 events in 1489 subjects who did not report nausea. In the PBO arm, there were 12 events of vomiting in 85 subjects who reported nausea vs. 13 events in 1176 subjects who did not report nausea. Most vomiting events were mild or moderate in severity. Severe vomiting events affected <1% of subjects in either nausea subgroup of either treatment arm. Subjects who reported nausea were also more likely to report headache, dizziness and diarrhoea than those who did not report nausea in both the NB and PBO arms.
Table 4.
Incidence of adverse events reported in >5% of subjects in either treatment arm (safety population)
| Adverse event, n (%) | NB | PBO | ||||
|---|---|---|---|---|---|---|
| Total (n = 2149) | Nausea (n = 660) | No nausea (n = 1489) | Total (n = 1261) | Nausea (n = 85) | No nausea (n = 1176) | |
| Nausea | 660 (30.7) | 660 (100) | 0 | 85 (6.7) | 85 (100) | 0 |
| Constipation | 420 (19.5) | 136 (20.6) | 284 (19.1) | 95 (7.5) | 13 (15.3) | 82 (7.0) |
| Headache | 391 (18.2) | 188 (28.5) | 203 (13.6) | 131 (10.4) | 16 (18.8) | 115 (9.8) |
| Dizziness | 207 (9.6) | 115 (17.4) | 92 (6.2) | 42 (3.3) | 10 (11.8) | 32 (2.7) |
| Vomiting | 204 (9.5) | 139 (21.1) | 65 (4.4) | 37 (2.9) | 14 (16.5) | 23 (2.0) |
| Insomnia | 191 (8.9) | 74 (11.2) | 117 (7.9) | 74 (5.9) | 11 (12.9) | 63 (5.4) |
| Upper respiratory tract infection | 181 (8.4) | 55 (8.3) | 126 (8.5) | 135 (10.7) | 8 (9.4) | 127 (10.8) |
| Dry mouth | 180 (8.4) | 59 (8.9) | 121 (8.1) | 30 (2.4) | 4 (4.7) | 26 (2.2) |
| Nasopharyngitis | 147 (6.8) | 46 (7.0) | 101 (6.8) | 86 (6.8) | 13 (15.3) | 73 (6.2) |
| Diarrhoea | 124 (5.8) | 58 (8.8) | 66 (4.4) | 61 (4.8) | 6 (7.1) | 55 (4.7) |
NB, naltrexone/bupropion extended release; PBO, placebo.
Discussion
In pivotal studies, treatment with NB was associated with statistically significant weight loss compared with PBO 8, 9, 10, 11. Nausea was the most common AE in subjects receiving NB and was also the most frequently reported AE leading to withdrawal 8, 9, 10, 11. This pooled analysis of the COR‐I, COR‐II and COR‐BMOD studies aimed to characterize the incidence, intensity and time course of nausea and, most importantly, to determine the potential role of nausea in weight loss among subjects receiving NB and PBO. The strengths of the analysis were the inclusion of >3400 subjects and the use of spontaneous AE reporting.
The analysis yielded several key findings. Although subjects receiving NB lost more weight and were more likely to report nausea than those receiving PBO, weight loss was independent of nausea. Specifically, weight change was not significantly different between subjects reporting and not reporting nausea at any treatment time point within either treatment arm. There was also no significant difference in the proportions of subjects in both the reporting nausea and not reporting nausea groups achieving the weight loss thresholds of 5, 10 or 15%. Among the subjects who reported nausea, neither the intensity (mild vs. moderate) nor the duration (<10 vs. ≥10 days) of nausea significantly influenced weight loss in either treatment arm. Subjects receiving NB and PBO who reported nausea were more likely to discontinue because of an AE than those not reporting nausea. However, discontinuations due to nausea were less common than discontinuations due to other AEs.
Nausea is a known side effect of naltrexone 13 and likely results the from local effects of naltrexone on opioid signalling in the gastrointestinal tract 4. In clinical studies of bupropion, nausea was also reported in higher proportions of subjects taking bupropion vs. PBO 14, 15. This pooled analysis indicates that nausea was nearly always mild or moderate in intensity and that the onset of nausea was generally limited to the dose escalation period in subjects receiving either NB or PBO. Nausea was generally either self‐limiting or manageable using standard or other therapies as deemed appropriate by the investigator.
The results of this pooled analysis contrast with those of Lean and co‐workers, who found in a phase 2 study of liraglutide that patients receiving liraglutide who reported nausea had more weight loss than those who did not report nausea 16. The study was conducted in different countries, and there were notable geographic variations in the reporting rates of nausea, which the authors attributed to possible differences in the way investigators enquired about and reported AEs in different centres and countries. Furthermore, subjects in the study were informed of the potential gastrointestinal side effects of liraglutide, which may have resulted in over‐reporting of these side effects during open‐label treatment. Differences in the liraglutide study and the studies included in this analysis may account for the disparate findings. First, the data analysed here were pooled from studies conducted entirely within the United States; therefore, there were no language issues. Secondly, all patients, whether they received the active drug or PBO, received the same instructions regarding potential AEs.
The three studies included in this analysis required that the escalation to the final dose of NB occur according to specific 4‐ or 5‐week protocols. Similarly, both the United States and European Union labelling recommend that the titration of NB dose occur over a 4‐week period following the initiation of treatment 14, 17. Although there are no data from randomized clinical studies supporting a slower dose escalation as a means of limiting nausea, the clinical experience of the physician authors suggests that slowing the dose escalation (e.g. maintaining once daily dosing through week 2) might be an option for improving tolerability. If symptoms persisted, use of simethicone or bismuth subsalicylate before taking study medication have been helpful 8, 9, 11. Based on clinical experience, the physician authors recommend that patients be reminded to consume adequate fluids and avoid excess caffeine consumption to prevent dehydration during NB initiation. It should be noted that one of the studies 9 also used a lower dose of 16 mg naltrexone/360 bupropion (data for this dose were not included in this analysis). In that study, the incidence of nausea was similar in subjects who received 16 or 32 mg naltrexone, although weight loss was less in subjects who received the 16‐mg than the 32‐mg dose 9.
In conclusion, nausea is the most frequently reported AE in subjects treated with NB. However, nausea did not appear to contribute to weight loss in either NB‐ or PBO‐treated subjects. Nausea lasting 1–2 weeks can be expected to occur in approximately one in three subjects during the initiation of NB therapy; however, it is almost always mild to moderate in severity and is transient in most subjects. Clinicians should discuss the potential for nausea with patients prior to initiation of NB therapy, and may suggest a variety of approaches that may help patients manage nausea and adhere to therapy.
Conflicts of Interest Statement
KuH serves on Takeda's speaker's bureau. KaH, CD and HL are employees of Takeda Pharmaceuticals USA, Inc. AEH is an employee and shareholder of Orexigen Therapeutics. JPF serves on Takeda's speaker's bureau.
Acknowledgments
KuH and AEH analysed and interpreted data and contributed to the writing of the manuscript. KaH contributed to the design of the post hoc analysis, data collection and interpretation, generation of figures and writing of the manuscript. JPF and CD contributed to data interpretation and writing of the manuscript. HL contributed to the collection, analysis and interpretation of data and generation of figures. All authors reviewed and approved the final version of the manuscript.
Clinical studies were funded by Orexigen Therapeutics, Inc. (La Jolla, CA, USA); the manuscript was supported by Takeda Pharmaceuticals International, Inc. (Deerfield, IL, USA) and Orexigen. Editorial support was provided by Melinda Ramsey and Mariana Ovnic of Complete Publication Solutions, LLC (North Wales, PA, USA).
The copyright line for this article was changed on 2 November 2016 after original online publication
References
- 1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014; 311: 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jensen MD, Ryan DH, Apovian CM et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63: 2985–3023. [DOI] [PubMed] [Google Scholar]
- 3. NIH and NHLBI Obesity Education Initiative . Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults. 1998. [WWW document]. URL http://www.nhlbi.nih.gov/guidelines/obesity/ob_gdlns.pdf. [PubMed]
- 4. Billes SK, Sinnayah P, Cowley MA. Naltrexone/bupropion for obesity: an investigational combination pharmacotherapy for weight loss. Pharmacol Res 2014; 84: 1–11. (accessed August 2015). [DOI] [PubMed] [Google Scholar]
- 5. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci 2005; 8: 571–578. [DOI] [PubMed] [Google Scholar]
- 6. Sinnayah P, Wallingford N, Evans A, Cowley MA. Bupropion and naltrexone interact synergistically to decrease food intake in mice. Paper Presented at the North American Association for the Study of Obesity Annual Scientific Meeting, New Orleans, LA, October 20–24, 2007.
- 7. Greenway FL, Whitehouse MJ, Guttadauria M et al. Rational design of a combination medication for the treatment of obesity. Obesity (Silver Spring) 2009; 17: 30–39. [DOI] [PubMed] [Google Scholar]
- 8. Apovian CM, Aronne L, Rubino D et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity‐related risk factors (COR‐II). Obesity (Silver Spring) 2013; 21: 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greenway FL, Fujioka K, Plodkowski RA et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR‐I): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2010; 376: 595–605. [DOI] [PubMed] [Google Scholar]
- 10. Hollander P, Gupta AK, Plodkowski R et al. Effects of naltrexone sustained‐release/bupropion sustained‐release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36: 4022–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wadden TA, Foreyt JP, Foster GD et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR‐BMOD trial. Obesity (Silver Spring) 2011; 19: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glucophage (Metformin Hydrochloride) Prescribing Information. Bristol‐Myers Squibb Company: Princeton, NJ, 2009. [Google Scholar]
- 13. Naltrexone Hydrochloride Tablets Prescribing Information. Mallinckrodt Inc: Hazelwood, MO, 2014. [Google Scholar]
- 14. Contrave (Naltrexone HCl and Bupropion HCl) Prescribing Information. Takeda Pharmaceuticals America, Inc.: Deerfield, IL, 2014. [Google Scholar]
- 15. Zyban (Bupropion Hydrochloride) Prescribing Information. GlaxoSmithKline: Research Triangle Park, NC, 2014. [Google Scholar]
- 16. Lean ME, Carraro R, Finer N et al. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non‐diabetic adults. Int J Obes 2014; 38: 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bower G, Toma T, Harling L et al. Bariatric surgery and non‐alcoholic fatty liver disease: a systematic review of liver biochemistry and histology. Obes Surg 2015; 25: 2280–2289. [DOI] [PubMed] [Google Scholar]
- 18. Brownell KD. The LEARN Program for Weight Control, 7th edn. American Health Publishing: Dallas, TX, 1998. [Google Scholar]
- 19. Knowler WC, Barrett‐Connor E, Fowler SE et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]