

Abstract
The Biology and Disease-driven Human Proteome Project (B/D-HPP) is aimed at supporting and enhancing the broad use of state-of-the-art proteomic methods to characterize and quantify proteins for in-depth understanding of the molecular mechanisms of biological processes and human disease. Based on a foundation of the pre-existing HUPO initiatives begun in 2002, the B/D-HPP is designed to provide standardized methods and resources for mass spectrometry and specific protein affinity reagents and facilitate accessibility of these resources to the broader life sciences research and clinical communities. Currently there are 22 B/D-HPP initiatives and 3 closely related HPP resource pillars. The B/D-HPP groups are working to define sets of protein targets that are highly relevant to each particular field to deliver relevant assays for the measurement of these selected targets and to disseminate and make publicly accessible the information and tools generated. Major developments are the 2016 publications of the Human SRM Atlas and of “popular protein sets” for six organ systems. Here we present the current activities and plans of the BD-HPP initiatives as highlighted in numerous B/D-HPP workshops at the 14th annual HUPO 2015 World Congress of Proteomics in Vancouver, Canada.
Keywords: Biology and Disease-driven Human Proteome Project, B/D-HPP, selected reaction monitoring–MS, SRM–MS, mass spectrometry, MS
Graphical abstract
INTRODUCTION, ORGANIZATION, AND GOALS
Under the aegis of the Human Proteome Organization (HUPO), the Human Proteome Project (HPP) was formed in 2010 and launched over the subsequent years to promote advances in our understanding of the human proteome. The HPP is a focal point for many proteomic research laboratories around the world and enhances the quality and interconnectedness of proteomics data resources (https://www.hupo.org/human-proteome-project/).1–3 The HPP is composed of three resource pillars, the C-HPP (chromosome-centric) initiative and the Biology and Disease-driven B/D-HPP. The B/D-HPP aims to develop targeted and high-throughput proteomics analyses, address research challenges of biological and disease networks, and generate multiplex assays of proteins especially suited for particular cells, tissues, and organs in health and across many diseases (http://www.thehpp.org/BD-HPP.php)
The initial concept of the B/D-HPP was to enhance the slow uptake of proteomics compared with other ‘omics fields in the research and clinical communities. Although protein biochemistry (including generation of mutant proteins) is a component of most published biology or disease-based scientific papers, the vast majority of this global effort remains focused on a small subset of proteins,4 most prominently the kinase families. One reason is the shortage of standardized tools, technologies, and informatics pipelines tailored for biomedical and clinical research.4 Thus, the goal of the multifaceted B/D-HPP is to produce these resources and create communities able to promote the development and adoption of proteomics to address biological and disease questions and to provide a framework for interaction within biology- and disease-focused research groups, including engagement of early career scientists in our field and outreach to the broader scientific communities.
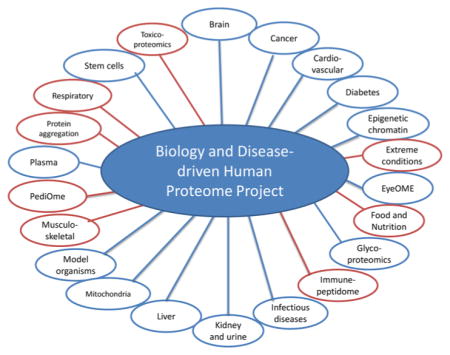
The B/D-HPP is an alliance of independent groups of researchers who are focused on specific diseases or molecular processes (see Figure 1). The B/D-HPP initiatives as of 2013 were brain, cancer, cardiovascular, diabetes, epigenetics and chromatin, eye, glycoproteomics, infectious disease, kidney and urine, liver, mitochondria, model organisms, plasma, and stem cells. Nearly all remain active (see Supplementary Table 1). Since then there has been grass roots expansion of B/D-HPP to embrace initiatives in the areas of extreme conditions, food and nutrition, immunopeptidome, musculo-skeletal, pediatric, protein aggregation, respiratory, and toxicoproteomics. Each B/D-HPP group has a chair and cochairs from different geographic regions of the world; they are responsible for establishing the specific goals and milestones for each group and for participating in workshops and scientific sessions at each annual HUPO Congress as well as their own meetings. (See Supplementary Table 1 for a list of the leaders of each B-D-HPP group and the three primary resource pillars). The goal for each B/D-HPP group is to form a network with their participating researchers to cross-fertilize ideas and share data, facilitate adoption of technologies, and address challenges within their specific disease or molecular process domain. This includes the adoption and promotion of the HUPO standards in all aspects of MS and bioinformatics (including peptide and protein identification). The HPP also encourages links between the B/D and C-HPP groups, now facilitated by the designation by the C-HPP of clusters, as shown on the cover of this Special Issue for cancers, reproductive health, membrane proteins, neurodegenerative (and protein misfolding) disorders and the in vitro transcription/translation technology platform.
Figure 1.

Composite schematic of the various groups within the Biology and Disease-driven Human Proteome Project (B/D-HPP). Blue represents the original groups, while red represents new groups that have started since 2013.
The B/D-HPP education and outreach program provides an annual Mentoring Day and a series of awards for early investigators: predoctoral, postdoctoral and clinical fellows, and early faculty. Information about the B/D-HPP is available at www.thehpp.org. Finally, a B/D-HPP newsletter has been launched, alongside the long-standing extensive newsletter series of the C-HPP.
THE HUMAN SRM ATLAS: THE OVERRIDING PROJECT OF THE B/D-HPP
Since the launching years, led by the Aebersold lab in Zurich and the Moritz lab in Seattle, and building on early work by Anderson et al.5 and by Carr,6 the B/D-HPP has pursued the goal of enhancing the capabilities of mass-spectrometry-based proteomics and bringing its applications to all fields of biology and disease through a major program of development of targeted proteomics. This year the release of the comprehensive Human SRM Atlas7 reports data on 166 174 proteotypic peptides that specifically identify 99.7% of the 20 277 predicted and annotated human proteins by the widely accessible, sensitive, and robust targeted MS method, selected reaction monitoring (SRM). These assays detect, verify, and quantitate such peptides from proteins of interest in specific pathways as well as their splice isoforms, sequence variants, and post-translational modifications. The team demonstrates the utility of the SRM approach by examining the network response to inhibition of cholesterol synthesis in liver cells and to docetaxel treatment of prostate cancer cells. The data are freely accessible at http://www.srmatlas.org.
Recent advances in data-independent acquisition mass spectrometry technologies such as SWATH-MS enable a deeper recording of the peptide contents of samples, including peptides with modifications. Keller et al.8 published a novel approach that applies the power of SWATH-MS analysis to the automated pursuit of modified peptides. A new SWATHProphet (PTM) functionality added to the open-source SWATHProphet software permits identification of precursor ions consistent with a modification along with the mass and localization of the modification in the peptide sequence. The method is sensitive and does not require anticipation of the modifications in advance. Keller8 detected a wide assortment of modified peptides, many unanticipated, in phospho-enriched human tissue culture cell samples, as well as urine containing unpurified synthetic peptides. These methods are likely to prove transformative in cell biology.
Two streams of work came together from B/D-HPP and HPP Bioinformatics Resource Pillar collaboration on “priority proteins” and “popular proteins”. The result is publicly available, comprehensive resources for SRM-targeted proteomics for proteins associated with particular diseases, including type 2 diabetes mellitus, ovarian cancers, breast cancers, colon cancers, chromatin proteins, type 1 diabetes, ascending aortic aneurysm, the heart, and the eye (follow www.thehpp.org to https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/proteinListSelector). Finally, a cross-B/D activity has been the analysis with bibliometric searches of the literature to produce sets of “popular proteins” (highly published) for six organ systems: cardiovascular, cerebral, hepatic, renal, pulmonary, and intestinal.9 For each protein in each protein set, SRM assays are specified, so that many users can plan appropriate studies.9
B/D-HPP WORKSHOPS AT 2015 HUPO WORLD CONGRESS IN VANCOUVER
Here we present highlights of the scientific and educational activities at the HUPO Congress in Vancouver in September 2015 organized by the B/D-HPP and the three HPP resource pillars, namely, Knowledgebase, Antibody Profiling, and MS (see Supplemental Table 2 for list of speakers). Workshop summaries are in alphabetical order.
Affinity-Based Protein Capture Resource Pillar and the Human Protein Atlas (HPA) (led by E. Lundberg, A. Bandrowski, C. Lindskog, M. Skogs, T. L. Alm, H. Rodriguez, D. Shankar, and M. Uhlen)
The original Human Antibody Initiative has continued to be a key component of the HPP and HUPO itself. This consortium impacts all B/D-HPP groups and the broader scientific community. One goal of the September 2015 meeting and a preceding major workshop at the EuPA annual meeting (Milano in June 2015) was to address the problems of antigen–antibody validation, proper identification of reagents used, and the need for greater reproducibility in reported findings throughout the scientific community. The Affinity Binders Knockdown Initiative aims to create a pipeline of well-validated antibodies and standard operating procedures for antibody validation. The results will be disseminated through the Antibodypedia (www.antibodypedia.org).
There was a major release of validated antibodies from the HPA in the January 2015 Science10 along with extensive annotations of protein type (secreted, membrane-spanning, housekeeping, regulatory), druggability, tissue specificity, presence in cancer or other cell lines, and, if relevant, the proteins relationship with metabolism. These classifications were based on immunohistochemical studies of 44 tissues and RNA sequencing of 32 tissues. These findings about protein expression are widely applicable at organ, tissue, and single-cell levels and contribute to the finding and annotation of missing proteins.10
Bioinformatics Workshop (led by E. Deutsch)
A major goal of the HPP Bioinformatics group at the HUPO 2015 World Congress was to bring together bioinformaticians, HPP leadership, and investigators to meet the need to generate more stringent quality assurance guidelines for the HPP and the larger community. This goal is grounded in the recognition that there are variable practices across laboratories, different criteria for protein identification with various search engines and databases, the experience of lax analytical filters in the large-scale reports by Kim et al.11 and Wilhelm et al.,12 and the many challenges to enhance the methodology for very large data sets. Specific guidelines and a checklist for authors and reviewers were generated, discussed in depth at the HPP post-Congress Workshop, and then refined for release in this 2016 HPP Special Issue of the Journal of Proteome Research (JPR) (see www.thehpp.org/guidelines).13
Clearly the current and future HPP data guidelines are an issue of high importance. Because pre-existing guidelines and their enforcement have limitations, the HPP’s new guidelines specifically address how best to make more reliable the claims of confidently detecting missing proteins and novel translation products. The major subtopics included: current HPP data guidelines, data deposition in ProteomeXchange, the 1% protein-level FDR requirement, manual inspection of spectra supporting extraordinary claims, consideration of alternate explanations of the data, use of synthetic reference peptides, and the use of SRM to confirm shotgun results. Each subtopic was introduced separately, followed by active discussion by the workshop participants. The outcome is the new HPP Data Interpretation Guidelines (version 2.1) (https://www.hupo.org/2015/12/news/establishing-new-hpp-guidelines/).13 JPR determined that all of the manuscripts published in the 2016 Special Issue must comply with these standards. The B/D-HPP recommends that these Guidelines and Checklist become adopted broadly by all proteomics researchers and by other proteomics and life sciences journals.
Proteomics Standards Initiative and ProteomeXchange Consortium (led by E. Deutsch, H. Hermjakob, and J. A. Vizcaíno)
The goal of the Vancouver workshop was to focus on the implementation of the current set of proteomics standards and to review the steps that have been taken over the year. The program started with an overview and recent advances of the Proteomics Standards Initiative (PSI) and highlighted recent publications14–21 on the current set of standards and the progress made at the 2015 PSI workshop in Seattle, USA which included the PSI Extended FASTA File (PEFF) format.
The PSI Molecular Interactions Working Group presented an update of the PSI-MI XML format for encoding molecular interactions in a rich XML schema, which is widely used among the molecular interactions databases. PSI-MI XML version 3.0, which enhances the format to describe more than two protein interaction partners and conditional interactions, is under development. The PSI Proteomics Informatics Working Group promoted the adoption of three recently completed formats: mzIdentML, for encoding proteins, peptides, and peptide–spectrum matches in XML; mzQuantML, a highly flexible XML format for encoding abundance measurements; and mzTab, a simplified tab-delimited text format for encoding the relevant summary information from mzIdentML and mzQuantML files. The current activity also includes development of proBED and proBAM, which are standardized formats for encoding the output of proteomics experiments in the widely used genomics BED and BAM formats, which can be read by many genomics tools. These new formats are aimed at proteogenomics workflows.
Finally, an overview of the ProteomeXchange Consortium of current proteomics data repositories was presented. Current members are PRIDE at the European Bioinformatics Institute (EBI), PeptideAtlas at the Institute for Systems Biology (ISB), and MassIVE at the University of California San Diego (UCSD). There are now >3000 data sets in ProteomeXchange. There are plans to include additional repositories, such as jPOST in Japan. Data sets deposited in PRIDE (MS/MS) and MassIVE are routinely downloaded for standardized reanalysis with complementary pipelines by PeptideAtlas and by GPMDB. The 2016 PSI Spring Meeting was held in Ghent, Belgium (Web site http://psidev.info/).
Brain Proteome Project (led by H. E. Meyer, G. Schmitz, Y. M. Park, A. Häggmark, A. Urbani, P. Marin, K. Marcus, P. Nilsson, and D. Martins-De-Souza)
The HUPO Brain Proteome Project (HBPP) is an international interdisciplinary initiative focused on the investigation of the human brain (see www.hbpp.org). The brain is a complex organ comprised of a large number of different tissue layers and cells. The goal of the brain proteome project is to understand the roles of the proteome in the functions of the human brain. To date, the B/D-HPP researchers have used a broad spectrum of methods and have fostered strong cooperation between scientists from different fields (affinity proteomics, bioinformatics, biostatistics, proteomics, analytical biotechnology, clinical science, neuro-biology, biochemistry, neuroanatomy, and neuropathology). At the Vancouver Congress the initiative focused on projects related to understanding the biogenesis of the most common forms of dementia: Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), and other neurodegenerative diseases. Insights from biomarker research of neurodegenerative diseases and the underlying disease mechanisms, especially in the hippocampus, were highlighted. This was the 25th Workshop of the HBPP (see www.hbpp.org).
Cancer Proteome Project (led by H. Zhang, C. R. Jimenez, E. Nice, M. S. Baker, J. Qin, A. Umar, K. Abbott, and Y. Chen)
The goal of the Cancer-HPP is to characterize different cancer proteomes, determine the correlation of transcriptome and proteome, identify high-priority proteins for each tumor type, and generate and disseminate assays and resources to support the analysis of complex biological networks or clinical specimens underlying different disease processes. The use of targeted antibody and MS/MS-based methods allows quantitative analysis of high-priority target proteins. This group has proposed the formation of an international cancer proteomic effort (similar to The Cancer Genome Atlas (TCGA) project) to identify and validate cancer proteins for different cancer types using MS-based methods (see CPTAC-HPP section).
In Vancouver, data from membrane proteome studies on colorectal cancers provided insights for subtype classification and novel biomarker definition. The primary role of post-translational modifications was underscored with phosphoproteomics studies that identified tissue signatures in nonsmall cell lung cancers and with glycomic analyses showing a unique N-linked glycan structure prevalent in ovarian cancers. The clinical perspectives of breast cancer proteomics were also discussed. Finally, the Chinese Human Proteome Project (CNHPP) presented extensive work to define the proteomic landscape of liver cancers.
Clinical Proteomic Tumor Analysis Consortium (CPTAC) HPP (led by H. Rodriguez, C. Kinsinger, B. Zhang, S. Thomas, and J. Whiteaker)
The goal of the CPTAC-HPP Workshop was to address the many challenges and progress in developing precise quantitative tools for protein analysis in the discovery and targeted arena.22 NCI’s public portal (http://assays.cancer.gov) contains nearly 800 fit-for-purpose targeted SRM-MS assays developed in coordination with the U.S. Food and Drug Administration (FDA) and the American Association for Clinical Chemistry. Next, there is a need to adapt and ensure that these assays can be used easily across many cell and tissue types. Much like antibody-based assays, transparency around expectation, validation, and limitations will be required for these MS-based tools.
Cardiovascular Proteome Project (led by P. Srinivas, S. J. Parker, A. W. Herren, T. Klein, M. Lam, and M. E. McComb)
The goal of the cardiovascular proteome project is to promote the use of technology broadly across the heart and vascular research domains. The group has determined which proteins are most studied in cardiovascular research as a means of identifying proteins and pathways that could be a focus for developing targeted quantitative assays. The cardiovascular initiative published a paper in 2015 entitled “Prioritizing Proteomics Assay Development for Clinical Translation” about the most studied of the <10 000 proteins that have been documented in UniProt to be expressed in the heart.23 To determine the “popularity” of a protein objectively, we searched a large-scale bibliometric analysis of the 24 million research papers curated on PubMed for “heart or cardiac” using a new software tool (BD2KPubMed) for both human and mouse. A surprising result is that few proteins made it to the top-20 proteins list of both mouse and human; instead, completely different proteins, networks, and pathways were being investigated. This suggests that there is an unrecognized gap between basic mechanistic science and human based studies (which are enriched in the use of biomarkers). This observation raises the key question of how the proteomics field should focus on development of quantitative protein assays that are relevant to clinical investigations, basic research, and biomarker discovery. A challenge for MS-based assay method developers is how to strategize development to prioritize important and popular proteins that can contribute to overcoming a bottleneck in technology dissemination. The next step is to broaden this work into other cell and organ types that comprise or influence the cardiovascular systems in health and disease, as reported more recently by Lam et al.9
Human Diabetes Proteome Project (led by P. Bergsten, J. C. Sanchez, D. Goodlett, and I. Grundberg)
The goal of the HUPO Human Diabetes Proteome Project (HDPP) is to enhance the understanding of mechanisms in development of diabetes via performing and analyzing large-scale network biology-based experiments. At previous semiannual HDPP workshops, scientists interested in pursuing diabetes research related to the aim of HDPP have been invited to join the initiative. This has resulted in a vibrant community of scientists who work in diverse areas of diabetes. One collective goal has been to define sets of proteins of interest for understanding diabetes and identify how these sets could be measured and analyzed in an efficient way. Information from these workshops has been disseminated to the broader community by the HDPP homepage (http://www.hdpp.info). The aim of the eighth HDPP workshop in Vancouver was to define sets of proteins of interest coming from experiments using classical ‘omics strategies and then determine how they can be translated through robust assay development to studies of large cohorts of patients. The HDPP consortium has defined a list of 25 top candidate biomarkers for diagnosis in plasma and a second list of 24 other proteins ranked by popularity in the context of diabetes research. The HDPP will disseminate approved measurement methodologies for the 24 targets, which could be a common project for HDPP partners. The group decided on objectives for 2015–2016 and published them in Translational Proteomics.24
Three studies were highlighted in Vancouver. First, the Finnish Type 1 Diabetes (TID) Prediction and Prevention (DIPP) Study conducted in Turku aims to detect changes in serum proteomes of children with HLA-conferred T1D susceptibility that would allow therapeutic intervention before ~70% of pancreatic islet cells are destroyed. Based on top scoring pair analysis, group-specific longitudinal changes were detected, allowing classification of the subjects developing T1D with a success rate of 91% using just two proteins, one from the complement system and one involved in lipid metabolism. Second is a European project focusing on beta cell function in Juvenile Diabetes and Obesity (BETA-JUDO). Integrated proteomics, transcriptomics and lipidomics studies were performed in a patient cohort and in isolated islets of Langerhans to assess insulin hypersecretion as an early factor driving emergence of obesity and type 2 diabetes. In the presence of increased fasting glucose concentrations palmitate promotes increased cholesterol formation in pancreatic human islets. Finally, a proximity extension assay (PEA) was introduced, which multiplexes 96 immunoassays in panels for human protein biomarkers. PEA uses 96 oligonucleotide–antibody pairs that contain unique DNA sequences allowing hybridization only to each other. Subsequent proximity extension creates 96 unique DNA reporter sequences, which are amplified by real-time PCR. This technology has identified etiological factors and predictors of impaired glucose metabolism and diabetes using a multiplex inflammation panel.
Human Proteomics at Extreme Conditions (led by E. N. Nikolaev, G. Arapidi, I. Larina, L. Pastushkova, I. Popov, L. C. Cameron, A. Bassini, M. G. Rodchenkova, A. Percy, and B. Domon)
This second workshop aimed to set central directions for the group and to discuss recent research on the molecular bases of physiological responses in space or while practicing sports under extreme conditions. Measurements of 140 proteotypic peptides showed protein alterations in 18 cosmonauts after a long duration flight, suggesting relevant physiological adaptations. Proteomic and metabolomic changes in breath condensate and urine of cosmonauts returned to normal within 1 to 2 weeks after a space flight. Parallel longitudinal studies of the urine proteome of participants in the MARS 500 program (simulation of a space flight to Mars) may be a resource for future biomarker studies. About 1000 proteins (5000 peptides) were measured in plasma from professional athletes under high physical stress and exhaustion. Also, results were reported from a metabolic stress analysis during sport and exercise; a metabolite database is being developed. Finally, an electrophoresis-based method to detect erythropoiesis-stimulating agents has facilitated detection of many doping cases leading to disqualification of a large number of athletes in renowned competitions, including the Tour de France.
EyeOME (Eye Proteome Project) (R. Semba, M. Ueffing, H. Chung, L. Zhou, S. Dammeier, S. M. Hauck, S. K. Bhattacharya, F. Grus, and V. B. Mahajan)
This group aims to grow the scientific community focused on understanding the biological pathways in the multiple anatomical compartments of the eye (see special issue of Proteomics on the Eyeome25). One major activity over the past year was determining the most popular proteins found in human and mouse eye studies (a total of 13 525 and 23 895 publications, respectively).26 While large numbers of proteins have been reported (4050 and 4717 proteins, respectively), stringent criteria, like the HPP Guidelines, were not applied to these lists. Moreover, most proteins had five or fewer citations (88.7 and 81.7%, respectively). Not surprisingly, the top 50 most intensively studied proteins for both species were very specific to the eye; they are photoreceptors or involved in photo-transduction, inflammation, angiogenesis, neurodevelopment, and/or lens transparency.
Food and Nutrition Proteomics Project (led by P. Roncada, M. Baker, L. Martens, and L. Dayon)
The central roles of food proteins in nutritional science are indisputable. The well-known affirmation that “we are what we eat” and that food can be medicine is not just philosophy but a sustainable strategy to reduce the prevalence of noncommunicable diseases. The global composition of diet, from microbiome to nutrient, including life style, can affect every step from gene expression to protein synthesis to degradation, with profound modulation of metabolic functions. This B/D-HPP group uses proteomics to help develop a novel sustainable personalized medicine through nutritional intervention. This includes a special focus on food safety, security, and quality issues, providing new insights and technologies from microbiome and consortia, detection of animal species in the food, modifications of proteins, identification of food allergens, and food authenticity. Proteins are associated with disorders from allergy to foodborne pathogens, including prions. There is a large European initiative on Food Allergens and New Food Allergens (COST ACTION IMPARAS: www.imparas.eu). The Truffle proteomics project was presented as a “model food” to illustrate how research in proteomics can be translated into optimized processes for production of premium food products.
Bioinformatics approaches and pipelines can be focused on tools for metaproteomic analysis to study microbial communities and their dynamics. The functional role of microbiota during the onset of obesity is a hot topic. The completed EU project Diogenes (http://www.diogenes-eu.org/) targeted the obesity problem from a dietary perspective to search for new routes of prevention.
Glycoproteome Project (led by H. Narimatsu, D. Kolarich, H. Kaji, H. Zhang, S. Tao, X. Zou, and N. H. Packer)
One of the earliest initiatives in the B/D-HPP consortium is the Glycoproteome Project. Its goal remains focused on education and the adoption and development of tools for the detailed knowledge of glycans and glycoproteomics. This year the group showcased a number of elegant new methods and coordinated the proteomics and glycomics inputs for the “Glycoproteome Atlas” (http://wlab.ethz.ch/cspa).27 The Atlas combines glycomics, the detailed structural analysis of glycans released from the glycoproteins, and glycoproteomics, the comprehensive analysis of polypeptide sites of post-translational modifications involving sugars. Previous Glycoproteome Project pilot studies focused on glycomics technologies. Future studies will focus on the development and advancement of glycoproteomics technologies.
Infectious Diseases Proteome Project (led by C. Gil, I. M. Cristea, F. Schmidt, S. Srivastava, J. LaBaer, C. Costello, and M. Fuentes)
A number of experimental workflows were discussed and shown to be successfully applied in host–pathogen interaction experiments (e.g., between S. aureus and various host cells). A DNASU plasmid repository has been established at the Biodesign Institute at Arizona State University as a nonprofit service core providing sequence-verified, highly annotated plasmids to researchers through an easy-to-use, accessible Web site (https://dnasu.org/DNASU/). There are gene collections of human, Saccharomyces cerevisiae, Vibrio cholera, Pseudomonas aeruginosa, Francisella tulariensis, viruses, and other species. These collections can be used to develop nucleic acid programmable protein arrays. For example, a study of all 262 outer membrane and exported P. aeruginosa proteins with a NAPPA array identified 12 proteins that trigger an adaptive immune response in acutely infected patients with cystic fibrosis. The association between viral infections and T1D was explored by profiling antiviral antibodies with a high-throughput immuno-proteomics approach in new-onset T1D patients. Genomics, proteomics, and bioinformatics analysis, using the in vitro model “Candida albicans-macrophages,” discovered that fungi inside macrophages are dying by apoptosis. A Candida albicans Peptide-Atlas is now available.
Broader issues were also discussed: (1) collaboration among scientists working in Infectious Disease proteomics; (2) development of SRM methods for detection and quantification of human and microbial proteins; (3) detection and quantification of these proteins by affinity or antibodies-based technologies; (4) building a Web page to freely access data; and (5) diffusion of HID-HPP activities (reports, special issues). A Web page will be created. Two special issues in Molecular and Cellular Proteomics and in Translational Proteomics devoted to Infectious diseases were announced.
Human Kidney and Urine Proteome Project (HKUPP) (led by T. Yamamoto, J. Arthur, and A. Vlahou)
Reporting of the identification and characterization of the various proteomes of the kidneys as well as urine was the primary goal of this Vancouver workshop. The proteome of nephron segments in the kidney was further analyzed by using laser microdissection of each nephron part demarcated with immunohistochemistry from FFPE human kidney sections, on-site direct digestion (OSDD) for peptide preparation, and LC–MS. Proteomes of the glomerulus, proximal tubule, distal tubule, and collecting duct were obtained and compared with each other and also with those of the cortex and medulla. From the comparison of these proteomes, nephron segment-unique proteins were selected and analyzed by pathway analysis tools for understanding unique functions of the nephron segments.
Laser-microdissection-assisted LC–MS also was applied to analyze glomerular proteomes of kidney biopsy sections. Unique glomerular protein profiles of different kidney diseases were collected. Human urine proteomes have been intensively analyzed by MS mostly for discovery of kidney disease biomarkers. However, the large amounts of plasma proteins excreted via the glomerulus into the urine of kidney disease patients, depending on the glomerular injury, have prevented discovery of urine biomarkers derived from the kidney tissue. Proteomics studies of the urine from kidney disease patients have generally not been successful. To overcome this problem, the nephron segment-unique proteins selected by the laser-microdissection-assisted MS are being examined to see whether they might serve as biomarkers of nephron-unique events in the urine from kidney disease patients, using specific antibodies. A few proteins have become biomarker candidates associated with nephron segment injuries.
Human Liver Proteome Project (led by F. J. Corrales, P. Zhang, Y. Jiang, P. Yang, J. E. Lee, and C. Ding)
The liver is a central organ that controls metabolic homeostasis, provides essential substances to the organism, and detoxifies xenobiotics. There are still many open questions about liver function and disease that largely limit the availability of clinical options for the treatment of chronic liver ailments. The goal of the HLPP session was to define cell-specific proteome profiles within the liver, factors responsible for maintaining the differentiated phenotype of hepatocytes, and mechanisms by which regulation of the protein methylation pattern might contribute to liver injury.
Hepatocellular cancer (HCC) proteomic studies performed at the National Centre for Protein Sciences, Beijing, have provided detailed transcriptomic and proteomics data to map relevant cellular pathways driving HCC metastasis.28 Clustering of tumors based on protein abundance identified six different subtypes that correlated with aggressiveness. An extensive characterization of the acetylome in HCC samples upon fractionation to nucleus, cytosol, and mitochondria found that most metabolic enzymes were acetylated during metabolic adaptation of transformed liver cells. Impairment of glycolysis and activation of gluconeogenesis in HCC cells can be mediated by increased acetylation and ubiquitination of key enzymes.
Data supporting a connection between changes in the methyl-proteome and liver cancer were also discussed. Deficient methylthioadenosine phosphorylase (MTAP) in liver cancer cells leads to accumulation of methylthioadenosine (MTA) and impairment of protein methylation reactions. MTAP deficiency leads to MTA accumulation and deregulation of central cellular pathways, increasing proliferation and decreasing the susceptibility to chemotherapeutic drugs.29 A standard sample for proteomic studies in liver cancer cells consisting of a mixed extract of seven SILAC-labeled hepatoma cell types was presented. The effect of sorafenib in hepatoma cells was investigated, and no major effects were found at the proteome level; this limited effect may explain the low efficacy of the drug.
Mitochondrial Proteome Project (led by M. Babu, E. Lundberg, I. Bongarzone, M. Ronci, and H. Vissers)
The mitochondrial HPP project, an initiative started as an outreach activity of the Italian Proteome Association, has extended to collaborators from other countries. The goal of the workshop was to bring together the broader community. After a summary of previous activities, four talks spanned interactomics, metabolism, methodology, and spatial proteomics. The coverage of the mitochondrial proteome by MS was updated. Mitochondrial interaction networks in neurodegenerative disorders and alterations in triple-negative breast cancers were reported.
iMOP (Multiorganism Proteome Project) (led by A. Stensballe, L. J. Foster, M. Semanjski, and N. Jehmlich)
iMOP, the initiative on multiorganism proteomes, aims to broaden the application of proteome research in nonhuman species. The members of iMOP are working with classical animal models to study human diseases but also focus on a wide range of species that greatly affect human health, including farm animals and crop species, pathogens, and the microbiome of humans and farm animals. The Vancouver workshop included presentations on animal models for studying inflammatory bowel disease and proteogenomic approaches to improve the health state of honeybees, HipA-mediated mechanisms in E. coli, and studies of mucosal microbiota from rat colon. The results presented and the final plenary discussion highlighted the relevance of the iMOP community to improve proteome research in nonhuman species.
Skeletal Muscle Proteome Project (led by L. Ferrucci and K. Hojlund)
Maintenance of the contractile function and energy metabolism of skeletal muscle is essential for autonomy and quality of life in humans. There are many situations and diseases that affect the function of muscle, including muscular dystrophies and aging cachexia that accompany debilitating diseases, such as chronic infections, cancers, kidney failure, and heart failure, as well as diabetes, autoimmune diseases, and osteoarthritis. The major goals of this group are to produce a comprehensive proteomic analysis of skeletal muscle biopsies and to increase our understanding of the mechanisms leading to different clinical forms of muscle pathology and improve diagnosis. Of note, skeletal muscle is underrepresented in PeptideAtlas.30 Also, knowledge of changes in muscle proteins with aging in individuals free of major diseases and drug treatments is quite limited.
The group provided an overview of the potential values and uses of skeletal muscle proteomics in aging, type 2 diabetes, and other diseases. The design and preliminary data from the Genetic and Epigenetic Signatures of Translational Aging Laboratory Testing (GESTALT) were presented. This study by the Intramural Program of the National Institute on Aging in Baltimore will characterize muscle proteins with aging in a group of healthy men and women over a wide age range (20–100 years). Finally, studies of the proteomes and phosphoproteomes of human skeletal muscle and isolated muscle mitochondria as well as quantitative studies of changes in the muscle proteome associated with obesity and type 2 diabetes were presented. These examples should stimulate a wider interest in proteomic characterization of muscle in humans and more collaborative research between international groups.
Human Plasma Proteome Project (HPPP) (led by J. Schwenk, E. Deutsch, H. Keshishian, G. Rosenberger, M. Harel, D. Juncker, C. Fredolini, and R. Popp)
Some of the goals of the HPPP have been met through major advances in proteome coverage from human plasma samples using more extensive depletion of the most abundant proteins, iTRAQ labeling, and high-resolution MS/MS spectra from modern instruments. For example, over 4000 proteins in each patient sample were identified and quantified in a study of heart attacks. OpenSWATH and OpenSWATH-PTM are valuable tools to define plasma proteomes and identify modified versions of target peptides. Microparticles and the PROMIS-Quan (PROteomics of MIcroparticles with Super-SILAC Quantification) tool to study diseases such as prostate cancer or melanoma are novel methods of identifying and quantifying biomarker candidates in human plasma. Protein capture platforms, alone or in combination with MS, were also presented. A SNAP-chip antibody sandwich-assay microarray for detecting and quantifying proteins in plasma is being applied in breast cancer studies. Concepts and workflow to combine affinity arrays with MS revealed the potential for large-scale biomarker studies to determine on/off target binding of antibodies in serum and plasma and develop clinical assays. The performance of immuno-enrichment steps for MALDI analysis was compared with existing clinical assays. The session concluded with a retrospective on the Human Plasma PeptideAtlas; recent updates have expanded the list of high-confidence plasma proteins above 4000 entries.
PediOME Proteome Project (Pediatrics) (led by A. D. Everett, H. Steen, D. Goodlett, and Y. Hathout)
The goal of the PediOME initiative is to explore children’s health through the application of proteomics. A special issue on Pediatric Proteomics in Proteomics: Clinical Applications was published in December 2014; a thematic manuscript series is being planned for Clinical Proteomics. The workshop keynote addressed the central role of proteomics studies in early biomarker detection and the important contribution that proteomics can make to the pediatric clinic. A specific challenge for the PediOME is to overcome sample volume limits associated with pediatric research. New developments in urine sample preparation and the value of data-independent acquisition (DIA) versus data-dependent acquisition for the identification of proteins were discussed. A fast and robust DIA method for comprehensive mapping of the urinary proteome that enables large-scale urine proteomics studies was introduced, with identification of a set of proteins in urine that could be diagnostic for a tuberculosis infection. Other non-MS based methods, specifically the aptamer-based SOMAscan, were used to identify potential diagnostic biomarkers that might stratify patients with muscular dystrophy.
Proteomics of Protein Misfolding and Aggregation Diseases (led by M. Rezeli, C. E. Costello, Y. Feng, H. Langen, and M. Pirmoradian)
Protein misfolding and aggregation diseases (PMADs), exemplified by Parkinson’s and Alzheimer’s diseases and systemic amyloidoses, are characterized by an abnormal deposition of protein aggregates of regular three-dimensional structure (amyloid). The goal of the B/D PMAD group is to develop proteomics assays for proteins that are relevant to the study, diagnosis, and therapy of protein aggregation diseases. These assays are tested and refined on a set of relevant patient samples for clinical applications and on samples from model organisms and cell culture for basic research. Besides developing assays for measuring protein abundances, a peculiarity of this initiative is the development of proteomics assays for “aberrant protein conformations”, those typically generated in PMADs. The workshop addressed assays for PMAD targets and their validation.
Toxicoproteomics Project (led by L. Sleno, O. Poetz, S. Naryzhny, B. P. Konstantinov, M. F. Templin, and M. Mikus)
The goal of the first HUPO Toxicoproteomics Workshop was to address three different areas that cover this field from discovery to applications in drug-development: discovery of reactive drug metabolites and drug–protein adducts, toxicological mechanisms of nongenotoxic carcinogens, and detection of drug-induced organ injury in body fluids (safety biomarkers). The session opened with a short discussion about the definition and relevance of toxicoproteomics, followed by a presentation of LC–MS/MS-based methods for the detection of covalently modified proteins, demonstrating the relevance of proteomics for the discovery of protein modifications by reactive drug metabolites and consequences thereof. A global analysis of cellular and plasma proteins using a combination of virtual (in silico) and experimental 2-DE with high-resolution nanoliquid chromatography-MS was presented. Complete proteomes and single proteoforms were contrasted in biomarker studies and protein signature analyses. A functional proteomic study introduced a novel combination of Western-blot methods with bead-array technology. Hundreds of Western blots could be performed with laser capture microscopy material; this method elucidated resistance mechanisms from analyses of liver and ovarian tumors. Detection of safety biomarkers was a major topic. Application of antibody-based proteomics to screen for biomarkers in plasma samples from persons with drug-induced liver injury illustrated the long journey from biomarker candidate identification to a partial biomarker validation. Finally, an MS-based immunoassay approach for safety biomarker analyses in preclinical animal models was applied to screen urine for kidney (DIKI) injury protein biomarkers across species. Data from safety toxicity studies conducted in monkeys strongly supported the development of translational DIKI biomarkers. The session closed with a discussion about potential benefits of combining the different methods presented.
EDUCATIONAL ACTIVITIES OF THE B/D-HPP
A major commitment of the B/D-HPP is the engagement of early career scientists. Our priority is the transmission of B/D-HPP ideals across generations of proteomic researchers, providing young scientists with a platform to unfold their own vision of proteomic science. To achieve this intent, two novel activities were launched in Vancouver. First, an entire Mentoring Day program was initiated, which will be an annual feature of the International HUPO Congress. The main goal of the Mentoring Day is to build bridges between junior and senior scientists from academia, industry, and government funding agencies using lectures and interactive workshop-like activities to promote the interaction. The theme of this first Mentoring Day in Vancouver was providing a perception of proteomics over time in society. Second, an inaugural manuscript competition specifically for early career scientists was held. The competition is intended for young scientists to emphasize their original projects with cutting-edge proteomics impacting conceptual ideas in different disease areas. The finalists of the competition presented their work at a dedicated session of the international HUPO meeting. An annual feature of HPP at the HUPO Congress is the nomination by B/D-HPP initiatives of physician-scientists for travel grants to come participate in the Congress, mingle with the B/D group of their interest, learn about practical proteomics applications for their field of research, and potentially develop collaborative or mentored relationships for their personal career development.
Announcements of each of these three educational activities as well as graduate student poster awards appeared on the HUPO Web site in the run-up to the Taipei 2016 Congress and will be promoted again for Dublin 2017 and Orlando 2018 HUPO Congresses.
B/D-HPP PERSPECTIVES
The number of B/D-HPP initiatives continues to increase, and the maturation of more established groups is now pushing new boundaries in technology. The B/D-HPP continues to extend its mandate through (a) community building, (b) encouraging, supporting, and enabling dissemination of innovative technologies, (c) establishing and supporting adoption of peptide and protein identification and quantification standards, (d) collaborating across B/D and C-HPP teams, and (e) increasing the ability of proteomic researchers to change the paradigm of science and medicine through widespread integration of proteomics in multiomics and physiological studies.
Excellence in science remains central to the mandate, enhanced by the development of young investigators and platforms for innovation and the establishment of standards for our field, as required for the world to move toward precision and personalized medicine.
Supplementary Material
Supplementary Table 1. Highlights of the Biology and Disease-driven Human Proteome Project, 2015–2016. (XLSX)
Supplementary Table 2. 2015 HUPO World Congress list of speakers. (XLSX)
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00444.
References
- 1.Legrain P, Aebersold R, Archakov A, Bairoch A, Bala K, Beretta L, Bergeron J, Borchers CH, Corthals GL, Costello CE, Deutsch EW, Domon B, Hancock W, He F, Hochstrasser D, Marko-Varga G, Salekdeh GH, Sechi S, Snyder M, Srivastava S, Uhlen M, Wu CH, Yamamoto T, Paik YK, Omenn GS. The Human Proteome Project: current state and future direction. Mol Cell Proteomics. 2011;10(7):M111.009993. doi: 10.1074/mcp.M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Omenn GS, Lane L, Lundberg EK, Beavis RC, Nesvizhskii AI, Deutsch EW. Metrics for the Human Proteome Project 2015: progress on the human proteome and guidelines for high-confidence protein identification. J Proteome Res. 2015;14(9):3452–60. doi: 10.1021/acs.jproteome.5b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Omenn GS, Lane L, Lundberg EK, Beavis RC, Overall CM, Deutsch EW. Metrics for the Human Proteome Project 2016: Progress on identifying and characterizing the human proteome, including post-translational modifications. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011;470(7333):163–5. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
- 5.Anderson NL, Anderson NG, Pearson TW, Borchers CH, Paulovich AG, Patterson SD, Gillette M, Aebersold R, Carr SA. A human proteome detection and quantitation project. Mol Cell Proteomics. 2009;8(5):883–6. doi: 10.1074/mcp.R800015-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2012;10(1):28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kusebauch U, Campbell DS, Deutsch EW, Chu CS, Spicer DA, Brusniak MY, Slagel J, Sun Z, Stevens J, Grimes B, Shteynberg D, Hoopmann MR, Blattmann P, Ratushny AV, Rinner O, Picotti P, Carapito C, Huang CY, Kapousouz M, Lam H, Tran T, Demir E, Aitchison JD, Sander C, Hood L, Aebersold R, Moritz RL. Human SRMAtlas: A resource of targeted assays to quantify the complete human proteome. Cell. 2016;166(3):766–78. doi: 10.1016/j.cell.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller A, Bader SL, Kusebauch U, Shteynberg D, Hood L, Moritz RL. Opening a SWATH window on posttranslational modifications: Automated pursuit of modified peptides. Mol Cell Proteomics. 2016;15(3):1151–63. doi: 10.1074/mcp.M115.054478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam M, Venkatraman V, Xing Y, Lau E, Cao Q, Ng D, Su A, Ge J, Van Eyk J, Ping P. Data-driven approach to determine popular proteins for targeted proteomics translation. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 11.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature. 2014;509(7502):575–81. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta-Huspenina J, Boese JH, Bantscheff M, Gerstmair A, Faerber F, Kuster B. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–7. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 13.Deutsch EW, Overall CM, Van Eyk J, Baker M, Paik YK, Weintraub S, Lane L, Martens L, Vandenbrouck Y, Kusebauch U, Hancock W, Hermjakob H, Aebersold R, Moritz RL, Omenn GS. Human Proteome Project mass spectrometry data interpretation guidelines 2.1. J Proteome Res. 2016 doi: 10.1021/acs.jproteo-me.6b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez-Riverol Y, Uszkoreit J, Sanchez A, Ternent T, Del Toro N, Hermjakob H, Vizcaino JA, Wang R. MS-data-core-api: an open-source, metadata-oriented library for computational proteomics. Bioinformatics. 2015;31(17):2903–5. doi: 10.1093/bioinformatics/btv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Riverol Y, Xu QW, Wang R, Uszkoreit J, Griss J, Sanchez A, Reisinger F, Csordas A, Ternent T, Del-Toro N, Dianes JA, Eisenacher M, Hermjakob H, Vizcaino JA. PRIDE Inspector Toolsuite: Moving toward a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol Cell Proteomics. 2016;15(1):305–17. doi: 10.1074/mcp.O115.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan J, Saha S, Barker G, Heesom KJ, Ghali F, Jones AR, Matthews DA, Bessant C. Galaxy Integrated Omics: Web-based standards-compliant workflows for proteomics informed by transcriptomics. Mol Cell Proteomics. 2015;14(11):3087–93. doi: 10.1074/mcp.O115.048777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kettner C, Field D, Sansone SA, Taylor C, Aerts J, Binns N, Blake A, Britten CM, de Marco A, Fostel J, Gaudet P, Gonzalez-Beltran A, Hardy N, Hellemans J, Hermjakob H, Juty N, Leebens-Mack J, Maguire E, Neumann S, Orchard S, Parkinson H, Piel W, Ranganathan S, Rocca-Serra P, Santarsiero A, Shotton D, Sterk P, Untergasser A, Whetzel PL. Meeting report from the second “Minimum Information for Biological and Biomedical Investigations” (MIBBI) workshop. Stand Genomic Sci. 2010;3(3):259–66. doi: 10.4056/sigs.147362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi D, Lawless C, Teleman J, Levander F, Holman SW, Hubbard S, Jones AR. Representation of selected-reaction monitoring data in the mzQuantML data standard. Proteomics. 2015;15(15):2592–6. doi: 10.1002/pmic.201400281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi D, Zhang H, Fan J, Perkins S, Pisconti A, Simpson DM, Bessant C, Hubbard S, Jones AR. The mzqLibrary–an open source Java library supporting the HUPO-PSI quantitative proteomics standard. Proteomics. 2015;15(18):3152–62. doi: 10.1002/pmic.201400535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocca-Serra P, Salek RM, Arita M, Correa E, Dayalan S, Gonzalez-Beltran A, Ebbels T, Goodacre R, Hastings J, Haug K, Koulman A, Nikolski M, Oresic M, Sansone SA, Schober D, Smith J, Steinbeck C, Viant MR, Neumann S. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics. 2016;12(1):14. doi: 10.1007/s11306-015-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deutsch EW, Albar JP, Binz PA, Eisenacher M, Jones AR, Mayer G, Omenn GS, Orchard S, Vizcaino JA, Hermjakob H. Development of data representation standards by the Human Proteome Organization Proteomics Standards Initiative. J Am Med Inform Assoc. 2015;22(3):495–506. doi: 10.1093/jamia/ocv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudnick PA, Markey SP, Roth J, Mirokhin Y, Yan X, Tchekhovskoi DV, Edwards NJ, Thangudu RR, Ketchum KA, Kinsinger CR, Mesri M, Rodriguez H, Stein SE. A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) common data analysis pipeline. J Proteome Res. 2016;15(3):1023–32. doi: 10.1021/acs.jproteome.5b01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lam MP, Venkatraman V, Cao Q, Wang D, Dincer TU, Lau E, Su AI, Xing Y, Ge J, Ping P, Van Eyk JE. Prioritizing proteomics assay development for clinical translation. J Am Coll Cardiol. 2015;66(2):202–4. doi: 10.1016/j.jacc.2015.04.072. [DOI] [PubMed] [Google Scholar]
- 24.Schvartz D, Bergsten P, Baek KH, Barba, De La Rosa A, Cantley J, Dayon L, Finamore F, PF, Gaudet P, Goo YA, Moulder R, Goodletti D, Johnson JD, Konvalinka A, Mulder H, Priego-Capote F, Sechi S, Snyder M, Tiss A, Wiederkehr A, Xenarios I, Kussmann M, Sanchez JC. The Human Diabetes Proteome Project (HDPP): The 2014 update. Translational Proteomics. 2015;8:1–7. [Google Scholar]
- 25.Semba RD, Enghild JJ. Proteomics and the eye. Proteomics: Clin Appl. 2014;8(3–4):127–9. doi: 10.1002/prca.201470024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semba RD, Lam M, Sun K, Zhang P, Schaumberg DA, Ferrucci L, Ping P, Van Eyk JE. Priorities and trends in the study of proteins in eye research, 1924–2014. Proteomics: Clin Appl. 2015;9(11–12):1105–22. doi: 10.1002/prca.201500006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bausch-Fluck D, Hofmann A, Bock T, Frei AP, Cerciello F, Jacobs A, Moest H, Omasits U, Gundry RL, Yoon C, Schiess R, Schmidt A, Mirkowska P, Hartlova A, Van Eyk JE, Bourquin JP, Aebersold R, Boheler KR, Zandstra P, Wollscheid B. A mass spectrometric-derived cell surface protein atlas. PLoS One. 2015;10(3):e0121314. doi: 10.1371/journal.pone.0121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen H, Zhong F, Zhang Y, Yu H, Liu Y, Qin L, He F, Tang Z, Yang P. Transcriptome and proteome of human hepatocellular carcinoma reveal shared metastatic pathways with significant genes. Proteomics. 2015;15(11):1793–800. doi: 10.1002/pmic.201400275. [DOI] [PubMed] [Google Scholar]
- 29.Bigaud E, Corrales FJ. Methylthioadenosine (MTA) Regulates Liver Cells Proteome and Methylproteome: Implications in Liver Biology and Disease. Mol Cell Proteomics. 2016;15(5):1498–510. doi: 10.1074/mcp.M115.055772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrah T, Deutsch EW, Omenn GS, Sun Z, Watts JD, Yamamoto T, Shteynberg D, Harris MM, Moritz RL. State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the Biology- and Disease-driven Human Proteome Project. J Proteome Res. 2014;13(1):60–75. doi: 10.1021/pr4010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Highlights of the Biology and Disease-driven Human Proteome Project, 2015–2016. (XLSX)
Supplementary Table 2. 2015 HUPO World Congress list of speakers. (XLSX)