

Abstract
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disorder that is caused by a CAG expansion in the Huntingtin (HTT) gene, leading to HTT inclusion formation in the brain. The mutant huntingtin protein (mHTT) is ubiquitously expressed and therefore nuclear inclusions could be present in all brain cells. The effects of nuclear inclusion formation have been mainly studied in neurons, while the effect on glia has been comparatively disregarded. Astrocytes, microglia, and oligodendrocytes are glial cells that are essential for normal brain function and are implicated in several neurological diseases. Here we examined the number of nuclear mHTT inclusions in both neurons and various types of glia in the two brain areas that are the most affected in HD, frontal cortex, and striatum. We compared nuclear mHTT inclusion body formation in three HD mouse models that express either full‐length HTT or an N‐terminal exon1 fragment of mHTT, and we observed nuclear inclusions in neurons, astrocytes, oligodendrocytes, and microglia. When studying the frequency of cells with nuclear inclusions in mice, we found that half of the population of neurons contained nuclear inclusions at the disease end stage, whereas the proportion of GFAP‐positive astrocytes and oligodendrocytes having a nuclear inclusion was much lower, while microglia hardly showed any nuclear inclusions. Nuclear inclusions were also present in neurons and all studied glial cell types in human patient material. This is the first report to compare nuclear mHTT inclusions in glia and neurons in different HD mouse models and HD patient brains. GLIA 2016;65:50–61
Keywords: Huntington's disease, inclusions, astrocytes, microglia, oligodendrocytes, neurons
Introduction
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disorder. Patients suffer from chorea, random involuntary movements, and several psychiatric and cognitive symptoms such as mood changes, depression, and dementia. The prevalence of HD is 10.6–13.7 individuals per 100,000 in the Western world (Evans et al., 2013; Fisher and Hayden, 2014; Morrison et al., 2011). HD is caused by a CAG expansion in the Huntingtin (HTT) gene resulting in a polyglutamine‐expansion in the mutant HTT protein (mHTT), and the formation of mHTT inclusions in the nucleus and the cytoplasm of cells. While the appearance of inclusions is an important hallmark of the disease, it is debated whether these structures are toxic because the presence of inclusions is inversely correlated with neuronal cell death in a cell model (Arrasate et al., 2004; Nucifora et al., 2012).
HD‐related research is primarily focusing on alterations in the brain, and neurons have been studied mostly as they seem to be the most affected cell type in HD. Glial cells have been comparatively disregarded despite their critical role in brain function. Both astrocytes and microglia directly regulate synaptic communication: astrocytes take up neurotransmitters from the synaptic cleft and release gliotransmitters that facilitate neuronal communication, and both microglia and astrocytes are involved in synaptic pruning and are responsive to neurotransmitters (Chung et al., 2013; Kettenmann et al., 2013; Pekny et al., 2016; Schafer et al., 2013). In HD patients, astrocytes and microglia become activated, as shown by the upregulation of glial fibrillary acidic protein (GFAP) and thymosin β4, respectively, and the degree of activation correlates with disease progression (Faideau et al., 2010; Sapp et al., 2001). In mHTT gene carriers, microglia activation occurs before the first symptoms become apparent as has been shown using positron emission tomography (PK PET) (Tai et al., 2007). Also, increased numbers of oligodendrocytes and GFAP‐positive astrocytes are observed as early as the first pathological stages in HD patients become manifest (Faideau et al., 2010; Myers et al., 1991). Transgenic PLP‐HTT mice, expressing mHTT exclusively in oligodendrocytes, show a progressive neurological phenotype and a reduced myelin protein expression, which makes it clear that oligodendrocytes can be affected by mHTT expression (Huang et al., 2015). Also, normal astrocyte functions are disturbed, as shown by decreases in glutamate transporter levels, functional glutamate transport, and the secretion of the neurotropic molecule CCL5/RANTES (Chou et al., 2008; Shin et al., 2005). In addition, alterations in potassium homeostasis occur in HD astrocytes due to a decrease in the inward rectifier potassium channel Kir4.1 levels (Tong et al., 2014). Astrocyte dysfunction contributes to HD, as astrocyte‐specific mHTT expression exacerbated the HD phenotype in drosophila and mice (Bradford et al., 2010; Kretzschmar et al., 2005). Microglia also change from their normal scanning mode into an immune activated phenotype and start to accumulate ferritin (Simmons et al., 2007). When expressing mHTT, microglia secrete proinflammatory cytokines and their migration toward chemotactic stimuli is impaired, probably because of defective actin remodeling (Kwan et al., 2012; Silvestroni et al., 2009).
Despite the accumulating evidence that glial cells are affected in HD mouse models and human HD patients, there are only a few studies reporting the presence of mHTT inclusions in astrocytes and oligodendrocytes (Huang et al., 2015; Shin et al., 2005; Tong et al., 2014), and the presence of inclusions in microglia has so far not been described. Here we have assessed the proportion of different types of glia and neurons with nuclear mHTT inclusions in the brain areas that are most affected in the disease: frontal cortex and striatum. We studied this in three commonly used HD mouse models that show differences in disease progression. The R6/2 model (Mangiarini et al., 1996) expresses the N‐terminal mHTT‐exon 1 protein, and has a much faster progression of symptoms and pathology when compared with the HdhQ150 and zQ175 HD mouse models that express full‐length mHTT (Lin et al., 2001; Menalled et al., 2012). Time points selected for investigations were based on the phenotype seen in the mice at a given age. Nuclear inclusions were also present in neurons and all studied glial cell types in human patient material. We found that proportion and size of nuclear inclusions differs between the different cell types, with neuronal inclusions being generally larger and more abundant than nuclear inclusions in glia.
Materials and Methods
Mouse Models
In this study, 4, 9, and 14‐week‐old R6/2 mice (Mangiarini et al., 1996), 2‐, 16‐, and 22‐month‐old homozygous HdhQ150 mice (Lin et al., 2001), 3‐, 6‐, 8‐, and 12‐month‐old heterozygous zQ175 mice (Menalled et al., 2012), and wild‐type littermates were used. The R6/2 mice analyzed had an average CAG expansion of 210 ± 4 (SD). The HdhQ150 homozygous mice used in this study had expanded CAG repeat sizes of 190 ± 16 (SD) on the large allele and 169 ± 12 (SD) on the smaller allele. For the zQ175 line, we analyzed tissue with a mean CAG repeat expansion of 193 ± 4 (SD). The end‐stage phenotypes for R6/2 mice (∼13 weeks) and homozygous HdhQ150 mice (∼22 months) are highly comparable (Kuhn et al., 2007; Labbadia et al., 2011; Mielcarek et al., 2014, 2015; Moffitt et al., 2009; Woodman et al., 2007), including the gross distribution of inclusions in the CNS (Woodman et al., 2007) and periphery (Moffitt et al., 2009). The main difference between the two lines is the age of phenotype onset and rate of disease progression (Lin et al., 2001). The zQ175 model originated from a spontaneous mutation in the chimeric knock‐in model with 140 CAGs (Menalled et al., 2003). In this model, HTT exon 1 is of human origin, whereas the rest of the Htt gene is murine. Progression of the disease occurs more rapidly in both heterozygous and homozygous zQ175 mice (Menalled et al., 2012) as compared with the HdhQ150 model, and the end stage of disease occurs at approximately 20 months in zQ175 heterozygotes.
Isolation of Astrocyte and Microglia Cell Populations
Wild‐type mice—6 and 11 weeks old—were used in this experiment (n = 12). The mice were anesthetized by i.p. injection of 100 µL sodium pentobarbital and perfused with 0.9% NaCl (pH 5.5). All experiments were performed according to the experimental animal guidelines of the University Medical Center Groningen. Animals were housed under normal conditions in a 12‐h light/dark cycle and fed ad libitum in the central experimental animal facility of the University of Groningen. After dissecting the brains from the skull, the cortex was isolated and cut into small pieces in ice‐cold dissection medium (Hanks bovine salt serum [HBSS], PAA, Cat.nr. H15‐010; 1.3% d‐(+)‐glucose solution, Sigma, Cat.nr. G8769; 1.5% 4‐(2‐hydroxyethyl)−1‐piperazineethanesulfonic acid [HEPES], PAA, 311‐001). Subsequently, the tissue was incubated in dissection medium supplemented with papain (8.7 U/mL) and 0.5 µg/mL DNAse1 at 37°C for 60 min. Enzyme digestion was stopped by adding fetal bovine serum (FBS) to an end concentration of 10%. Tissue fragments were triturated and 4 mL percoll (90% percoll, 150 mM NaCl) was added to a volume of 3 mL dissection medium and centrifuged at 200g with low brake at 4°C for 20 min. The top phase was discarded, and the Percoll layer containing the cells and the myelin layer were collected and diluted five times using dissection medium, followed by centrifugation at 200g at 4°C for 10 min. To avoid oligodendrocyte contamination, cell pellets were resuspended in MACS buffer (2 mM Ethylenediaminetetraacetic acid [EDTA] and 0.5% bovine serum albumin [BSA] in PBS pH 7.2) and incubated with myelin removal beads (Miltenyi Biotec; 130‐096‐733) at 4°C for 15 min. Subsequently, the samples were centrifuged at 200g at 4°C for 5 min, supernatant was discarded and cells were resuspended in MACS buffer. Cells were then loaded on MACS columns, which were washed thrice with MACS buffer to elute the cells for FACS sorting.
FACS Procedure
The obtained cell suspension was incubated with CD16/CD32 (1:100, eBioscience) to block Fc gamma receptors and prevent monocyte isolation. After washing, cells were resuspended and incubated with anti‐GLT‐1 (1:100, 0.6 mg/mL (Orre et al., 2014b) in staining medium (HBSS, PAA, 1.3% d‐(+)‐glucose solution, Sigma, Cat.nr. G8769; 1.5% HEPES, PAA, 311‐001, 0.1 mM EDTA) at 4°C for 30 min. After washing, cells were incubated with the secondary antibody anti‐rabbit‐Alexa488 (1:200) and the conjugated antibodies CD45‐PeCy7 (1:200 eBioscience) and CD11B‐PE (1:150, eBioscience) for 30 min at 4°C. Cells were washed with staining medium and 4′,6‐diamidino‐2‐fenylindool (DAPI) staining was used to sort living cells. Using an MoFlo XDP sorter (Beckman Coulter), GLT+ astrocytes were sorted based on a GLT+/CD11B− expression and microglia were sorted on CD11B+/CD45+ expression. The sorted cells were subjected to RNA isolation.
RNA Isolation and QPCR
RNA was isolated from the cells by adding TRIzol (400 μL, Thermo Fisher) to the cell pellets, followed by addition of chloroform (80 μL). Samples were centrifuged, the top phase was collected and mixed with an equal volume of isopropanol and 1 μL glycogen (20 mg/μL, Invitrogen). Samples were stored overnight at −20°C. The next day, the samples were centrifuged (max. speed for 1 h at 4°C) and pellets were washed twice with 75% ethanol. The total sample was used as template for cDNA synthesis with a mix of oligo(dT) and random hexamer primers according to manufacturer's instructions (Quantitect‐Qiagen). After DNAse treatment, the RNA was incubated with reverse transcriptase for 30 min at 42°C. The samples were diluted 1:20 to serve as a template for real‐time quantitative polymerase chain reaction (QPCR) analysis. QPCR was used for the quantification of expression of endogenous Htt levels using the primers 5′‐ATCTCAGCCAGTCTGGTGCT‐3′ and 5′‐CCCACTGTTCTGGAGGTGTT‐3′. Hprt, Gapdh, Actg1, and Acta1 were used for normalization.
Volcano plot analysis of proteomics data of Sharma et al. 2015
The Volcano plots (Supp. Info., Fig. 1B) were generated by using data of Sharma et al. (2015). In this study, neurons, microglia, astrocytes, and oligodendrocytes were isolated from 9‐week‐old C57BL/6 mice, which were used for proteomics analysis to determine cell‐type‐specific expression patterns. The data were used to plot HTT expression in each cell type in relation to their specific markers. In the Volcano plot, Log2 protein intensity per cell type was plotted against the median abundance of all cell types together.
Mouse Tissue Preparation for Immunofluorescence Microscopy
All experimental procedures performed on mice were conducted under a project license from the Home Office and approved by the King's College London Ethical Review Process Committee in the UK. Hemizygous R6/2 mice were bred by backcrossing R6/2 males to (CBA × C57BL/6) F1 females (B6CBAF1/OlaHsd, Harlan Olac, Bicester, UK). HdhQ150 homozygous mice on a (CBA × C57BL/6) F1 background were obtained by intercrossing HdhQ150 heterozygous CBA/Ca and C57BL/6J congenic lines as described previously (Woodman et al., 2007). zQ175 heterozygous mice were maintained by backcrossing to C57BL/6J females (Stock 000664, Charles River). All animals were fed ad libitum (Special Diet Services, Witham, UK) and mice were subjected to a 12‐h light/dark cycle.
The mice were anesthetized by i.p. injection of 100 µL sodium pentobarbital (juvenile mice < 5 weeks 50 µL) and perfused with 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (NaH2PO4, pH 7.4). After dissecting the brains from the skull, the brains were postfixed in 4% PFA at 4°C overnight. Brains were stored in 0.1 M NaH2PO4/0.5% PFA until further use. Brains were washed with phosphate‐buffered saline (PBS, 154 mM NaCl, 1.1 mM KH2PO, 5.6 mM Na2HPO4, pH 7.4 Lonza) and incubated in 20% sucrose in PBS overnight. Subsequently, brains were frozen on dry ice and stored at −80°C. Brains were cut coronally in 10‐µm‐thick sections that were mounted on Starfrost microscope slides and stored at −20°C until further use. Two mice brains were analyzed for each time point, and the immunostaining experiments were replicated at least once for each mouse.
Immunofluorescent Staining of Mouse Tissue Sections
Sections were postfixed with 4% PFA for 15 min and washed with PBS. Afterwards, the sections were heated for antigen retrieval in 10 mM sodium citrate + 0.05% tween‐20 at 85–95°C for 10 min. Subsequently, sections were rinsed in PBS, blocked and permeabilized with 1% BSA, 2% FBS, and 0.4% Triton X‐100 in PBS for 1 h, and incubated overnight with primary antibodies within the same buffer. Antibodies used are listed in Table 1. Sections were subsequently washed with PBS and incubated with fluorescent secondary antibodies donkey‐anti‐mouse, ‐rabbit, or ‐goat with Alexa488, Cy3, or Cy5 tags to visualize immunostaining (1:700 Jackson ImmunoResearch Laboratories). Finally, sections were washed in PBS and embedded in Vectashield, including DAPI to stain nuclei (Vector Laboratories). All procedures were performed at room temperature.
Table 1.
Antibodies
| Company | Species | Dilution (mouse) | Dilution (human) | FACS dilution | |
|---|---|---|---|---|---|
| NeuN | Millipore (MAB377) | Mouse monoclonal | 1:500 | 1:500 | |
| S100B | DAKO (Z0311) | Rabbit polyclonal | 1:4,500 | 1:600 | |
| GFAP | DAKO (Z0334) | Rabbit polyclonal | 1:2,000 | 1:1,000 | |
| IBA1 | Wako (019‐19471) | Rabbit polyclonal | 1:4,000 | 1:1,000 | |
| OLIG2 | Chemicon (Ab9610) | Rabbit polyclonal | 1:500 | 1:200 | |
| S829 | (Sathasivam et al., 2001) | Sheep polyclonal | 1:800 | 1:500 | |
| GLT‐1 | (Orre et al., 2014b) | Rabbit polyclonal | 1:100 | ||
| CD11B‐PE | eBioscience | Rat monoclonal | 1:150 | ||
| CD45‐PECy7 | eBioscience | Mouse monoclonal | 1:200 |
Human HD Patient Material and Immunofluorescent Staining
Frontal cortex and striatum of 6 human HD patient cases were used for analysis. Details are presented in Table 2 (Donor 1‐6). Brain tissue was formalin fixed and paraffin embedded. Sections of 6 µm were used for immunostaining. Sections were deparaffinized and washed with PBS. The staining procedure was similar as described for the mouse tissue immunostaining using the same antibodies (Table 1).
Table 2.
Clinopathological Table
| Donor ID | Gender | Age of onset | Age | CAG repeats | VS grade | Brain area | Source |
|---|---|---|---|---|---|---|---|
| 1 | m | >61 | 68 | 19/40 | 3 | Frontal cortex | W. den Dunnen (UMCG) |
| 2 | m | >33 | 51 | 25/49 | 4 | Frontal cortex | W. den Dunnen (UMCG) |
| 3 | m | >55 | 64 | 17/41 | 2 | Frontal cortex | W. den Dunnen (UMCG) |
| 4 | f | ∼10 | 11 (JHD) | 22/84 | n.d. | Caudate+ accumbens/frontal cortex | W. van Roon (LUMC) |
| 5 | f | ∼6 | 20 (JHD) | 17/86 | 3 | Caudate putamen/frontal cortex | W. van Roon (LUMC) |
| 6 | m | n.d. | 37 (JHD) | 15/68 | 3 | Caudate putamen/frontal cortex | W. van Roon (LUMC) |
| 7 | m | Control | 51 | n.d. | – | Caudate+ accumbens/frontal cortex | W. van Roon (LUMC) |
| 8 | m | Control | 54 | n.d. | – | Striatum/frontal cortex | W. van Roon (LUMC) |
| 9 | f | Control | 61 | n.d. | – | Striatum/frontal cortex | W. van Roon (LUMC) |
| 10 | f | n.d. | 57 | 23/43 | 4 | Striatum/cortex | W. van Roon (LUMC) |
| 11 | f | 47 | 67 | 15/42 | 1 | Cortex | W. van Roon (LUMC) |
| 12 | m | >70 | 80 | 17/42 | 2 | Cortex | W. van Roon (LUMC) |
| 13 | m | 44 | 68 | 9/44 | 3 | Cortex | W. van Roon (LUMC) |
| 14 | m | 15 | 40 (JHD) | 18/53 | 3 | Cortex | W. van Roon (LUMC) |
M = male; f = female; VS grade = Vonsattel grade; n.d. = no data available.
Image Analysis
To determine the nuclear inclusion proportion per cell type, images were taken with a Leica SP8 confocal microscope using a 63× oil objective and Leica LAS X imaging software. A minimum of five images was taken from each sample, the number of cells differed per cell type ranging from 3 to 50 cells per field. Per image, the total number of cell‐marker‐positive cells and the number of cells with inclusions were counted. Per cell type a minimum of 200 cells were included.
To quantify the number of cells in HD mice and their wild‐type littermates, images were acquired using a Leica DM RA microscope equipped with a Plan Apo 25× 1.40 oil objective (Carl Zeiss), a cooled charge‐coupled device camera (KX1400; Apogee Instruments, Logan, UT, USA), and Image‐Pro Plus software. DAPI‐stained nuclei were counted by a custom‐made Matlab script. With this script, the total nuclear surface area was measured and the number of distinct objects was counted. The number of positive cells was counted manually by persons that were unaware of the mouse genotypes.
Preparation of Insoluble Protein Fractions
Postmortem human brain tissue slides were homogenized with a bullet blender (Next Advance, strength 8, 3 min) in a buffer containing 150 mM sucrose, 15 mM HEPES pH7.9, 60 mM KCl, 0.5 mM EDTA pH8, and 0.1 mM ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid pH8 using a 1:5 weight‐to‐volume ratio. After addition of Triton X‐100 to a final concentration of 1%, samples were kept on ice for 1 h. Insoluble protein pellets were prepared by centrifugation (max. speed for 10 min), and washed thrice with 60 mM Tris. Pellets were resuspended in 15% SDS and incubated overnight at 95°C. Protein concentration of resuspended pellets was determined with the bicinchoninic (BCA) assay kit (Thermo Fisher Scientific, Waltham, USA).
Filter‐Trap Assay
One hundred micrograms of protein suspension in 300 µL of 15% SDS was blotted onto a cellulose acetate membrane using a vacuum applicator. Blots were washed twice with 0.2% SDS and fixed in 0.5% glutaraldehyde. Blots were blocked with 4% nonfat milk (Nutricia, Schiphol, The Netherlands) in Tris‐buffered saline with Tween‐20 and probed with primary anti huntingtin antibody 3702‐1 (Epitomics, Burlinggame CA, USA), and secondary antibody horse radish peroxidase conjugated goat‐anti‐rabbit (Santa Cruz). Blots were visualized with enhanced chemoluminescence (ECL, #32106, ThermoFisher) and Hyperfilm ECL (#28906837, GE healthcare, Little Chalfond, United Kingdom). Signal intensity was quantified with imageJ‐software.
Statistics
Two‐tailed unpaired Student's t‐test was used to assess differences in RNA expression levels, P < 0.05 was accepted as significant. One‐way ANOVA with a Student–Newman–Keuls post hoc test was used to assess differences in inclusion diameters in the various cell types. The Marascuillo procedure was used to compare proportions of inclusions in different cell types and to assess the number of cells in wild‐type and HD mice, for all tests significance level was set at 0.05. Supporting Information, Tables 1–4 display the results of all statistical tests in detail.
Results
Huntingtin is Expressed in Neurons and Glia
Before quantifying the frequency of nuclear mHTT inclusions in neuronal and glial cells, we examined the expression levels of huntingtin in the various cell types of the mouse brain. By data analysis of published transcriptomics and proteomics studies, huntingtin mRNA and protein levels in neurons, astrocytes, oligodendrocytes, and microglia could be determined (Supp. Info., Fig. 1). Whereas mRNA levels were slightly higher in neurons compared with glia in both human and mice (Supp. Info., Fig. 1A, data obtained from (Zhang et al., 2016)), HTT protein levels were similar in oligodendrocytes, astrocytes, neurons, and microglia in mice (Sharma et al., 2015). Compared with the median protein expression levels of all these cell types combined (Log2 fold change, x‐axis), cell‐specific markers were differentially expressed, whereas HTT protein levels (Log2 protein expression intensities, y‐axis) were comparable in all cell types (Supp. Info., Fig. 1B, analyzed data published by Sharma et al., 2015). Htt mRNA expression in astrocytes and microglia was confirmed by QPCR from cells isolated from wild‐type mice using a protocol developed earlier in our lab (Orre et al., 2014b) by dissecting out the cortex and isolating dissociated GLT+ astrocytes and CD11B+/CD45+ microglia by FACS. Htt is expressed in astrocytes and microglia in young mice of 6 and 11 weeks old (Supp. Info., Fig. 1C). These results confirm earlier data on Htt RNA expression in isolated astrocytes and microglia in adult (3 months) and aged (15 months) mice (Orre et al., 2014b).
Differences in Nuclear mHTT Inclusion Formation between Neurons and Glial Cells in R6/2 Mice
While both cytoplasmic and nuclear inclusions were present in R6/2 mice, we deliberately quantified only the nuclear inclusions in neurons, astrocytes, oligodendrocytes, and microglia as it is often difficult to determine whether cytoplasmic inclusions were present in particular cells due to the complexity of brain tissue, and cell‐specific markers do not always label the entire cytoplasm. To determine whether the frequency of nuclear mHTT inclusions differs between neurons and the various glial cell types, we performed immunofluorescent stainings on brain sections from R6/2 mice (Mangiarini et al., 1996). The pathogenesis in R6/2 mice progresses rapidly; inclusions have been observed at postnatal day 0 (P0) and their number increases with age, with the first phenotypes being present as early as P30 and the disease reaching its end stage within 4 months (Stack et al., 2005). To visualize nuclear mHTT inclusions, we used the S829 antibody that was raised against the N‐terminus of polyQ‐expanded HTT exon 1 (Sathasivam et al., 2001), in combination with at least one cell‐type‐specific marker. As GFAP mainly labels reactive astrocytes, we also used S100B as a glial marker to visualize also nonreactive astrocytes.
We analyzed the number of nuclear inclusions in neurons, astrocytes, oligodendrocytes, and microglia in the frontal cortex and striatum of mice aged 4, 9, and 14 weeks. Nuclear inclusions were found in neurons, S100B‐positive glial cells, GFAP‐positive astrocytes, oligodendrocytes, and microglia (Fig. 1A), yet the proportion of these inclusions differed between cell types. While the number of nuclear inclusions increased with age in all cell types in both the frontal cortex and the striatum, neurons contained the highest proportion of nuclear inclusions (around 45% of neurons) at 14 weeks in the frontal cortex. In the striatum, the fraction of S100B‐positive glial cells with nuclear inclusions was lower compared with neurons (around 35%), whereas GFAP‐positive astrocytes and oligodendrocytes showed an even lower percentage (5–10% at 14 weeks). Strikingly, IBA1‐positive microglia hardly showed any nuclear inclusions at all, with 14‐week‐old mice showing only nuclear inclusions in about 1% of the cells (Fig. 1B and Supp. Info., Table 1). In general, nuclear inclusions had a diameter of 1 µm in the cortex and 1–1.5 µm in the striatum in the different types of glial cells. The nuclear inclusions were significantly larger in neurons compared with glia: about 2 µm at 14 weeks (Fig. 1C). In the striatum, neuronal nuclear inclusions were also larger than those in glia at 14 weeks (Fig. 1C). The differences in the proportion of nuclear inclusions between the different cell types was not due to cell death as no differences were observed in the number of cells in wild‐type brains and R6/2 brains (Fig. 1D and Supp. Info., Table 2). Together, these data show that in the R6/2 HD model, nuclear mHTT inclusions are more abundant and larger in neurons compared with glia, with S100B‐positive glial cells showing a higher proportion of nuclear inclusions than GFAP‐positive astrocytes or oligodendrocytes, and that microglia hardly show any nuclear inclusion formation.
Figure 1.
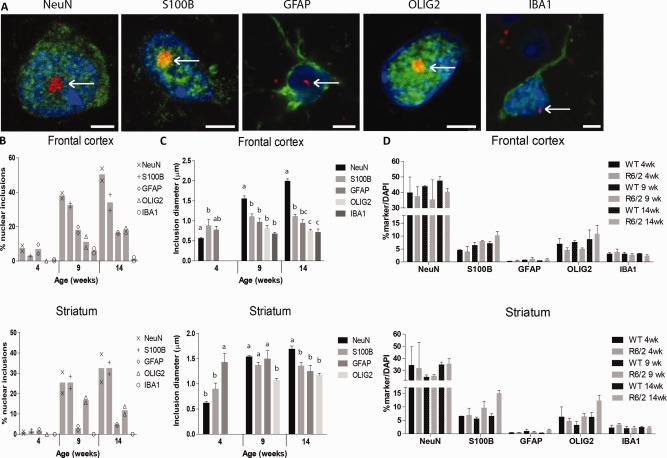
Nuclear mHTT inclusions in R6/2 striatum and cortex. (A) Exemplary images of mHTT inclusions (S829, red) were present in the nuclei (DAPI, blue) of neurons (NeuN, green, striatum), S100B‐positive glia (green, striatum), GFAP‐positive astrocytes (green, frontal cortex), oligodendrocytes (OLIG2, green, striatum), and microglia (IBA1, green, frontal cortex). Representative pictures of each cell, morphology similar in cortex and striatum. Scale bar is 3 µm. (B) The percentage of cells with a nuclear inclusion in the cortex and striatum. (C) The change in the diameter of nuclear inclusions with disease progression in the various cell types. Different letters represent significantly different groups as determined by one‐way ANOVA with Student–Newman–Keuls post hoc test. (D) The percentage of nuclei co‐staining for various cell type markers. Cell numbers were similar between wild‐type and R6/2 mice (N = 11,943–27,037). WT = wild‐type. [Color figure can be viewed at wileyonlinelibrary.com]
Differences in Nuclear mHTT Inclusions Between Neurons and Glial Cells in Knock‐In HD Mouse Models
We also examined the proportion of nuclear inclusions in the various cell types in two knock‐in HD mouse models that express the full‐length mHTT protein. The first was the HdhQ150 model, where approximately 150 CAGs were knocked into the mouse Htt gene (Lin et al., 2001). The second full‐length mHTT model was the zQ175 model, which is chimeric for human HTT exon 1, with approximately 175 CAGs, and the remainder of the mouse Htt gene (Menalled et al., 2012). Similar to the R6/2 model, nuclear inclusions were found in all cell types studied in HdhQ150 mice (Fig. 2A), and the percentage of cells that had nuclear inclusions was comparable to the R6/2 model. Neurons contained the highest frequency of nuclear inclusions, closely followed by S100B‐positive glial cells. Around 10% of GFAP‐positive astrocytes and oligodendrocytes contained nuclear inclusions, while microglia only incidentally contained nuclear inclusions (Fig. 2B and Supp. Info., Table 1). Average nuclear inclusion diameter in the HdhQ150 model was 1.3 µm in S100B‐positive glial cells, 1 µm in GFAP‐positive astrocytes and oligodendrocytes, and 2 µm in neurons at the disease end stage (Fig. 2C). We observed a significant 50% increase in GFAP expression in the striatum (2, 16, and 22 months) and a 75% increase in the frontal cortex (16 and 22 months) (Fig. 2D and Supp. Info., Table 3), consistent with what was described before (Lin et al., 2001). In addition, the number of S100B‐positive glial cells in both frontal cortex (16 months: +33%; 22 months: +50%) and striatum (22 months: +93%) and the number of microglia in the frontal cortex (22 months: +30%) were significantly increased at end‐stage disease in HdhQ150 mice.
Figure 2.
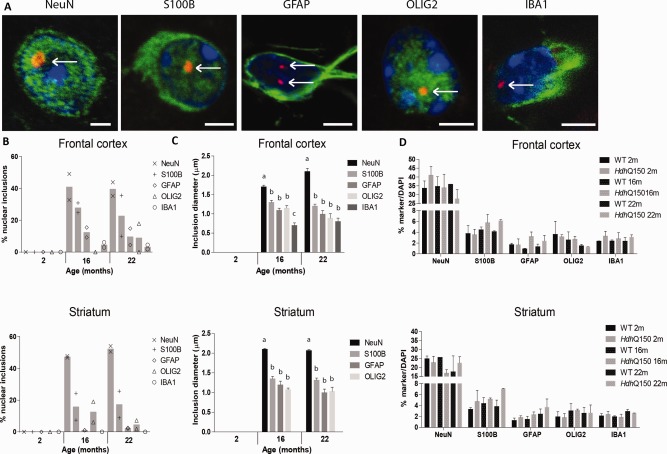
Nuclear mHTT inclusions in the homozygous HdhQ150 mouse striatum and cortex. (A) Exemplary images of mHTT inclusions (S829, red) were present in nuclei (DAPI, blue) of neurons (NeuN, green, striatum), S100B‐positive glia (green, striatum), GFAP‐positive astrocytes (green, striatum), oligodendrocytes (OLIG2, green, striatum), and microglia (IBA1, green, frontal cortex). Representative pictures of each cell, morphology similar in cortex and striatum. Scale bar is 3 µm. (B) The percentage of cells with a nuclear inclusion in the cortex and striatum. (C) The change in diameter of nuclear inclusions with disease progression in the various cell types. Different letters represent significantly different groups as determined by one‐way ANOVA with Student–Newman–Keuls post hoc test. (D) The percentage of nuclei co‐staining for various cell type markers. Quantification of total cell numbers showed an increase in S100B‐postive glia and GFAP‐positive astrocytes in HdhQ150 brains as compared with wild‐type (minimum N = 10,005–20,879). WT = wild‐type. [Color figure can be viewed at wileyonlinelibrary.com]
Consistent with our R6/2 and HdhQ150 data, we observed nuclear inclusions in the zQ175 sections in neurons and all types of glia analyzed (Fig. 3A). The proportion of cells containing a nuclear inclusion increased with disease progression in both frontal cortex and striatum, and was comparable to the proportions observed in the other HD mouse models (Fig. 3B and Supp. Info., Table 1). Neurons and S100B‐positive glial cells contained the highest proportion of nuclear inclusions (around 35% of all cells) in both frontal cortex and striatum, followed by the GFAP‐positive astrocytes and oligodendrocytes (around 5% of all cells), whereas nuclear inclusions were hardly present in microglia. Although the abundance of zQ175 nuclear mHTT inclusions was similar to R6/2 and HdhQ150, all nuclear inclusions were slightly smaller in this model (Fig. 3C), most likely reflecting the fact that tissue from end‐stage disease was not available. We observed significantly lower numbers of neuronal cells in both frontal cortex and striatum in 12‐month‐old zQ175 mice (frontal cortex: −27%; striatum: −41%); however, in glia no major decrease in numbers were detected. So, similar to R6/2 and HdhQ150, also in zQ175 mice the lower proportion of nuclear inclusions in glial cells compared to neurons is not due to glial cell death (Fig. 3D and Supp. Info., Table 4).
Figure 3.
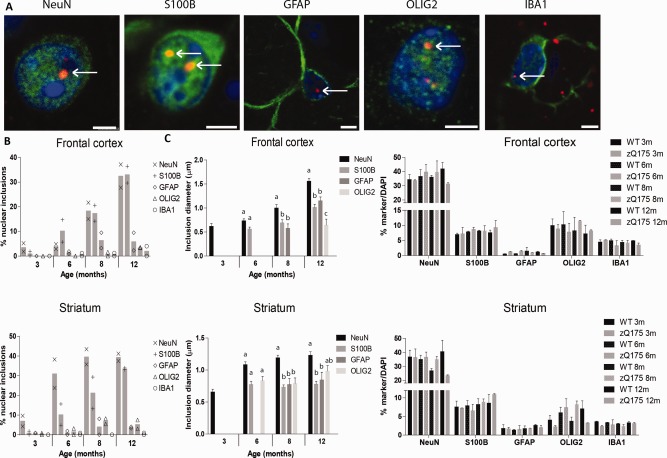
Nuclear mHTT inclusions in the zQ175 model. (A) Exemplary images of HTT inclusions (S829, red) were present in the nuclei (DAPI, blue) of neurons (NeuN, green, frontal cortex), S100B‐positive glia (green, striatum), GFAP‐positive astrocytes (green, frontal cortex), oligodendrocytes (OLIG2, green, striatum), and microglia (IBA1, green, frontal cortex). Representative pictures of each cell, morphology similar in cortex and striatum. Scale bar is 3 µm. (B) The percentage of cells with a nuclear inclusion in the cortex and striatum. (C) The change in diameter of nuclear inclusions with disease progression in the various cell types. Different letters represent significantly different groups as determined by one‐way ANOVA with Student–Newman–Keuls post hoc test. (D) The percentage of nuclei co‐staining for various cell type markers. Quantification of cell numbers in wild‐type mice and zQ175 mice (minimum N = 9,629–17,518). WT = wild‐type. [Color figure can be viewed at wileyonlinelibrary.com]
Comparison of HD Mouse Models with Human HD Patient Material
Next to the three HD mouse models, we performed also double labeling of HTT and cell‐specific markers in the frontal cortex and striatum from HD postmortem brains. The percentage of cells containing a nuclear inclusion (max. 0.3% in neurons) was much lower in human patients compared with the HD mouse models (max 40–50% in neurons) (Fig. 4B). In juvenile HD (JHD) patient brains, we found nuclear inclusions in all cell types (Fig. 4A), and in a higher percentage of cells than in the adult onset HD brains (Fig. 4C). This is consistent with increased insoluble HTT protein due to the increase in Q length (Supp. Info., Fig. 2), although filter‐trap analysis showed that JHD 86Q sample produced less aggregates than the 55Q sample. This could be related to subject‐to‐subject variations or to the young age of the Juvenile HD patient with 86Q who died at the age of 11, whereas the other patients were adults with the average age of 60. Also in JHD, more nuclear inclusions were found in the frontal cortex than in the striatum (Fig. 4C and Supp. Info., Fig. 2).
Figure 4.
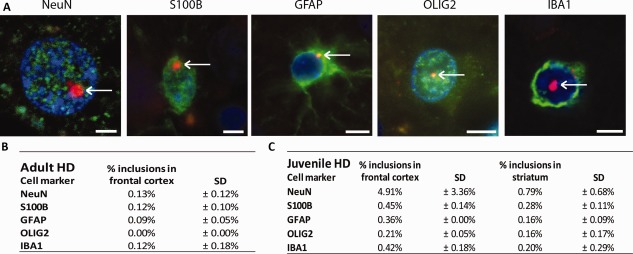
Nuclear mHTT inclusions in Huntington's disease postmortem human brain. (A) Exemplary images of nuclear mHTT inclusions (S829, red) were present in the nuclei (DAPI, blue) of neurons (NeuN, green, frontal cortex), S100B‐positive glia (green, frontal cortex), GFAP‐positive astrocytes (green, striatum, frontal cortex), and microglia (IBA1, green, frontal cortex) oligodendrocytes (OLIG2, green, frontal cortex). Representative pictures of each cell, morphology similar in cortex and striatum. Scale bar is 3 µm. (B) The proportion of the various cell types that contain a nuclear inclusion in the frontal cortex of adult‐onset patients. (C) The proportion of the various cell types that contain a nuclear inclusion in the frontal cortex and striatum of juvenile‐onset patients. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
While mHTT aggregation has been mainly observed and studied in neurons, we investigated nuclear mHTT inclusions in glial and neuronal cells in three commonly used HD mouse models, the R6/2 transgenic model and the HdhQ150 and zQ175 knock‐in models, and in adult and juvenile HD patient postmortem brains. We showed that nuclear inclusions are present in neurons, astrocytes, oligodendrocytes, and microglia but with very different frequencies. Interestingly, the three mouse models showed a similar percentage of nuclear mHTT inclusion bearing cells in the cortex and striatum, despite the expression of full‐length mHTT or only mHTT exon1. At late stages of the disease, nuclear inclusions were found in 30–50% of the neurons, 30% of the S100B‐positive glial cells, 4–10% of the GFAP‐positive astrocytes, 3–10% of the oligodendrocytes, and 0–2% of the microglia. Both the proportion of cells containing nuclear inclusions and the nuclear inclusion size was lower in glial cells compared with neurons. In neurons, nuclear inclusions were on average 2 µm in diameter, whereas glial nuclear inclusions measured only 1 µm on average. Interestingly, only neuronal nuclear inclusions changed in size with disease progression, increasing from 0.6 µm to a maximum of 2 µm at the end stage. This is in agreement with earlier findings in R6/2 mice, zQ175 mice, and human patients (Carty et al., 2015; Gutekunst et al., 1999; Li et al., 1999). This suggests that nuclear inclusions persist for a long period of time and increase in size, most likely due to sequestration of newly synthesized mHTT but also the sequestering of other proteins. As the nuclear inclusions in all cell types were also positively immunostained for ubiquitin (data not shown), this indicates that the used S829 antibody against mHtt recognizes nuclear inclusions in all cell types with similar efficiency. Interestingly, an increase in nuclear inclusion size with disease progression was not observed in glia, suggesting a better capability of these cells to constrain mHTT inclusion size. As we did not find indications of cell death in glia, the stable nuclear inclusion size and lower nuclear inclusion proportion in glia suggests that the various glial cell types are better capable to slow down mHTT aggregation. In addition, it has been suggested that the cellular compartment influences inclusion structure due to interaction with chaperones and other factors, so the composition of nuclear inclusions may be different in glia. Indeed, it has been recently shown that brain material derived from HD patients contains a number of structurally different types of inclusions, and that amorphous inclusions are less toxic than fibrillary inclusions (André et al., 2013; Hoffner and Djian, 2015). We deliberately quantified only the nuclear inclusions in the studied cell types as the used markers for co‐staining did not label the entire cytoplasm, making it too difficult to determine whether cytoplasmic inclusions were indeed present in particular cells that were immunostained for GFAP, NeuN, S100B, OLIG2, or IBA1.
Nuclear mHTT Inclusions in Astrocytes
Interestingly, there is a large diversion in the occurrence of nuclear inclusions in S100B‐positive glial cells (20–35%) compared with GFAP‐positive astrocytes (2–10%). S100B is not exclusively expressed in astrocytes, but also in oligodendrocytes, NG2 cells, and neurons (Vives et al., 2003). Immunostaining experiments combining S100B with astrocyte markers GS and ALdh1L1 showed that 50% of both GS and ALdh1L1 cells were positive for S100B cells and vice versa. Co‐staining S100B with NeuN showed no co‐staining of S100B with NeuN, excluding neuronal staining by S100B (data not shown). S100B and GFAP may represent different cell populations, as GFAP is an astrocyte‐specific protein but it is not expressed by every astrocyte in mouse brains. GFAP is a marker for astrogliosis, a process where astrocytes become activated in response to brain damage as a result of trauma, stroke, or in neurodegenerative diseases (Hol and Pekny, 2015; Middeldorp and Hol, 2011; Pekny et al., 2016). Reactive astrocytes change their gene expression profile toward a more proinflammatory phenotype, including potential changes in protein homeostasis. Upregulation of immunoproteasome components (catalytic subunits and proteasome activator PA28αβ) and several heat‐shock proteins (HSP70, HSPB1, and HSPB3) has been observed in reactive astrocytes in the cortex of a mouse model for Alzheimer's Disease (Orre et al., 2014a). As reactive astrocytes have an increased proteasome activity, which correlates with GFAP upregulation, this could explain the reduced proportion of nuclear mHTT inclusions in GFAP‐positive astrocytes (Middeldorp et al., 2009; Orre et al., 2013). As the number of GFAP‐positive cells were not increased, it is unclear whether in our models true reactive gliosis is observed.
Mechanisms behind Lower Number of Nuclear Inclusions in Microglia
The number of inclusions is most likely related to mHtt protein levels, which is regulated by both mHTT synthesis and degradation. mHTT expression has been determined at the mRNA level in astrocytes, neurons, oligodendrocytes, and microglia for both human and mice (Supp. Info., Fig. S1A). The two studied full‐length mHTT mouse models (HdhQ150, zQ175) have the mouse Htt promotor/enhancer region, resulting in similar levels of Htt expression compared with endogenous Htt expression in wild‐type mice. As the expression level of mHTT is similar in neurons and glia, this study is indicative of a cell‐type‐specific sensitivity for inclusion formation with microglia being the least sensitive, while neurons are most vulnerable. The surprisingly low proportion of nuclear mHTT inclusions in microglia suggests that these cells may be able to prevent the aggregation process. One explanation could be that microglia are able to proliferate under neurodegenerative conditions, in contrast to neurons and astrocytes, as was demonstrated in both Alzheimer's disease and HD (Kamphuis et al., 2012; Kraft et al., 2012). This would result in reduced mHTT levels in their progeny, thereby reducing the likelihood of inclusion formation. The observed asymmetrical diversion of inclusions to one of the daughter cells (Rujano et al., 2006) could also result in higher number of daughter cells without inclusions. However, cell proliferation cannot fully explain the discrepancy in the inclusion proportion because we only observed major increases in the number of astrocytes and microglia in the HdhQ150 model but not the other models used, and astrocytes only proliferate in the adult brain to a limited extend (Kamphuis et al., 2012). Besides, no clear microglia activation could be observed in the mouse models. Next to the possibility that microglia containing nuclear inclusions die and are replaced by new cells, differences in protein degradation pathways may underlie the observed dissimilarities in inclusion frequency in mice. In HD patients, we observed activation of astrocytes and microglia (data not shown), which is consistent with previous findings (Faideau et al., 2010; Sapp et al., 2001). Immune activation is in AD associated with higher levels and increased activity of immunoproteasome subunits in astrocytes and microglia (Orre et al., 2013) and autophagosomes in microglia (Su et al., 2016). These changes can lead to a more efficient degradation of aggregation‐prone proteins and thus might underlie the lower proportion of nuclear inclusions in glia. In human HD patients, the number of inclusions was much lower compared with mice, as previously described (Seidel et al., 2016). However, in HD patients, HTT is also present as neuropil inclusions/granular cytoplasmic structures (DiFiglia et al., 1997; Seidel et al., 2016). In juvenile HD cases (JHD), more nuclear inclusions were present (Fig. 4C) as previously described (DiFiglia et al., 1997), whereas the used mouse models have even longer polyglutamine repeats compared with human patients. So, while these HD mouse models may not be representative for the human situation in terms of the absolute number of nuclear inclusions, these models provide a valuable insight in the relative differences between multiple cell types in relation to inclusion formation.
Conflict of Interest
The authors have no conflict of interest.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors thank Jan Ruijter for the statistical advice, the core facility Cellular Imaging at the AMC, and especially Ron Hoebe for the technical assistance and Harm Kampinga for the provision of the R6/2 mice for the glia isolations. A.J. was funded by an AMC PhD scholarship.
Contributor Information
Elly M. Hol, Email: e.m.hol-2@umcutrecht.nl
Eric A. Reits, Email: e.a.reits@amc.uva.nl
References
- André W, Sandt C, Dumas P, Djian P, Hoffner G. 2013. Structure of inclusions of Huntington's disease brain revealed by synchrotron infrared microspectroscopy: Polymorphism and relevance to cytotoxicity. Anal Chem 85:3765–3773. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. [DOI] [PubMed] [Google Scholar]
- Bradford J, Shin J, Roberts M, Wang C, Sheng G, Li S, Li X. 2010. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem 285:10653–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty N, Berson N, Tillack K, Thiede C, Scholz D, Kottig K, Sedaghat Y, Gabrysiak C, Yohrling G, von der Kammer H, Ebneth A, Mack V, Munoz‐Sanjuan I, Kwak S. 2015. Characterization of HTT inclusion size, location, and timing in the zQ175 mouse model of Huntington's disease: an in vivo high‐content imaging study. PLoS One 10:e0123527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SY, Weng JY, Lai HL, Liao F, Sun SH, Tu PH, Dickson DW, Chern Y. 2008. Expanded‐polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J Neurosci 28:3277–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA. 2013. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993. [DOI] [PubMed] [Google Scholar]
- Evans SJ, Douglas I, Rawlins MD, Wexler NS, Tabrizi SJ, Smeeth L. 2013. Prevalence of adult Huntington's disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry 84:1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Deglon N, Ferrante RJ, Bonvento G. 2010. In vivo expression of polyglutamine‐expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet 19:3053–3067. doi: 10.1093/hmg/ddq212. Epub 2010 May 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher ER, Hayden MR. 2014. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord 29:105–114. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. 1999. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 19:2522–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffner G, Djian P. 2015. Polyglutamine aggregation in Huntington disease: Does structure determine toxicity? Mol Neurobiol 52:1297–1314. [DOI] [PubMed] [Google Scholar]
- Hol EM, Pekny M. 2015. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol 32:121–130. [DOI] [PubMed] [Google Scholar]
- Huang B, Wei W, Wang G, Gaertig M, Feng Y, Wang W, Li X, Li S. 2015. Mutant huntingtin downregulates myelin regulatory factor‐mediated myelin gene expression and affects mature oligodendrocytes. Neuron 85:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM. 2012. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer's disease mouse model. GLIA 60:615–629. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F, Verkhratsky A. 2013. Microglia: New roles for the synaptic stripper. Neuron 77:10–18. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Kaltenbach LS, Lo DC, Harry GJ. 2012. Activated microglia proliferate at neurites of mutant huntingtin‐expressing neurons. Neurobiol Aging 33:621.e17–621.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar D, Tschäpe J, Bettencourt Da Cruz A, Asan E, Poeck B, Strauss R, Pflugfelder GO. 2005. Glial and neuronal expression of polyglutamine proteins induce behavioral changes and aggregate formation in Drosophila. GLIA 49:59–72. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin R, Shelbourne P, Delorenzi M, Augood SJ, Faull RL, Olson JM, Bates GP, Jones L, Luthi‐Carter R. 2007. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild‐type huntingtin dosage. Hum Mol Genet 16:1845–1861. [DOI] [PubMed] [Google Scholar]
- Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C, Moller T, Stella N, Akassoglou K, Tabrizi SJ, Muchowski PJ. 2012. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest 122:4737–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi‐Carter R, Paganetti P, Bates GP. 2011. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 121:3306–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, Li XJ. 1999. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington's disease transgenic mice. Hum Mol Genet 8:1227–1236. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen‐Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. 2001. Neurological abnormalities in a knock‐in mouse model of Huntington's disease. Hum Mol Genet 10:137–144. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. 1996. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson‐Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. 2012. Comprehensive behavioral and molecular characterization of a new knock‐in mouse model of Huntington's disease: zQ175. PLoS One 7:e49838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. 2003. Time course of early motor and neuropathological anomalies in a knock‐in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol 465:11–26. [DOI] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM. 2011. GFAP in health and disease. Prog Neurobiol 93:421–443. [DOI] [PubMed] [Google Scholar]
- Middeldorp J, Kamphuis W, Sluijs JA, Achoui D, Leenaars CHC, Feenstra MGP, van Tijn P, Fischer DF, Berkers C, Ovaa H, Quinlan RA, Hol EM. 2009. Intermediate filament transcription in astrocytes is repressed by proteasome inhibition. FASEB J 23:2710–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Inuabasi L, Bondulich MK, Muller T, Osborne GF, Franklin SA, Smith DL, Neueder A, Rosinski J, Rattray I, Protti A, Bates GP. 2014. Dysfunction of the CNS‐heart axis in mouse models of Huntington's disease. PLoS Genet 10:e1004550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Toczek M, Smeets CJ, Franklin SA, Bondulich MK, Jolinon N, Muller T, Ahmed M, Dick JR, Piotrowska I, Greensmith L, Smolenski RT, Bates GP. 2015. HDAC4‐myogenin axis as an important marker of HD‐related skeletal muscle atrophy. PLoS Genet 11:e1005021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt H, McPhail GD, Woodman B, Hobbs C, Bates GP. 2009. Formation of polyglutamine inclusions in a wide range of non‐CNS tissues in the HdhQ150 knock‐in mouse model of Huntington's disease. PLoS One 4:e8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison PJ, Harding‐Lester S, Bradley A. 2011. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 80:281–286. [DOI] [PubMed] [Google Scholar]
- Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, Richardson EP, Bird ED. 1991. Decreased Neuronal and Increased Oligodendroglial Densities in Huntington's Disease Caudate Nucleus. J Neuropathol Exp Neurol 50:729–742. [DOI] [PubMed] [Google Scholar]
- Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, Yang G, Ratovitski T, Delannoy M, Muchowski PJ, Finkbeiner S, Legleiter J, Ross CA, Poirier MA. 2012. Identification of novel potentially toxic oligomers formed in vitro from mammalian‐derived expanded huntingtin exon‐1 protein. J Biol Chem 287:16017–16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Jansen AH, Kooijman L, Bossers K, Hol EM. 2014a. Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35:2746–2760. [DOI] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Dooves S, Kooijman L, Chan ET, Kirk CJ, Dimayuga Smith V, Koot S, Mamber C, Jansen AH, Ovaa H, Hol EM. 2013. Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain 136:1415–1431. [DOI] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I, Klooster J, Bossers K, Hol EM. 2014b. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging 35:1–14. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, Hol EM, Sofroniew MV, Verkhratsky A. 2016. Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345. [DOI] [PubMed] [Google Scholar]
- Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MA, van der Want JJ, de Vos RA, Brunt ER, Sibon OC, Kampinga HH. 2006. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol 4:e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. 2001. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 60:161–172. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Woodman B, Mahal A, Bertaux F, Wanker EE, Shima DT, Bates GP. 2001. Centrosome disorganization in fibroblast cultures derived from R6/2 Huntington's disease (HD) transgenic mice and HD patients. Hum Mol Genet 10:2425–2435. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Stevens B. 2013. The? quad‐partite? synapse: Microglia‐synapse interactions in the developing and mature CNS. GLIA 61:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Siswanto S, Fredrich M, Bouzrou M, Brunt ER, van Leeuwen FW, Kampinga HH, Korf HW, Rub U, den Dunnen WF. 2016. Polyglutamine aggregation in Huntington's disease and spinocerebellar ataxia type 3: similar mechanisms in aggregate formation. Neuropathol Appl Neurobiol 42:153–166. [DOI] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique‐Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M. 2015. Cell type‐ and brain region‐resolved mouse brain proteome. Nat Neurosci 18:1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. 2005. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171:1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestroni A, Faull RL, Strand AD, Moller T. 2009. Distinct neuroinflammatory profile in post‐mortem human Huntington's disease. Neuroreport 20:1098–1103. [DOI] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. 2007. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. GLIA 55:1074–1084. [DOI] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. 2005. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol 490:354–370. [DOI] [PubMed] [Google Scholar]
- Su P, Zhang J, Wang D, Zhao F, Cao Z, Aschner M, Luo W. 2016. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 319:155–167. [DOI] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. 2007. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain 130:1759–1766. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. 2014. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci 17:694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives V, Alonso G, Solal AC, Joubert D, Legraverend C. 2003. Visualization of S100B‐positive neurons and glia in the central nervous system of EGFP transgenic mice. J Comp Neurol 457:404–419. [DOI] [PubMed] [Google Scholar]
- Woodman B, Butler R, Landles C, Lupton MK, Tse J, Hockly E, Moffitt H, Sathasivam K, Bates GP. 2007. The Hdh(Q150/Q150) knock‐in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull 72:83–97. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. 2014. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan S, Clarke L, Caneda C, Plaza C, Blumenthal P, Vogel H, Steinberg G, Edwards MB, Li G, Duncan IIIJ, Cheshier S, Shuer L, Chang E, Grant G, Gephart M, Barres B. 2016. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information