

Abstract
Hyperhomocysteinemia (HHcy) has been observed to promote hypertension, but the mechanisms are unclear. Toll-like receptor 4 (TLR-4) is a cellular membrane protein that is ubiquitously expressed in all cell types of the vasculature. TLR-4 activation has been known to promote inflammation that has been associated with the pathogenesis of hypertension. In this study we hypothesize that HHcy induces hypertension by TLR-4 activation, which promotes inflammatory cytokine (IL-1β, IL-6, and TNF-α) upregulation and initiation of mitochondria-dependent apoptosis, leading to cell death and chronic vascular inflammation. To test this hypothesis, we used C57BL/6J (WT) mice, cystathionine β-synthase (CBS)-deficient (CBS+/−) mice with genetic mild HHcy, C3H/HeJ (C3H) mice with TLR-4 mutation, and mice with combined genetic HHcy and TLR-4 mutation (CBS+/−/C3H). Ultrasonography of the superior mesenteric artery (SMA) detected an increase in wall-to-lumen ratio, resistive index (RI), and pulsatility index (PI). Tail cuff blood pressure (BP) measurement revealed elevated BP in CBS+/− mice. RI, PI, and wall-to-lumen ratio of the SMA in CBS+/−/C3H mice were similar to the control group, and BP was significantly alleviated. TLR-4, IL-1β, IL-6, and TNF-α expression were upregulated in the SMA of CBS+/− mice and reduced in the SMA of CBS+/−/C3H mice. Molecules involved in the mitochondria-mediated cell death pathway (BAX, caspase-9, and caspase-3) were upregulated in CBS+/− mice and attenuated in CBS+/−/C3H mice. We conclude that HHcy promotes TLR-4-driven chronic vascular inflammation and mitochondria-mediated cell death, inducing hypertension. TLR-4 mutation attenuates vascular inflammation and cell death, which suppress hypertension.
Keywords: homocysteine, vascular inflammation, mitochondria-mediated cell death, peripheral resistance, inward vascular remodeling
primary, or essential, hypertension is the most common type of hypertension, with unclarified etiology that affects 95% of all hypertensive patients. It has been reported that 75 million adults in the United Sates and 1 billion people worldwide are affected by hypertension (48). Persistent elevation of blood pressure (BP) has been known to be a risk factor for development of stroke, myocardial infarction, and chronic kidney and vascular diseases (9, 44). A high level of homocysteine (Hcy), or hyperhomocysteinemia (HHcy), is an independent risk factor of hypertension (30, 40, 54). Hcy is a product of methionine metabolism that is cleared in the body by remethylation and transsulfuration pathways (55). Hcy remethylation is mediated by methionine synthase, where vitamin B12 (cobalamin) is used as a cofactor and 5-methyltetrahydrofolate is utilized as the methyl donor (42). 5-Methyltetrahydrofolate, or the active form of folate (B9), is synthesized from 5,10-methylenetetrahydrofolate by methylenetetrahydrofolate reductase. In transsulfuration, which occurs in the small intestine, liver, pancreas, and kidney, a cofactor, vitamin B6 (pyridoxal phosphate), is required to convert Hcy to cystathionine by cystathionine β-synthase (CBS) (61). Cystathionine is hydrolyzed by vitamin B6-dependent cystathionine γ-lyase (CSE) to cysteine, which is used as a precursor for synthesis of the antioxidant glutathione or the vasodilator hydrogen sulfide (63). Nutritional deficiencies in vitamin cofactors (B12, B9, and B6) and mutations of methylenetetrahydrofolate reductase, CBS, and CSE enzymes are the common causes of HHcy (7, 20, 22, 38, 47).
HHcy-mediated vascular dysfunction and remodeling, as the hallmarks of hypertension, developed as the result of a complex of mechanisms, including instigation of oxidative stress, mitochondrial apoptosis, and inflammation. Hcy undergoes autoxidation and has the ability to directly produce superoxide and hydrogen peroxide because of a highly reactive thiol group (46). The presence of nitric oxide in the oxidative environment leads to peroxynitrite formation, which further exacerbates oxidative stress, reducing nitric oxide bioavailability (59). In addition, our group and other studies have shown that, in HHcy, there is an increase in expression and activity of NADPH oxidase (NOX) that mediates superoxide production (17, 21). Elevation of the level of Hcy disrupts the oxidant-antioxidant balance by diminishing the activity and expression of the antioxidant enzyme glutathione peroxidase (3). Apart from oxidative stress, HHcy plays a role in the instigation of mitochondrial disorders.
Mitochondrial dysfunction and mitochondria-dependent apoptosis have been shown to promote endothelial cell loss, leading to endothelial dysfunction (1, 57), which contributes to the pathogenesis of hypertension (15, 18). The intrinsic pathway, or mitochondria-dependent apoptosis (27, 62), is triggered by various factors, reactive oxygen species (ROS), DNA damage, Ca2+ overload, hypoxia, and oxidized LDL, and is regulated by a B cell lymphoma 2 (BCL-2) family of proteins, which are classified as proapoptotic (BAX) and antiapoptotic (BCL-2) proteins. BAX is expressed in the cytosol and, upon activation, is translocated to mitochondria, where it initiates mitochondria outer membrane permeabilization followed by cytochrome c release and apoptosome (cytochrome c-apoptotic protease-activating factor 1 complex) formation, which plays a role in caspase-9 and caspase-3 activation. The effector caspase-3 facilitates DNA and protein fragmentation, which promotes cell death. The third event mediated by HHcy, in addition to oxidative stress and mitochondrial dysfunction, is vascular inflammation.
Chronic vascular inflammation has been considered a mechanism of the initiation and exacerbation of hypertension (25, 35, 52). Inflammation is the immediate response of the immune system to the presence of pathogens, which is characterized by augmentation of proinflammatory cytokine (IL-1β, IL-6, and TNF-α) and chemokine [monocyte chemoattractant protein 1 (MCP-1)] secretion, which regulates the immune response and the migration of immune cells to target organs. Several in vitro studies have shown upregulation of inflammatory markers, including activation of NF-κB, an inflammatory cytokine transcription factor, in HHcy (11, 29). Zhang et al. reported that plasma IL-6, TNF-α, and MCP-1 levels are positively correlated with Hcy levels (66). Scherer et al. also observed mild HHcy-mediated augmentation of inflammatory cytokine (IL-1β, IL-6, TNF-α, and MCP-1) production in serum and different organs (51). Xia et al. showed HHcy-induced inflammasome formation and activation, which contribute to IL-1β maturation, triggering the early innate immunity cascade (60). Although HHcy is known to promote proinflammatory cytokine production, the precise mechanism of inflammatory response initiation remains unclarified. Pathogen recognition receptors and, in particular, Toll-like receptors (TLRs) are the antigen sensors that play a role in innate immune system activation and have recently gained significant attention in the field of hypertension. Thirteen TLRs have been described in mammals (TLR-1 to -10 in humans and TLR-11 to -13 in mice): cell surface TLRs that sense the presence of bacteria and fungus (TLR-1, -2, -4, and -5) and TLRs that are localized to intracellular membranes and recognize viral or microbial nucleic acid (TLR-3, -7, -8, and -9) (35). The role of TLR-4, which is ubiquitous within the vasculature (endothelial cells and vascular smooth muscle cells), has been recently highlighted in sterile inflammation. TLR-4 activation with downstream NF-κB-mediated cytokine upregulation has been implicated in the pathogenesis of hypertension (14, 36). Hence, TLR-4 inhibition has been shown to reduce inflammatory cytokine elevation and attenuate hypertension (8, 13). Therefore, we hypothesize that HHcy induces hypertension by TLR-4 activation, which promotes inflammatory cytokine (IL-1β, IL-6, and TNF-α) upregulation and initiation of mitochondria-dependent apoptosis, leading to cell death and chronic vascular inflammation. In addition, we aimed to elucidate the role of TLR-4 mutation in attenuation of HHcy-mediated vascular inflammation and mitochondria-dependent cell death.
MATERIALS AND METHODS
Animal models.
C57BL/6J [wild-type (WT)] mice, CBS-deficient (CBS+/−) mice with genetic mild HHcy, mice with mutation of TLR-4 [C3H/HeJ (C3H)], and mice with combined genetic HHcy and TLR-4 mutation (CBS+/−/C3H) were used in the present study. The animals were 13–14 wk old; their body weight was 25–30 g. The mice were purchased from Jackson Laboratory (Bar Harbor, ME). All standard procedures and experiments involving animals conformed with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Louisville.
Genotyping.
The background of CBS+/− heterozygous mice was confirmed according to the Jackson Laboratory protocol for genotyping. Briefly, samples from the tails of the mice were collected for DNA extraction. DNA was used to amplify with CBS primer sets by PCR. The PCR product samples were run on 1.2% agarose gel [prepared in Tris base, acetic acid, and EDTA (TAE buffer), pH 8.4] with ethidium bromide. The images were recorded in a gel documentation system (Bio-Rad, Hercules, CA). To confirm TLR-4 mutation in C3H/HeJ mice, the respective protocol of restriction fragment length polymorphism (RFLP)-PCR was used (31). Briefly, DNA extracted from the tails of the animals was amplified with a specific TLR-4 set of primers (Table 1). PCR products were digested with NLaIII restriction enzyme (RE) overnight at 37°C. The RE digestion products were loaded on a 10% polyacrylamide gel and run at 80 V. The gel was incubated with 1× TAE buffer containing ethidium bromide. The images were made in a gel documentation system (Bio-Rad). The mutant TLR-4 gene includes the CATG sequence, which is cut by the RE, yielding 96- and 108-bp bands, while the absence of the TLR-4 mutation is confirmed by undigested products at 204 bp. To confirm the genetic background of CBS+/−/C3H mice, we first determined CBS gene deficiency in DNA samples that were selected for further detection of TLR-4 mutation.
Table 1.
Sequence-specific oligonucleotide primers
| Nucleotide Sequence |
||
|---|---|---|
| Gene | Forward | Reverse |
| TLR-4 | 5′-TCAGAATGAGGACTGGGTGA | 5′-CTCGGCAACCACATAGAAACT |
| IL-1β | 5′-CACCTCTCAAGCAGAGCACAG | 5′-GGGTTCCATGGTGAAGTCAAC |
| IL-6 | 5′-TGGAGTCACAGAAGGAGTGGCTAAG | 5′-TCTGACCACAGTGAGGAATGTCCAC |
| TNF-α | 5′-TCGTAGCAAACCACCAAGTG | 5′-AGATAGCAAATCGGCTGACG |
| BAX | 5′-GGCTGGACACTGGACTTCCT | 5′-GGTGAGGACTCCAGCCACAA |
| Rn18S | 5′-AAATCAGTTATGGTTCCTTGGTC | 5′-GCTCTAGAATTACCACAGTTATCCAA |
BP measurement.
BP was recorded using a noninvasive tail cuff method (CODA, Kent Scientific, Torrington, CT). Prior to BP measurements, the animals were placed in the restraining chambers on a warm platform for 30 min for 3 consecutive days to ensure that they adapted to the procedure. BP was recorded in a proper environment (room temperature, lighting, and noise-free atmosphere) for 4 groups, with 10 animals in each group.
Vascular ultrasonography.
Ultrasound of the superior mesenteric artery (SMA) was performed with the Vevo 2100 system (Visual Sonics, Toronto, ON, Canada) under isoflurane anesthesia. Physiological parameters (heart rate and respiratory rate) were monitored during the procedure. The mouse was placed on a warmed (37°C) platform in the supine position, and the abdominal area was depilated. Imaging was performed using acoustic gel (Other-Sonic, Pharmaceutical Innovations, Newark, NJ), which was applied to the skin in the abdominal area, and a 13- to 24-MHz transducer (model MS550D, Vevo). To calculate wall-to-lumen ratio, wall thickness and lumen diameter were measured using SMA images in B mode. The SMA resistive index (RI) was calculated as RI = (PSV − EDV)/PSV, with peak systolic velocities (PSVs) and end-diastolic velocities (EDVs) measured in pulsed-wave Doppler mode. The SMA pulsatility index (PI) was measured in pulsed-wave Doppler mode and calculated as PI = (PSV − EDV)/MV, where MV is mean velocity. Ultrasound of the SMA was performed for four groups, with five animals in each group.
Quantitative RT-PCR.
To assess mRNA expression of different genes in the SMA, RNA was isolated with TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. RNA quantification and purity were assessed by spectrophotometry (Nanodrop 1000, Thermo Scientific, Waltham, MA). Aliquots (2 μg) of total RNA were reverse-transcribed into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol. Quantitative PCR (qPCR) was performed for different genes (BAX, IL-1β, IL-6, and TNF-α) in a final reaction volume of 20 μl containing 10 μl of PerfeCTa SYBR Green supermix, low-concentration ROX (Quanta Biosciences, Gaithersburg, MD), 6 μl of nuclease-free water, 2 μl of cDNA, and 40 pmol of forward and reverse primers. All sequence-specific oligonucleotide primers (Invitrogen, Carlsbad, CA) are presented in Table 1. Data are represented as fold expression, calculated as the cycle threshold difference between control and sample normalized with the housekeeping gene Rn18s.
Western blot analysis.
SMA protein content was extracted using RIPA buffer (Boston BioProducts, Ashland, MA), PMSF (Calbiochem, La Jolla, CA), and protease inhibitor (Sigma Aldrich, St. Louis, MO). The protein extract was incubated overnight at 4°C with shaking and centrifuged at 13,000 rpm for 20 min. The supernatant was collected in another tube for protein estimation using the Bradford dye method (Bio-Rad). The prepared protein lysate (60 μg) was heated at 95°C for 5 min and loaded on a polyacrylamide gel with SDS in running buffer and run at constant current (100 V). For protein transfer to a PVDF membrane, an electrotransfer apparatus (Bio-Rad) was used. After transfer, the membranes were blocked in 5% nonfat milk for 1 h at room temperature and incubated overnight with primary antibodies (Table 2) at 4°C. After they were washed with Tris-buffered saline-Tween 20 buffer, the membranes were incubated with secondary antibodies for 1 h at room temperature and washed again. The membranes were developed with the ECL Western blotting detection system (GE Healthcare, Piscataway, NJ), and all images were recorded in the gel documentation system ChemiDoc XRS (Bio-Rad, Richmond, CA). The membranes were stripped with stripping buffer (Boston BioProducts) and then blocked with 5% milk for 1 h at room temperature. After they were washed, the membranes were reprobed with anti-β-actin antibody (Table 2) as a loading control protein. The data were analyzed by Bio-Rad Image Lab densitometry software and normalized to β-actin bands.
Table 2.
Primary antibodies
| Protein | Source | Catalog No. |
|---|---|---|
| Caspase-9 | Abcam | ab32539 |
| Caspase-3 (cleaved) | Cell Signaling | 9661 |
| BAX | Abcam | ab182733 |
| TLR-4 | Abcam | ab22048 |
| TNF-α | Abcam | ab6671 |
| β-Actin | Sigma Life Science | A2228 |
Immunohistochemistry.
SMA tissue was immersed in tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC) in a disposable plastic tissue-embedding mold (Polysciences, Warrington, PA). The tissue blocks were kept at −80°C until they were cut into 5-μm-thick sections (Cryocut, model CM 1850, Leica Microsystems, Buffalo Grove, IL). Tissue sections were placed on polylysine-coated slides (Polysciences), incubated with permeabilization solution (0.2 g of bovine serum albumin and 3 μl of Triton X-100 in 10 ml of 1× PBS) for 1 h at room temperature, and then washed with 1× PBS. The sections were incubated with a 1:250 dilution of primary antibody (Table 2) overnight at 4°C. After the slides were washed with 1× PBS, they were incubated with a 1:500 dilution of fluorescently labeled secondary antibodies (goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Texas Red, Invitrogen, Waltham, MA) for 1 h at room temperature and then stained with a 1:10,000 dilution of 4′,6-diamidino-2-phenylindole (DAPI) for 20 min at room temperature. After they were washed, the slides were mounted with mounting medium and visualized using a laser scanning confocal microscope (Fluo View 1000, Olympus, Center Valley, PA). The images were analyzed by measurement of fluorescence intensity with Image-Pro Plus software (Media Cybernetics, Rockville, MD).
TUNEL assay.
The DeadEnd fluorometric terminal deoxynucleotidyl transferase dUTP nick-end-labeling (TUNEL) system (Promega, Madison, WI) measures nuclear DNA fragmentation, which is an important biochemical hallmark of apoptosis. The TUNEL system detects fragmented DNA by catalytically binding fluorescein-12-dUTP to 3′-OH DNA ends using recombinant terminal deoxynucleotidyl transferase. The TUNEL assay was performed on frozen SMA tissue sections using a commercially available kit (DeadEnd fluorometric TUNEL system). The assay was done according to the manufacturer's protocol for tissue staining, including a positive control preparation and nuclear staining with DAPI. The slides were visualized with a confocal microscope (Fluo View 1000, Olympus) using a green fluorescence filter to detect DNA fragmentation and a blue DAPI filter to detect the nucleus. The images were analyzed with Image-Pro Plus software (Media Cybernetics).
Statistical analysis.
Statistical analysis was performed with Primer of Biostatistics 7.0 (McGraw-Hill). Significance was determined by one-way analysis of variance followed by Holm's multiple-comparison test between the groups. The difference was considered significant when P < 0.05. Values are means ± SE (n ≥ 4).
RESULTS
Genotyping for WT, CBS+/−, C3H, and CBS+/−/C3H mice.
For genotype analysis, the Jackson Laboratory protocol was followed. CBS+/− and CBS+/−/C3H mice had two bands at 450 and 308 bp, while CBS+/+ mice had a single band at 308 bp, when primers specific for the CBS gene were used (Fig. 1A). TLR-4 mutant (C3H and CBS+/−/C3H) mice had bands at 96 and 108 bp, whereas nonmutant (WT and CBS+/−) mice had a single band at 204 bp (Fig. 1B), when RFLP-PCR was used with TLR-4 primers.
Fig. 1.
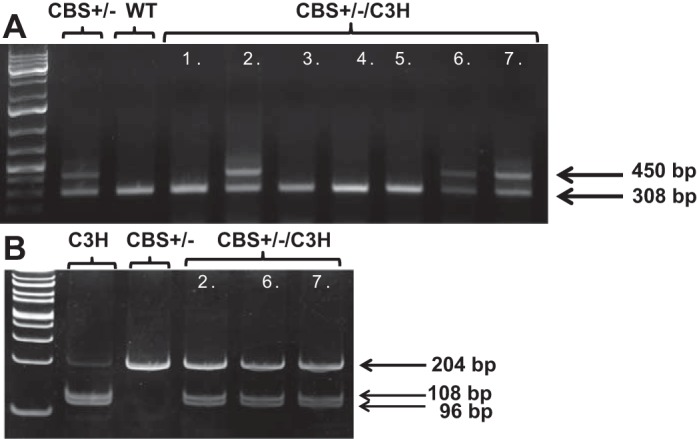
A: genotyping for the cystathionine β-synthase (CBS) gene. CBS+/− and CBS+/−/C3H mice had 2 bands, at 450 and 308 bp; wild-type (WT) mice had 1 band, at 308 bp. B: restriction fragment length polymorphism-PCR for the Toll-like receptor 4 (TLR-4) gene. TLR-4 mutant (C3H and CBS+/−/C3H) mice had 2 bands, at 96 and 108 bp; nonmutant (WT and CBS+/−) mice had 1 band, at 204 bp.
TLR-4 mutation suppresses HHcy-mediated hypertension.
Systolic, diastolic, and mean BP were significantly higher in HHcy (CBS+/−) than WT mice (Fig. 2). Systolic, diastolic, and mean BP were significantly lower in TLR-4-mutant (C3H) than WT mice (Fig. 2). Systolic, diastolic, and mean BP were significantly lower in mice with combined genetic HHcy and TLR-4 mutation (CBS+/−/C3H) than CBS+/− mice (Fig. 2).
Fig. 2.
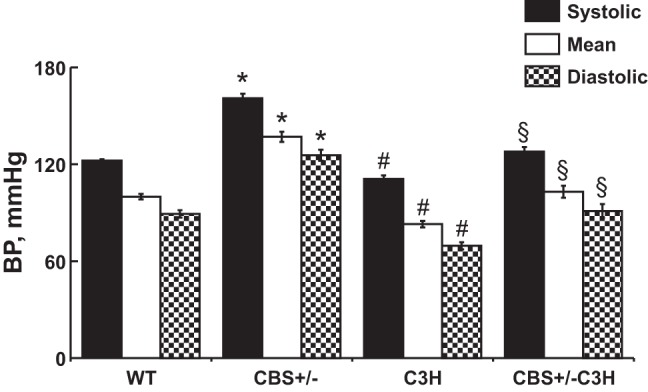
Systolic, mean, and diastolic blood pressure (BP). Hyperhomocysteinemic (HHcy) mice had significantly higher systolic, mean, and diastolic BP and mice with combined genetic HHcy and TLR-4 mutation (CBS+/−/C3H) had significantly lower systolic, mean, and diastolic BP. Values are means ± SE (n = 10). *P < 0.05, WT vs. CBS+/−. #P < 0.05, WT vs. C3H. §P < 0.05, CBS+/− vs. CBS+/−/C3H.
Wall-to-lumen ratio, RI, and PI.
To assess structural changes in the SMA, wall thickness and lumen diameter were measured and the SMA wall-to-lumen ratio was calculated. The SMA wall-to-lumen ratio was increased in CBS+/− mice compared with WT and C3H mice (Fig. 3A). The SMA wall-to-lumen ratio of the CBS+/−/C3H mice was similar to that of the control group (Fig. 3A). RI and PI were calculated from the blood flow velocities in the artery during the cardiac cycle and used to determine peripheral resistance. RI and PI of the SMA were increased in CBS+/− mice compared with WT and C3H mice (Fig. 3B). In mice with combined genetic HHcy and TLR-4 mutation, the SMA RI and PI were similar to the control group (Fig. 3B).
Fig. 3.

A: wall-to-lumen ratio. To assess structural changes in the superior mesenteric artery (SMA), wall thickness and lumen diameter were measured and the SMA wall-to-lumen ratio was calculated. The SMA wall-to-lumen ratio was increased in CBS+/− mice compared with WT and C3H mice. The SMA wall-to-lumen ratio of CBS+/−/C3H mice was similar to the control group. Values are means ± SE (n = 5). *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−. B: pulsatility index (PI) and resistive index (RI) of the SMA. PI and RI are calculated from blood flow velocities in the SMA during the cardiac cycle and used to determine a peripheral resistance. PI and RI of the SMA were increased in CBS+/− mice compared with WT and C3H mice. SMA PI and RI were similar to controls in mice with combined genetic HHcy and TLR-4 mutation. Values are means ± SE (n = 5). *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−.
TLR-4 mutation reduces vascular inflammation.
To examine inflammatory marker activation, we used immunohistochemistry (IHC) to analyze TLR-4 and TNF-α expression (Fig. 4) and qPCR to analyze IL-1β, IL-6, and TNF-α mRNA expression (Fig. 5). IHC showed increased intensity of TLR-4 and TNF-α in the SMA of CBS+/− mice (Fig. 4). TLR-4 and TNF-α intensities were reduced in mice with combined genetic HHcy and TLR-4 mutation (Fig. 4). IL-1β, IL-6, and TNF-α mRNA expression were elevated in the SMA of CBS+/− mice and reduced in the SMA of CBS+/−/C3H mice (Fig. 5).
Fig. 4.

A and B: immunohistochemistry (IHC) for TLR-4 and TNF-α. IHC showed increased intensity of TLR-4 and TNF-α in the SMA of CBS+/− mice. TLR-4 and TNF-α intensities were reduced in mice with combined genetic HHcy and TLR-4 mutation. Values are means ± SE (n = 4). *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−.
Fig. 5.

A and B: quantitative RT-PCR for IL-1β (n = 5) and IL-6 (n = 4) and TNF-α (n = 5). IL-1β, IL-6, and TNF-α mRNA expression were elevated in the SMA of CBS+/− mice and reduced in the SMA of CBS+/−/C3H mice. Values are means ± SE. *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−.
TLR-4 mutation attenuates mitochondria-mediated cell death.
To evaluate apoptotic marker activation, we analyzed BAX gene expression by qPCR (Fig. 6A), BAX protein expression by Western blotting (Fig. 6B), caspase-9 protein expression by Western blotting (Fig. 6C), and cleaved caspase-3 expression by IHC (Fig. 6D). BAX mRNA expression was significantly upregulated in CBS+/− mice compared with other groups (Fig. 6A). BAX mRNA expression was reduced in CBS-deficient mice with TLR-4 mutation (CBS+/−/C3H) compared with CBS+/− mice (Fig. 6A). BAX protein expression was increased in CBS+/− mice and reduced in CBS+/−/C3H mice (Fig. 6B). Caspase-9 protein expression was significantly increased in HHcy (CBS+/−) and CBS+/−/C3H mice compared with WT and TLR-4-mutant (C3H) mice (Fig. 6C). In addition, upregulation of cleaved caspase-3 expression in CBS+/− mice was confirmed by IHC (Fig. 6D). The intensity of cleaved caspase-3 was reduced in the SMA of CBS+/−/C3H mice compared with CBS+/− mice (Fig. 6D). These results suggest that induction of mitochondria-dependent apoptosis in HHcy and TLR-4 mutation alleviates mitochondria-mediated apoptosis.
Fig. 6.

A: quantitative RT-PCR for the BAX gene. BAX mRNA expression was significantly upregulated in CBS+/− mice compared with other groups. BAX mRNA expression was reduced in CBS-deficient mice with TLR-4 mutation (CBS+/−/C3H) compared with CBS+/− mice. Values are means ± SE (n = 4). *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−. B: Western blotting for BAX protein expression. BAX protein expression was increased in CBS+/− mice compared with the other groups. BAX protein expression was reduced in CBS-deficient mice with TLR-4 mutation (CBS+/−/C3H) compared with CBS+/− mice. C: Western blot for caspase-9 protein expression. Caspase-9 protein expression was increased in HHcy mice (CBS+/−) and CBS+/−/C3H mice compared with WT and TLR-4 mutant (C3H) mice. Values are means ± SE (n = 6). *P < 0.05, CBS+/− vs. C3H. #P < 0.05, C3H vs. CBS+/−/C3H. D: IHC showed elevated levels of cleaved caspase-3 in the SMA of CBS+/− mice. Intensity of cleaved caspase-3 was reduced in the SMA of CBS+/−/C3H mice compared with CBS+/− mice. Values are means ± SE (n = 4). *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−.
TLR-4 mutation mitigates HHcy-induced DNA fragmentation.
TUNEL assay was used to evaluate DNA fragmentation in the SMA of different groups (Fig. 7). DNA fragmentation was significantly augmented in the SMA of CBS+/− mice compared with WT and C3H mice (Fig. 7). TUNEL-positive cell count was reduced in the SMA of CBS+/−/C3H mice compared with CBS+/− mice (Fig. 7).
Fig. 7.

TLR-4 mutation mitigates HHcy-induced DNA fragmentation. TUNEL assay was used to evaluate DNA fragmentation (arrows) in the SMA. DNA fragmentation was significantly augmented in the SMA of CBS+/− mice compared with WT and C3H mice. TUNEL-positive cell count was reduced in the SMA of CBS+/−/C3H mice compared with CBS+/− mice. ctr, Control. Values are means ± SE (n = 4). *P < 0.05 vs. WT. #P < 0.05 vs. CBS+/−.
DISCUSSION
Elevated plasma Hcy has been shown to be a risk factor for peripheral artery disease and hypertension (30, 33, 38, 65). In our previous work we showed that HHcy induced endothelial cell injury and peripheral vascular remodeling with collagen deposition in the SMA (21). Consistent with our findings, several studies reported that HHcy promotes endothelial cell injury, vascular endothelial dysfunction, and vascular remodeling, which contribute to the pathogenesis of hypertension (34, 41, 50). The elevation of total peripheral resistance that is attributed to alterations in structural and physical properties of the resistance arteries is the hallmark of primary, or essential, hypertension. In the present study, using ultrasonography, we detected an increase in SMA wall-to-lumen ratio, RI, and PI in CBS+/− mice, which indicates inward vascular remodeling and an increase in peripheral resistance due to HHcy. The increase in peripheral vascular resistance has been associated with raised systolic, diastolic, and mean arterial BP in CBS+/− mice. RI, PI, and wall-to-lumen ratio of the SMA in mice with combined genetic HHcy and TLR-4 mutation (CBS+/−/C3H) were similar to the control group, which could explain attenuation of HHcy-mediated high BP.
A significant number of studies have described inflammation as one of the toxic effects of Hcy. Zhang et al. reported that plasma Hcy was positively correlated with plasma proinflammatory cytokine and chemokine (IL-6, TNF-α, and MCP-1) levels and promoted inflammatory monocyte differentiation (66). Zanin et al. showed that HHcy is involved in the synthesis and secretion of IL-1β via NF-κB in murine macrophages (64). Han et al. observed endothelial cell inflammatory injury through activation of NF-κB and cytokine IL-6 upregulation in HHcy (23). In agreement with previous findings, we found an upregulation of proinflammatory cytokine (IL-1β, IL-6, and TNF-α) expression in the SMA of CBS+/− mice, confirming chronic vascular inflammation induced by genetic mild HHcy. Interestingly, we observed an active presence of IL-1β in the SMA tissue of mice with the TLR-4 mutation (C3H) that could be explained by potential involvement of other Toll-like/IL-1 receptors. However, we have found that other cytokines (IL-6 and TNF-α) are reduced in the SMA tissue of C3H mice, suggesting that TLR-4 mutation attenuates vascular inflammation. In addition, we observed that mice with combined genetic HHcy and TLR-4 mutation exhibit reduced levels of proinflammatory cytokines (IL-1β, IL-6, and TNF-α) in the SMA compared with CBS+/− mice, further indicating that TLR-4 mutation prevents HHcy-induced chronic vascular inflammation. The role of the TLR-4-mediated signaling pathway has been recently highlighted in the pathophysiology of several cardiovascular diseases, including hypertension (5, 16, 19, 24). Pryshchep et al. observed the abundant expression of TLR-4 in all cell types of six different vascular beds (aorta, carotid, temporal, subclavian, iliac, and mesenteric arteries). It was confirmed that TLR-4 is expressed in endothelial and vascular smooth muscle cells in atherosclerosis, while in normal, noninflamed arteries, adventitial dendritic cells are the major sensors of pathogen-related motifs (45). McCarthy and Webb described the role of the interaction of musculoskeletal injury-induced endothelial TLR-4 and damage-associated molecular pattern molecules (high-mobility group box 1 and mitochondrial DNA) in the development of hypertension in football players (36). Dange et al. reported increased blood and brain TLR-4, TNF-α, and IL-1β expression in angiotensin II-induced hypertension and also showed that central blockade of TLR-4 delayed the progression of hypertension and improved cardiac function in hypertensive rats (12). Bobek et al. observed that TNF-α-infused mice developed proteinuric hypertension similar to human preeclampsia that was accompanied by upregulation of TLR-4 protein expression in the placenta (6). Li et al. reported TLR-4-induced matrix metalloproteinase (MMP)-9 elevation in human aortic smooth muscle cells and confirmed that TLR-4 siRNA silencing regulated MMP-9 expression, indicating the role of TLR-4-mediated signaling pathway in vascular remodeling (28). Balistreri et al., for the first time, described a rs4986790 TLR-4 polymorphism that confers a higher susceptibility for sporadic thoracic aorta aneurism and, together with rs1799752 angiotensin-converting enzyme, rs3918242 MMP-9, and rs2285053 MMP-2 single-nucleotide polymorphisms, is an independent risk factor for sporadic thoracic aorta aneurism. In addition, they observed that the cases with combined risk genotype showed higher levels of inflammatory mediators and plasma MMP-9 and MMP-2 levels that were accompanied by elastic fragmentation in tissue aorta samples (4, 49). In previous work we reported TLR-4, IL-1β, IL-6, and TNF-α upregulation in heart tissues of CBS+/− mice with genetic mild HHcy. The inflammatory markers were further increased when mice were fed a high-methionine diet, which exacerbated HHcy (10). In agreement with previous studies, we found upregulation of TLR-4 expression in the SMA of CBS+/− mice that connects the downstream inflammatory cytokine signaling pathway activation in mild HHcy. Mice with combined genetic HHcy and TLR-4 mutation exhibit less TLR-4 expression than CBS+/− mice, preventing downstream inflammatory cytokine elevation.
Mitochondria-dependent apoptosis has been implicated in the induction of vascular remodeling that contributes to pathogenesis of hypertension (37, 56, 67). A significant number of studies have described the role of HHcy in the initiation of mitochondria-dependent apoptosis through activation of ROS. Kim et al. showed that Hcy treatment of primary human bone marrow stromal cells led to activation of ROS, caspase-9, caspase-3, and cytochrome c release into the cytosol from mitochondria, suggesting that a mitochondria-initiated cell death pathway is a predominant mechanism of apoptosis in HHcy (26). Sipkens et al. observed a NOX2, NOX4, and Hcy concentration-dependent increase in caspase-3 in human umbilical vein endothelial cells (53). Moreira et al. reported that Hcy increases superoxide levels and cell death in human aortic endothelial cells, but vitamin B12 supplementation prevents oxidative stress-induced cell death (39). However, some studies have been stressing an inflammatory component as the critical mechanism of mitochondria-mediated cell death. Pan et al. reported that the transcription factor NF-κB regulates the expression of both proinflammatory genes and genes that contribute to mitochondria-dependent apoptosis (the BCL-2 gene family) (43). It has been implied that NF-κB activation of NF-κB occurs prior to DNA fragmentation and is accompanied by upregulation of proapoptotic proteins (BAX) (32, 58). Aoki et al. reported that NF-κB promotes endothelial cell death through translocation of BAX to mitochondria and downregulation of Bcl-2 (2). In the present study we found an upregulation of BAX, caspase-9, and caspase-3 expression in the SMA of CBS+/− mice with mild HHcy compared with controls. Mice with TLR-4 mutation alone expressed less BAX, caspase-9, and cleaved caspase-3 in the SMA. Mice with combined genetic HHcy and TLR-4 mutation exhibited downregulation of BAX and cleaved caspase-3 expression in the SMA. In addition, DNA fragmentation in the SMA was increased in the mouse model of mild HHcy, but mice with TLR-4 mutation (C3H and CBS+/−/C3H) showed decreased DNA fragmentation in the SMA. These findings suggest that the TLR-4-driven inflammatory signaling pathway contributes to initiation of mitochondria-mediated cell death; thus TLR-4 mutation plays a role in alleviation of mitochondrial apoptosis.
Summary.
Our study demonstrates that HHcy induces hypertension by TLR-4 activation followed by inflammatory cytokine elevation and initiation of mitochondrial apoptosis, which lead to cell death and chronic vascular inflammation (Fig. 8). TLR-4 mutation attenuates vascular inflammation and cell death, which suppress hypertension.
Fig. 8.

Schematic representation of the hypothesis. HHcy induces hypertension by TLR-4 activation followed by inflammatory cytokine (IL-1β, IL-6, and TNF-α) elevation and initiation of mitochondria-dependent apoptosis, which lead to cell death and chronic vascular inflammation. TLR-4 mutation attenuates vascular inflammation and cell death, which suppress hypertension. ΔΨm, mitochondrial membrane potential; Apaf-1, apoptotic protease-activating factor 1.
Conclusions and perspectives.
Experimental studies have demonstrated that elevated plasma Hcy is a risk factor for peripheral artery disease and hypertension. However, the precise mechanism of detrimental Hcy interaction with the vascular wall is incompletely understood. In the present study we have described different aspects of Hcy action in the development of peripheral vascular remodeling, which is a hallmark of hypertension. Our study, for the first time, has illustrated that Hcy acts through TLR-4, which is highly expressed in all cell types of the vascular bed. HHcy-mediated TLR-4 activation promotes chronic vascular inflammation with proinflammatory cytokine elevation, facilitating mitochondria-mediated cell death, which favors inward vascular remodeling. We have also demonstrated that TLR-4 mutation attenuated chronic vascular inflammation and mitochondria-induced cell injury, which prevented peripheral vascular remodeling and suppressed hypertension. Several experimental studies have illustrated the promising effect of TLR-4-targeted therapy in prevention of hypertension. However, further studies are required for detailed investigation of the role of the TLR-4-mediated signaling pathway in the pathogenesis of hypertension and other cardiovascular diseases. Future studies that will elucidate the mechanisms that underlie the TLR-4-mediated inflammatory pathway may offer novel concepts for hypertension therapy.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-74185 and HL-108621.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.F. and S.C.T. developed the concept and designed the research; A.F., N.J., and N.M. performed the experiments; A.F. analyzed the data; A.F., P.C., A.K., N.J., and G.H.K. interpreted the results of the experiments; A.F. and N.M. prepared the figures; A.F. drafted the manuscript; A.F., P.C., A.K., G.H.K., and S.C.T. edited and revised the manuscript; S.C.T. approved the final version of the manuscript.
ACKNOWLEDGMENTS
Part of this work was presented at the Council on Hypertension 2015 Scientific Sessions, September 16–19, 2015, Washington, DC, and the Annual Meeting of the International Academy of Cardiovascular Sciences, September 10–12, 2015, Omaha, NE.
REFERENCES
- 1.Ahsan A, Han G, Pan J, Liu S, Padhiar AA, Chu P, Sun Z, Zhang Z, Sun B, Wu J, Irshad A, Lin Y, Peng J, Tang Z. Phosphocreatine protects endothelial cells from oxidized low-density lipoprotein-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Apoptosis 20: 1563–1576, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Aoki M, Nata T, Morishita R, Matsushita H, Nakagami H, Yamamoto K, Yamazaki K, Nakabayashi M, Ogihara T, Kaneda Y. Endothelial apoptosis induced by oxidative stress through activation of NF-κB: antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension 38: 48–55, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ 11 Suppl 1: S56–S64, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Balistreri CR. Genetic contribution in sporadic thoracic aortic aneurysm? Emerging evidence of genetic variants related to TLR-4-mediated signaling pathway as risk determinants. Vasc Pharmacol 74: 1–10, 2015. [DOI] [PubMed] [Google Scholar]
- 5.Balistreri CR, Candore G, Colonna-Romano G, Lio D, Caruso M, Hoffmann E, Franceschi C, Caruso C. Role of Toll-like receptor 4 in acute myocardial infarction and longevity. JAMA 292: 2339–2340, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Bobek G, Surmon L, Mirabito KM, Makris A, Hennessy A. Placental regulation of inflammation and hypoxia after TNF-α infusion in mice. Am J Reprod Immunol 74: 407–418, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Boini KM, Xia M, Abais JM, Xu M, Li CX, Li PL. Acid sphingomyelinase gene knockout ameliorates hyperhomocysteinemic glomerular injury in mice lacking cystathionine-β-synthase. PLos One 7: e45020, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 122: 535–543, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carretero OA, Oparil S. Essential hypertension. I. Definition and etiology. Circulation 101: 329–335, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Chaturvedi P, Kamat PK, Kalani A, Familtseva A, Tyagi SC. High methionine diet poses cardiac threat: a molecular insight. J Cell Physiol 231: 1554–1561, 2016. [DOI] [PubMed] [Google Scholar]
- 11.Curro M, Gangemi C, Gugliandolo A, Risitano R, Ferlazzo N, Ientile R, Caccamo D. Transglutaminase 2 is involved in homocysteine-induced activation of human THP-1 monocytes. Free Radic Res 49: 299–308, 2015. [DOI] [PubMed] [Google Scholar]
- 12.Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, Francis J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc Res 103: 17–27, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Dange RB, Agarwal D, Teruyama R, Francis J. Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflamm 12: 31, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Batista PR, Palacios R, Martin A, Hernanz R, Medici CT, Silva MA, Rossi EM, Aguado A, Vassallo DV, Salaices M, Alonso MJ. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLos One 9: e104020, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng XU, Xia KE, Chen PO, Ali Sheikh MS, Yang DF, Li SM, Yang TL. Reversion of left ventricle remodeling in spontaneously hypertensive rats by valsartan is associated with the inhibition of caspase-3, -8 and -9 activities. Biomed Rep 3: 533–536, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Z, Liu S, Wang X, Khaidakov M, Fan Y, Deng X, Xiang D, Mehta JL. Lectin-like oxidized low-density lipoprotein receptor-1 regulates autophagy and Toll-like receptor 4 in the brain of hypertensive mice. J Hypertens 33: 525–533, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Edirimanne VE, Woo CW, Siow YL, Pierce GN, Xie JY, O K. Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Can J Physiol Pharmacol 85: 1236–1247, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Eirin A, Lerman A, Lerman LO. Mitochondria: a pathogenic paradigm in hypertensive renal disease. Hypertension 65: 264–270, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eissler R, Schmaderer C, Rusai K, Kuhne L, Sollinger D, Lahmer T, Witzke O, Lutz J, Heemann U, Baumann M. Hypertension augments cardiac Toll-like receptor 4 expression and activity. Hypertens Res 34: 551–558, 2011. [DOI] [PubMed] [Google Scholar]
- 20.Ekim M, Ekim H, Yilmaz YK, Kulah B, Polat MF, Gocmen AY. Study on relationships among deep vein thrombosis, homocysteine and related B group vitamins. Pakistan J Med Sci 31: 398–402, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Familtseva A, Kalani A, Chaturvedi P, Tyagi N, Metreveli N, Tyagi SC. Mitochondrial mitophagy in mesenteric artery remodeling in hyperhomocysteinemia. Physiol Rep 2: e00283, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao L, Kolanuvada B, Naik G, Zhang Y, Zhao M, Sun L, Alaie D, Petrillo RL. Hyperhomocysteinemia-induced upper extremity deep vein thrombosis and pulmonary embolism in a patient with methyltetrahydrofolate reductase mutation: a case report and literature review. Blood Coagul Fibrinolysis 27: 720–723, 2016. [DOI] [PubMed] [Google Scholar]
- 23.Han S, Wu H, Li W, Gao P. Protective effects of genistein in homocysteine-induced endothelial cell inflammatory injury. Mol Cell Biochem 403: 43–49, 2015. [DOI] [PubMed] [Google Scholar]
- 24.Ionita MG, Arslan F, de Kleijn DP, Pasterkamp G. Endogenous inflammatory molecules engage Toll-like receptors in cardiovascular disease. J Innate Immun 2: 307–315, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Itani HA, Harrison DG. Memories that last in hypertension. Am J Physiol Renal Physiol 308: F1197–F1199, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim DJ, Koh JM, Lee O, Kim NJ, Lee YS, Kim YS, Park JY, Lee KU, Kim GS. Homocysteine enhances apoptosis in human bone marrow stromal cells. Bone 39: 582–590, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Kvansakul M, Hinds MG. The Bcl-2 family: structures, interactions and targets for drug discovery. Apoptosis 20: 136–150, 2015. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Xu H, Liu S. Toll-like receptors 4 induces expression of matrix metalloproteinase-9 in human aortic smooth muscle cells. Mol Biol Rep 38: 1419–1423, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Li JJ, Li Q, Du HP, Wang YL, You SJ, Wang F, Xu XS, Cheng J, Cao YJ, Liu CF, Hu LF. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int J Mol Sci 16: 12560–12577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Guo X, Chen S, Zheng L, Yang H, Sun G, Yu S, Li W, Zhou L, Wang J, Hu W, Sun Y. Hyperhomocysteinemia independently associated with the risk of hypertension: a cross-sectional study from rural China. J Hum Hypertens 30: 508–512, 2016. [DOI] [PubMed] [Google Scholar]
- 31.Liang CF, Liu JT, Wang Y, Xu A, Vanhoutte PM. Toll-like receptor 4 mutation protects obese mice against endothelial dysfunction by decreasing NADPH oxidase isoforms 1 and 4. Arterioscler Thromb Vasc Biol 33: 777–784, 2013. [DOI] [PubMed] [Google Scholar]
- 32.Liang J, Luan Y, Lu B, Zhang H, Luo YN, Ge P. Protection of ischemic postconditioning against neuronal apoptosis induced by transient focal ischemia is associated with attenuation of NF-κB/p65 activation. PLos One 9: e96734, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao X, Xing X, Xu R, Gong Q, He Y, Li S, Wang H, Liu C, Ding X, Na R, Liu Z, Qu Y. Folic acid and vitamins D and B12 correlate with homocysteine in Chinese patients with type-2 diabetes mellitus, hypertension, or cardiovascular disease. Medicine 95: e2652, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazza A, Cuppini S, Schiavon L, Zuin M, Ravenni R, Balbi G, Montemurro D, Opocher G, Pelizzo MR, Colletti PM, Rubello D. Hyperhomocysteinemia is an independent predictor of sub-clinical carotid vascular damage in subjects with grade-1 hypertension. Endocrine 46: 340–346, 2014. [DOI] [PubMed] [Google Scholar]
- 35.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 306: H184–H196, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCarthy CG, Webb RC. The toll of the gridiron: damage-associated molecular patterns and hypertension in American football. FASEB J 30: 34–40, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molina MN, Ferder L, Manucha W. Emerging role of nitric oxide and heat shock proteins in insulin resistance. Curr Hypertens Rep 18: 1, 2016. [DOI] [PubMed] [Google Scholar]
- 38.Moll S, Varga EA. Homocysteine and MTHFR mutations. Circulation 132: e6–e9, 2015. [DOI] [PubMed] [Google Scholar]
- 39.Moreira ES, Brasch NE, Yun J. Vitamin B12 protects against superoxide-induced cell injury in human aortic endothelial cells. Free Radic Biol Med 51: 876–883, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narayanan N, Pushpakumar SB, Givvimani S, Kundu S, Metreveli N, James D, Bratcher AP, Tyagi SC. Epigenetic regulation of aortic remodeling in hyperhomocysteinemia. FASEB J 28: 3411–3422, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson J, Wu Y, Jiang X, Berretta R, Houser S, Choi E, Wang J, Huang J, Yang X, Wang H. Hyperhomocysteinemia suppresses bone marrow CD34+/VEGF receptor 2+ cells and inhibits progenitor cell mobilization and homing to injured vasculature—a role of β1-integrin in progenitor cell migration and adhesion. FASEB J 29: 3085–3099, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pacana T, Cazanave S, Verdianelli A, Patel V, Min HK, Mirshahi F, Quinlivan E, Sanyal AJ. Dysregulated hepatic methionine metabolism drives homocysteine elevation in diet-induced nonalcoholic fatty liver disease. PLos One 10: e0136822, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan W, Lin L, Zhang N, Yuan F, Hua X, Wang Y, Mo L. Neuroprotective effects of dexmedetomidine against hypoxia-induced nervous system injury are related to inhibition of NF-κB/COX-2 pathways. Cell Mol Neurobiol 36: 1179–1188, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petrie JR, Marso SP, Bain SC, Franek E, Jacob S, Masmiquel L, Leiter LA, Haluzik M, Satman I, Omar M, Shestakova M, Van Gaal L, Mann JF, Baeres FM, Zinman B, Poulter NR, LEADER Investigators. LEADER-4: blood pressure control in patients with type 2 diabetes and high cardiovascular risk: baseline data from the LEADER randomized trial. J Hypertens 34: 1140–1150, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation 118: 1276–1284, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pushpakumar SB, Kundu S, Metreveli N, Sen U. Folic acid mitigates angiotensin-II-induced blood pressure and renal remodeling. PLos One 8: e83813, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Renga B. Hydrogen sulfide generation in mammals: the molecular biology of cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE). Inflamm Allergy Drug Targets 10: 85–91, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics Committee, Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125: e2–e220, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruvolo G, Pisano C, Candore G, Lio D, Palmeri C, Maresi E, Balistreri CR. Can the TLR-4-mediated signaling pathway be “a key inflammatory promoter for sporadic TAA”? Mediators Inflamm 2014: 349476, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saha S, Chakraborty PK, Xiong X, Dwivedi SK, Mustafi SB, Leigh NR, Ramchandran R, Mukherjee P, Bhattacharya R. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. FASEB J 30: 441–456, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherer EB, Loureiro SO, Vuaden FC, da Cunha AA, Schmitz F, Kolling J, Savio LE, Bogo MR, Bonan CD, Netto CA, Wyse AT. Mild hyperhomocysteinemia increases brain acetylcholinesterase and proinflammatory cytokine levels in different tissues. Mol Neurobiol 50: 589–596, 2014. [DOI] [PubMed] [Google Scholar]
- 52.Shirai T, Hilhorst M, Harrison DG, Goronzy JJ, Weyand CM. Macrophages in vascular inflammation—from atherosclerosis to vasculitis. Autoimmunity 48: 139–151, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sipkens JA, Hahn N, van den Brand CS, Meischl C, Cillessen SA, Smith DE, Juffermans LJ, Musters RJ, Roos D, Jakobs C, Blom HJ, Smulders YM, Krijnen PA, Stehouwer CD, Rauwerda JA, van Hinsbergh VW, Niessen HW. Homocysteine-induced apoptosis in endothelial cells coincides with nuclear NOX2 and peri-nuclear NOX4 activity. Cell Biochem Biophys 67: 341–352, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sowmya S, Swathi Y, Yeo AL, Shoon ML, Moore PK, Bhatia M. Hydrogen sulfide: regulatory role on blood pressure in hyperhomocysteinemia. Vasc Pharmacol 53: 138–143, 2010. [DOI] [PubMed] [Google Scholar]
- 55.Steed MM, Tyagi SC. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal 15: 1927–1943, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun XQ, Zhang R, Zhang HD, Yuan P, Wang XJ, Zhao QH, Wang L, Jiang R, Jan Bogaard H, Jing ZC. Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria-dependent apoptosis. Hypertens Res 39: 302–311, 2016. [DOI] [PubMed] [Google Scholar]
- 57.Tsai HY, Lin CP, Huang PH, Li SY, Chen JS, Lin FY, Chen JW, Lin SJ. Coenzyme Q10 attenuates high glucose-induced endothelial progenitor cell dysfunction through AMP-activated protein kinase pathways. J Diabetes Res 2016: 6384759, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang C, Wang Z, Zhang X, Zhang X, Dong L, Xing Y, Li Y, Liu Z, Chen L, Qiao H, Wang L, Zhu C. Protection by silibinin against experimental ischemic stroke: up-regulated pAkt, pmTOR, HIF-1α and Bcl-2, down-regulated Bax, NF-κB expression. Neurosci Lett 529: 45–50, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Weiss N, Keller C, Hoffmann U, Loscalzo J. Endothelial dysfunction and atherothrombosis in mild hyperhomocysteinemia. Vasc Med 7: 227–239, 2002. [DOI] [PubMed] [Google Scholar]
- 60.Xia M, Conley SM, Li G, Li PL, Boini KM. Inhibition of hyperhomocysteinemia-induced inflammasome activation and glomerular sclerosis by NLRP3 gene deletion. Cell Physiol Biochem 34: 829–841, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang F, Tan HM, Wang H. Hyperhomocysteinemia and atherosclerosis. Sheng Li Xue Bao 57: 103–114, 2005. [PubMed] [Google Scholar]
- 62.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59, 2008. [DOI] [PubMed] [Google Scholar]
- 63.Yu XH, Cui LB, Wu K, Zheng XL, Cayabyab FS, Chen ZW, Tang CK. Hydrogen sulfide as a potent cardiovascular protective agent. Clin Chim Acta 437: 78–87, 2014. [DOI] [PubMed] [Google Scholar]
- 64.Zanin RF, Bergamin LS, Morrone FB, Coutinho-Silva R, de Souza Wyse AT, Battastini AM. Pathological concentrations of homocysteine increase IL-1β production in macrophages in a P2X7, NF-κB, and ERK-dependent manner. Purinergic Signal 11: 463–470, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeng Y, Li M, Chen Y, Wang S. Homocysteine, endothelin-1 and nitric oxide in patients with hypertensive disorders complicating pregnancy. Int J Clin Exp Pathol 8: 15275–15279, 2015. [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang D, Fang P, Jiang X, Nelson J, Moore JK, Kruger WD, Berretta RM, Houser SR, Yang X, Wang H. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ Res 111: 37–49, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Zou C, Yang S, Fu J. p120 catenin attenuates the angiotensin II-induced apoptosis of human umbilical vein endothelial cells by suppressing the mitochondrial pathway. Int J Mol Med 37: 623–630, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]