

ABSTRACT
Midbrain dopaminergic (mDA) neurons are generated in the ventral midbrain floor plate depending on Sonic Hedgehog (SHH) signaling for induction. Primary cilia transduce canonical SHH signals. Loss of intraflagellar transport protein IFT88, essential for ciliary function, disrupts SHH signaling in the ventral midbrain and results in the reduction in mDA progenitors and neurons. We investigate whether conditional inactivation of the kinesin motor protein KIF3A recapitulates phenotypes observed in conditional Ift88 mutants. Conditional Kif3a inactivation reduced the mDA progenitor domain size, but did not result in mDA neuron reduction, most likely because of a delayed loss of cilia and delayed inactivation of SHH signaling. We thereby define a precise spatiotemporal window within which primary cilia-dependent SHH signaling determines mDA fate.
KEYWORDS: dopaminergic neurons, IFT88, intraflagellar transport, KIF3A, midbrain, primary cilia, Sonic hedgehog, WNT
Midbrain dopaminergic (mDA) neurons are organized into 3 main nuclei, the substantia nigra pars compacta (SNc, A9 group), the ventral tegmental area (VTA, A10 group) and the retrorubral field (RRF, A8 group).1 mDA neurons are critically involved in motor and reward-related behaviors, and in the regulation of arousal and cognitive processes. Thus, the dysfunction of mDA neurons contributes to a number of psychiatric conditions including depression, schizophrenia and addiction.2–4 In addition, motor symptoms of Parkinson's disease (PD) are caused by the degeneration of SNc mDA neurons.5 To study the pathomechanisms and improve treatment options for such disorders, previous studies have explored the possibility of generating mDA neurons from induced pluripotent stem cells.6 However, this quest requires a detailed understanding of the factors and processes driving mDA neuron development. Even though this has been the subject of extensive research during the last 2 decades, the exact mechanisms underlying mDA neuron development are still not fully understood.
Fully differentiated mDA neurons express the enzymes of the dopamine synthesis pathway TH (tyrosine hydroxylase) and AADC (aromatic L-amino acid decarboxylase), the transporter proteins DAT (dopamine transporter) and VMAT2 (vesicular monoamine transporter 2), and a set of transcription factors including PITX3 (paired-like homeodomain 3), NURR1 (nuclear receptor related 1 protein), and LMX1A/B (LIM homeobox transcription factor 1 α/β).7 During embryonic development, mDA progenitors in the ventral midline (floor plate) express the transcription factors FOXA1/2 (forkhead box 1/2) and LMX1A/B as well as the secreted morphogen SHH (Sonic Hedgehog). The induction of mDA progenitors depends on efficient signaling of SHH, WNTs (Wingless-related MMTV integration site) and FGFs (fibroblast growth factors).
mDA neurons are not generated in the absence of SHH signaling; in Shh null mutant embryos the neural tube is dorsalized and as a consequence the dopaminergic progenitor domain in the ventral midline fails to form.8,9 SHH signaling is transduced by the transmembrane receptors Smoothened (SMO) and Patched (PTCH), with PTCH suppressing SMO activity in the absence of SHH. Under this condition, GLI3, a zinc finger transcription factor of the glioma-associated oncogene family, is proteolytically cleaved to its repressor form (GLI3R), which inhibits the transcription of SHH target genes. When SHH is present, the inhibition of SMO by PTCH is lifted; as a result, the formation of GLI repressor proteins is attenuated and a GLI activator forms, such as GLI2 activator (GLI2A). GLIA in turn stimulates the transcription of SHH target genes, e.g. Gli1 (a readout and a strong amplifier of SHH signaling) and Ptch.10
Previous studies have posited a significant role of the primary cilium in SHH signal transduction.11,12 Primary cilia are non-motile membrane protrusions that are present as a single copy in most mammalian cells and serve as an antenna for extracellular signals. Because primary cilia lack de novo protein synthesis, the required proteins are transported from the cytosol to the cilium tip by the intraflagellar transport (IFT) system. IFT occurs along 9 microtubule pairs, which are anchored to the basal body and span the entire length of the primary cilium. This transport system utilizes the motor proteins Kinesin-II (anterograde transport, consists of the 2 subunits KIF3A and KIF3B) and Dynein-II (retrograde transport, composed of DYNC2H1 and DYNC2L1).13 The cargo interacts with the respective motor protein via specialized IFT particles, some of which are used for anterograde transport (IFT A complex) while others facilitate retrograde transport (IFT B complex).
Importantly, the key proteins in the SHH signal cascade, PTCH, SMO and GLI, are trafficked within the primary cilium.14–16 Thus, the loss of IFT particles and motor proteins results in aberrant or even the complete loss of ciliary functions, culminating in the cell's failure to respond to SHH signaling, since neither GLI2A nor GLI3R are formed in the absence of cilia function.12,15,17 Therefore, the loss of primary cilia in the developing CNS results in phenotypes reminiscent of SHH pathway mutants.
Similarly to loss of SHH signaling, the complete inactivation of WNT1 dramatically reduces mDA progenitors and precursors, leading to a severe depletion of mDA neurons at later stages while inactivation of WNT signaling in mDA progenitors or differentiating mDA interferes with their progression from progenitor state to differentiated mDA neurons.18–20
WNT signals are conveyed via a complex containing a frizzled receptor and co-receptors, which recruits the phosphoprotein Dishevelled (DSH) upon activation by WNT ligands. In the canonical pathway, the recruitment of DSH leads to the translocation of β-catenin to the nucleus, where it activates target genes. In the non-canonical (planar cell polarity (PCP) or calcium) pathway, WNT signaling activates a small Rho GTPase or a G-protein, which stimulates actin remodeling and regulates calcium release from the endoplasmatic reticulum, respectively, via distinct signaling cascades.21 Several components of the WNT signal cascade localize to primary cilia,22 and an inhibitory effect of primary cilia on the WNT pathway was previously suggested, since the absence of functional primary cilia caused a hyper-responsiveness to canonical WNT signaling.23,24 On the other hand, canonical WNT signaling is not altered in Kif3a, Ift88 and Ift172 mutant mice.25 Thus, the role of the primary cilium in WNT signaling remains controversial.
Given the importance of both SHH and WNT signaling in mDA neurogenesis and the potential role of primary cilia as mediators of both signaling pathways, we explored the functional relevance of primary cilia in mDA specification in 2 separate experimental paradigms. For the first investigation, we summarize recently-published results26 in the following sections. We employed mouse mutants carrying a hypomorphic allele of the Ift gene Ift88 (cobblestone mutant),26 which results in the 70-80 % reduction of Ift88 mRNA and IFT88 protein levels, but which still produces a wildtype protein sequence.24 Even though the structural integrity of primary cilia appeared preserved in the cobblestone (cbbs) mutant embryo, the mDA progenitor domain was highly disorganized at embryonic day (E) 11.5, and contained peculiar rosette-like structures.26 Interestingly, many of these rosette-like structures expressed Shh and were intermingled with large clusters of SHH-responding (Gli1-expressing) cells, suggesting that despite the severe reduction in IFT88 levels and the presumed loss of ciliary function the SHH pathway was still activated in some ventral midbrain cells. In addition, Gli3-positive clusters were interspersed within the ventral midline region. Wnt1 and Axin2 (a readout for canonical WNT signaling)26 were not expressed in the ventral midbrain of cbbs mutants, indicating a complete disruption of canonical WNT signaling. Interestingly, in a few cbbs mutants the mDA progenitor domain was still induced as indicated by the expression of the floor plate markers Corin and Arx,27 within a FOXA2 positive domain. However, the Lmx1a-expressing domain was severely reduced in the cbbs embryos and was located at a distance to the ventricle, consistent with the dependence of Lmx1a expression on both SHH and WNT signaling.20 Further, the clear anatomical distinction between the LMX1A/FOXA2-expressing mDA progenitors and NKX6-1/FOXA2-positive red nucleus progenitors that is seen in control embryos was not maintained in cbbs mutants (Fig. 1). Our subsequent investigations of differentiated mDA neurons at E12.5 showed a dramatic reduction of neurons expressing TH. These TH-positive cells co-expressed further mDA markers, such as NURR1, PITX3, LMX1A and FOXA2 suggesting a proper specification toward an mDA neuronal fate. Interestingly, these few mDA neurons were often found surrounding the rosette-like structures. However, due to the early lethality of cbbs mutants,24 a follow-up investigation on the survival of differentiated mDA neurons was not possible.
Figure 1.
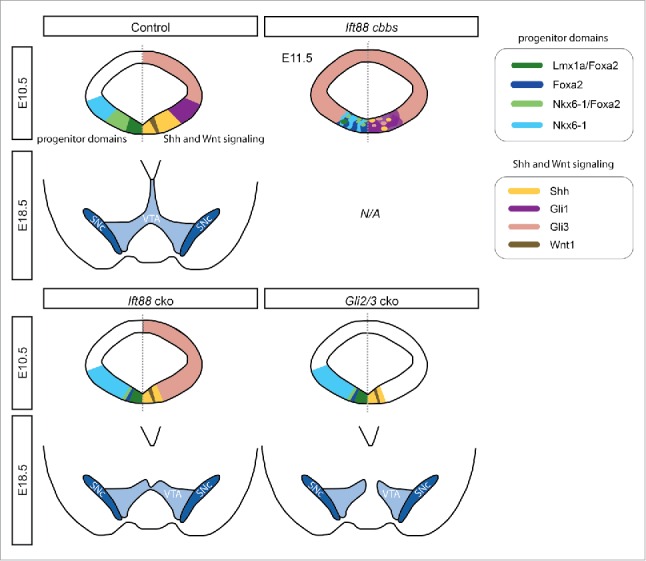
The reduced size of mDA progenitor domains results in decreased numbers of mDA neurons in cbbs, Ift88 and Gli2/3 cko mutant embryos. Schematic representation of mDA progenitor domains at E10.5 (upper panels) and the VTA and SNc at E18.5 (lower panels) of control, cbbs, Ift88 cko and Gli2/3 cko embryos as described previously.26 The E10.5 schematics show progenitor domains on the left and the expression of SHH and WNT signaling pathways components on the right. For the cbbs mutant, results are summarized for E11.5. E18.5 cbbs mutants were not analyzed, because the mutants are not viable beyond E14.5.
To overcome the residual expression of IFT88 in the cbbs mutant as well as the global defects in these mutants (e.g neural tube closure defects and early lethality), we conditionally inactivated Ift88 in the midbrain and anterior hindbrain after E8.5 in our previous study.26 Ift88 conditional knockout mice (Ift88 cko) were generated by crossing En1-Cre28 line to mice with floxed alleles of Ift88 (Ift88flox/flox).15 As the loss of primary cilia is thought to interrupt the formation of GLI2A and GLI3R and thus resulting in diminished or abolished SHH signaling, we compared the phenotype of Ift88 cko mice with mice in which the SHH pathway was switched-off by inactivating both Gli2 and Gli3 in a conditional manner. To this end, we used Gli2zfd/flox29,30 and Gli3xt/flox31,32 mice and crossed them with the En1-Cre line (referred to as Gli2/3 cko). mDA progenitors respond to SHH between E8.0 and E9.5, thus inactivation of SHH signaling at E8.5 results in the partial induction of the mDA progenitor domain.33,34
The conditional knockout of Ift88 resulted in the reduction of primary cilia at the ventricle of the ventral midbrain at E9.5, and their complete loss by E10.5. The loss of primary cilia starting from E9.5 was paralleled by an inactivation of SHH signaling, as shown by the absence of Gli1 expression in Ift88 cko embryos, and a ventral expansion of the Gli3 expression domain as compared to control embryos. Similarly, Gli1 expression was not induced in Gli2/3 cko embryos. In both Ift88 cko and Gli2/3 cko, the size of the expression domains of Shh, LMX1A, and FOXA2 was significantly reduced compared to control. Due to the reduction of the FOXA2 expression domain, the FOXA2/NKX6-1-positive progenitor domain was almost fully abolished in both Ift88 and Gli2/3 cko mutants, even though NKX6-1 expression was not obviously altered (Fig. 1). Further, mRNA levels of several Wnt ligands (Wnt1, Wnt5a, Wnt7a) were significantly reduced in microdissected E11.5 midbrains of Ift88 cko mutants as compared to control mice. In line with our finding of the reduced mDA progenitor domain size, we found a significantly lower number of TH-positive mDA neurons in Ift88 cko (at E13.5 and E18.5) and in Gli2/3 cko (at E12.5 and E18.5) as compared to control embryos. However, these TH-positive neurons had acquired the full mDA identity, as demonstrated by their co-expression of the mDA markers PITX3, NURR1, DAT, VMAT2 and AADC. Interestingly, the distribution of mDA neurons showed different patterns between control, Ift88 cko and Gli2/3 cko embryos at E18.5; the VTA and SN were overall less densely populated by TH-positive neurons and the lateral-medial expansion appeared reduced in Ift88 and Gli2/3 cko embryos compared to control. Moreover, medial parts of the VTA were greatly reduced in Ift88 cko mice, an effect that was even more prominent in Gli2/3 cko mice (Fig. 1).
PTCH-independent, constitutively active SHH signaling depends on primary cilia in adult neural stem cells.35 In our previous study,26 we examined whether this is also the case in mDA progenitors, and analyzed mice with the Smo-M2 allele. Upon Cre-mediated activation, a constitutively active version of SMO is expressed, which escapes the repression by PTCH, leading to the continuous localization of SMO at the membrane, irrespective of the presence or absence of SHH (En1Cre/+; R26SmoM/+, termed SmoM2 ca). Because of the constant activation of the SHH pathway and the excessive formation of GLI2A, SmoM2 ca embryos exhibited a ventralization of the midbrain. However, when the floxed allele of Ift88 was introduced into this mouse line (resulting in En1Cre/+; R26SmoM/+; Ift88flox/flox mice), the ventralization of the midbrain was reversed and resulted essentially in the same phenotype as in Ift88 cko embryos. Further, consistent with the trafficking of SMO or SMO-M2 within the primary cilium,16,36 our results emphasize that SMO activity depends on functional primary cilia and that the loss of primary cilia abolishes even constitutively-active SHH signaling. In summary, our previous work strongly suggests that the reduction in mDA progenitors and consequently in mDA neurons in Ift88 mutants that lack functional primary cilia, is largely due to the loss of SHH signaling (Fig. 1).26
In the second investigation, whose results we present here, our aim was to explore whether the observed phenotype in Ift88 cko mice is caused solely by the ciliary function of IFT88. To this end, we decided to inactivate Kif3a37 in the developing midbrain (En1Cre/+, Kif3aflox/flox, referred to as Kif3a cko). To investigate the loss of primary cilia upon loss of KIF3A expression in Kif3a cko brains, cilia were detected with immunofluorescence, using an anti-γ-tubulin antibody to recognize the basal body38 and an anti-ARL13B antibody to label the ciliary axoneme39 We observed a mild loss of primary cilia from the midbrain ventricular surface at E9.5 as evident by decreased immunoreactivity of ARL13B, while a complete loss ensued at E10.5. On the other hand, basal bodies were preserved after the loss of Kif3a, as indicated by the anti-γ-tubulin staining (Fig. 2A-D). SHH signaling was not completely inactivated at E9.5, since Gli1 expression could still be detected in the Kif3a cko midbrain; only by E10.5 Gli1 was no longer expressed (Fig. 2E, F, I, J). This is in contrast to Ift88 or Gli2/Gli3 cko mutant embryos, in which Gli1 was already absent in the E9.5 ventral midbrain26. Similarly, the ventral expansion of Gli3 expression observed in the Ift88 cko mutants was not apparent in the Kif3a cko midbrain, further indicating that the full inactivation of SHH signaling occurred later in the Kif3a cko than in the Ift88 cko embryos (Fig. 2G, H, K, L). However, similar to Ift88 and Gli2/3 cko mutants, the Shh/Foxa2 double-positive area (Fig. 2M-P, W), and the Lmx1a-positive domain (Fig. 2Q, R, X) were reduced in E10.5 Kif3a cko mutant midbrain in comparison to controls. On the other hand, the size of the Wnt1 expression domain (Fig. 2U, V) and the Corin-positive domain (Fig. 2S, T) did not appear altered between Kif3a cko and control embryos.
Figure 2.

In Kif3a cko embryos primary cilia and SHH signaling are lost at E10.5 resulting in a reduced size of the mDA progenitor domain. Antibody staining for the ciliary marker ARL13B (red) and γ-tubulin (green) (A-D) at the ventricular surface of the ventral midbrain shows that primary cilia are present at E9.5 in Kif3a cko mutants, but are completely lost at E10.5. Blue, Hoechst-labeled nuclei. (E-L) RNA in situ hybridization for Gli1 (E, F, I, J) and Gli3 (G, H, K, L) in E9.5 and E10.5 midbrain sections. While Gli1 was still expressed at E9.5, its expression disappeared by E10.5 in Kif3a cko mutants. Black arrowheads indicate the ventral borders of the expression domains. (M-V) RNA in situ hybridization for Shh (M,N), Foxa2 (O,P), Lmx1a (Q,R), Corin (S,T) and Wnt1 (U,V) in E10.5 ventral midbrain. Arrows: Patchy expression of Foxa2. (W,X) Size of the Foxa2-positive (W) and Lmx1a-positive (X) expression domain (normalized to the inner perimeter of the ventricle) at E10.5. The size of both the Foxa2 (n ≥ 3, t(10) = 2.92) and the Lmx1a (n ≥ 3, t(6) = 3.55) domain was significantly reduced in Kif3a cko mutants as compared to controls. *P < 0.05, **P < 0.01. Scale bars: A-D, 10 μm; E-L, 200 μm, M-V, 100 μm.
To investigate how the loss of SHH signaling after E9.5 affects the generation of mDA neurons in Kif3a cko mice, we analyzed mDA neurons in E13.5 and E18.5 Kif3a cko brains. In E13.5 Kif3a cko embryos, the TH-positive cell clusters were distributed differently than in controls, but the TH-positive cells co-expressed the mDA markers LMX1A and FOXA2. NKX6-1 was expressed in an adjacent domain (Fig. 3A-F). At E18.5, mDA neurons appeared to be correctly specified since they were positive for TH, PITX3, NURR1 and DAT (Fig. 3G-L). Together, these results suggest that mDA neurons undergo normal differentiation in Kif3a cko mice. We found no obvious reduction in the number of TH-positive cells in Kif3a cko mice as compared to control, neither at E13.5 (Fig. 3A-F, control: 681.5 ± 102.5 cells, Kif3a cko: 659.5 ± 23.5 cells, n = 2) nor at E18.5 (Fig. 3G-H', M).
Figure 3.
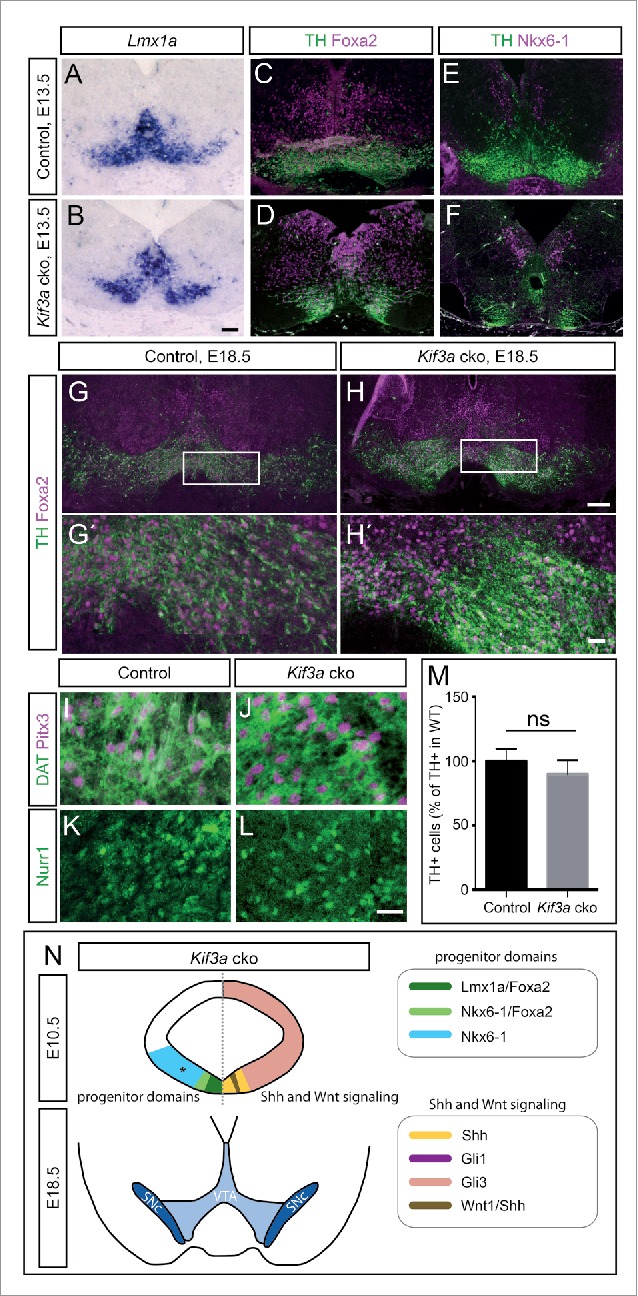
The number of TH-positive mDA neurons is not altered in Kif3a cko mutants. (A-D) mDA neurons in E13.5 Kif3a cko and control embryos as shown by (A,B) RNA in situ hybridization for Lmx1a and (C,D) immunofluorescent staining of TH and FOXA2. (E,F) TH and NKX6-1-expressing neurons in E13.5 Kif3a cko and control embryos. (G-L) mDA neurons in E18.5 midbrains of Kif3a cko and control embryos. Antibody staining for (G-H´) TH and FOXA2, (I,J) DAT and PITX3, and (K,L) NURR1 in ventral midbrains of Kif3a cko and control embryos. (G´,H´) Higher magnification of boxed areas in (G,H). (M) Quantification of TH-positive neurons in control versus Kif3a cko brains at E18.5. The number of TH-expressing cells in the ventral midbrain was not significantly different between Kif3a cko and control mice (control: n = 6, Kif3a cko: n = 4, t(8) = 1.54, p = 0.162). (N) Schematic representation of mDA progenitor domains at E10.5 (upper panels) and the VTA and SNc at E18.5 (lower panels) of Kif3a cko embryos. The E10.5 schematics show progenitor domains on the left and the expression of SHH and WNT signaling pathways components on the right. Asterisk indicates that NKX6-1 expression was only analyzed at E13.5. Scale bars: A-F, 100 μm, G,H, 200 μm, G´,H´, 40 μm, I-L, 20 μm.
In summary, we demonstrate here that the conditional inactivation of Kif3a in the midbrain results eventually in the loss of primary cilia and SHH signaling in the ventral midbrain, as seen in the Ift88 cko mutant embryos,26 but not in the reduction of mDA neurons (Fig. 3N). The dependence of SHH signaling on KIF3A function has previously been demonstrated, for example in cerebellum40 and hippocampus.35 Interestingly, in our study, the disappearance of functional primary cilia from the ventricular zone of the ventral midbrain was delayed in Kif3a cko embryos by about one day, as compared to Ift88 cko embryos, resulting in the shutdown of SHH signal transduction only after E9.5. This delayed disappearance of primary cilia may reflect the persistence of functional KIF3A protein, which may be caused by the following: relative protein and/or mRNA stabilities of KIF3A and IFT88, the relative importance of the KIF3A and IFT88 proteins for primary ciliary maintenance, and potentially even differences in the timing of the Cre-mediated genetic recombination event. In our previous study,26 we verified the loss of primary cilia in the ventral embryonic midbrain with scanning electron microscopy and with immunofluorescent stainings for ciliary axoneme. Although we restrict our analysis in this study to immunofluorescence detection of the primary cilium, we think that a parsimonious explanation of our observations would be that a loss of ARL13B is indeed reflective of a loss of primary cilia, as has been seen in other studies (e.g., developing cerebellum40,41) comparing loss of Ift88 to loss Kif3a gene expression. A primary cilia-dependent loss-of-function phenotype in both the Ift88 and the Kif3a cko mutants is also reflected in the similar biological readouts of the 2 phenotypes. For example, the delayed inactivation of SHH signaling in the Kif3a cko embryos still resulted in a reduced size of the Lmx1a-positive mDA progenitor domain and a smaller Foxa2 domain at E10.5 and overall changes in the anatomy of TH-positive neuronal clusters at E13.5 (Fig. 2O-R, Fig. 3A, B). However, the number of differentiated mDA neurons in Kif3a cko mice was not decreased compared to control (Fig. 3M). While this may seem to be a surprising result at first blush, the lack of effect on mDA neuron number is consistent with previous data. Genetic fate mapping of SHH-responding cells showed that mDA progenitors cease to respond to SHH signaling after E9.5.33,34 Moreover, conditional inactivation of SHH signaling after E9.0 in the Shh-positive progenitor domain results in only transient phenotypes and eventually normal numbers of TH-positive neurons.34,42 Thus, the induction and specification of mDA neurons may have been completed by the time primary cilia and SHH signaling are lost in Kif3a cko mice. Alternatively or in addition, mild defects in the number of mDA progenitors might be compensated for by adjustments in mDA progenitor proliferation and differentiation. These results, combined with our analysis of the Ift88 cko mutants,26 have demonstrated an exquisite spatiotemporal specificity in the coincident timing of the presence of functional primary cilia in mDA progenitors, and their consequent processing of developmentally-normal levels of SHH signaling.
In conclusion, these data and our previous study26 show that the induction of mDA progenitors, and consequently the generation of a normal number of mDA neurons during embryonic development, is critically dependent on primary cilia function and SHH signaling, but only before E9.5. These data also demonstrate that when studying the function of proteins involved in primary cilia function, the interpretation of conditional knock-out phenotypes should take into careful consideration the precise timing of the inactivation of a given "ciliary gene." This can have enormous consequences for the resulting loss of primary cilia function and thus the primary cilia-dependent signaling pathway(s), as evidenced by the specific mutant phenotype.
Materials and methods
Mouse lines and genotyping
The generation of the mouse lines Kif3aflox37 and En1Cre28 were described previously. Genotype analysis was performed by PCR on genomic DNA from embryonic tissue (tail or yolk sack samples) as described26,43 using the following additional primers for Kif3a: K1: 5’-TCT GTG AGT TTG TGA CCA GCC-3’ (flox), K2: 5’- AGG GCA GAC GGA AGG GTG G-3’ (common). 12 noon of the day of the vaginal plug was designated the date embryonic day 0.5 (E0.5) and embryonic stages before E12.5 were identified by somite analysis.26,44 All experiments were conducted according to the guidelines of the states of Baden-Württemberg and North Rhine-Westphalia, Germany.
Immunohistochemical analysis and in situ hybridization
Tissue collection and processing, immunohistochemistry and RNA in situ hybridization were performed as described.26 In brief, embryos or embryonic brains were dissected and collected in cold 0.1M PBS and fixed for periods adapted to the embryonic stage (30 min to overnight at 4°C) in 4% paraformaldehyde (PFA, in 0.1M PBS). After rinsing in 0.1M PBS, the embryos were treated either in an ascending sucrose series (10, 20, and 30% in 0.1M PBS) and mounted in Jung tissue freezing medium (Leica Biosystems, Wetzlar, Germany), or dehydrated and processed for paraffin embedding.
Immunohistochemical stainings of 10-14 μm cryosections or 7 μm paraffin sections were performed following standard procedures as described previsouly.26,33 Primary antibodies: mouse anti-γ-tubulin (clone GTU-88, Sigma-Aldrich, St. Louis, USA) 1:1000; rabbit anti-ARL13B (kind gift of Tamara Caspary, Emory University, Atlanta, USA) 1:1500; rabbit or mouse anti-TH (AB152, MAB318, Millipore, Billerica, Massachusetts, USA) 1:500; goat anti-FOXA2 (clone M-20; Santa Cruz Biotechnology, Santa Cruz, USA) 1:1000; rat anti-DAT (AB369, EMD Millipore) 1:1000; rabbit anti-NURR1 (sc-990, Santa Cruz) 1:250; mouse anti-NKX6-1 (clone F55A10 developed by OD Madsen; Developmental Studies Hybridoma Bank) 1:50; rabbit anti-PITX3 (38-2850, Thermo Fischer Scientific) 1:250. Secondary antibodies: Cy3-conjugated donkey anti-rabbit, anti-mouse or anti-goat (Cat.No. 11-165-152, 715-165-150 or 05-165-147, Jackson ImmunoResearch Laboratories, West Grove, USA) 1:200, Alexa 488 conjugated donkey anti-rabbit or anti-mouse IgG (Cat.No. AB150149, A21206 or A21202, Life Technologies, Carlsbad, USA) 1:500, Goat Alexa 546 and Alexa 488 anti-rabbit IgG, Alexa 488 anti-mouse IgG1, or Alexa 488 anti-mouse IgG (Cat.No. A11029, BD Biocsciences, San Jose, USA) 1:1000. For immunofluorescent detection of transcription factors (FOXA2, LMX1A, NKX6-1, NURR1, PITX3), sections were pre-treated with 1 mM EDTA (65°C, 10 min) and incubated with biotinylated anti-mouse or anti-goat secondary antibodies (1:200, Cat.No. 715-065-150 or 711-065-152, Jackson Immuno Research) followed by Cy3-conjugated Streptavidin (Cat.No. 016-160-084, Jackson ImmunoResearch Laboratories, West Grove, USA) at 1:1000.
For non-radioactive RNA in situ hybridization, paraffin sections were deparaffinized using Xylene, rehydrated in a descending ethanol series, treated with Proteinase K (Roche, Penzberg, Germany), and then acetylated. Cryosections were fixed in 4% PFA and washed in PBS. Subsequently, sections were washed in H2O, dehydrated in an ascending ethanol series, and incubated in chloroform. Following the overnight hybridization with the cRNA probes and immunodetections of digoxigenin with alkaline phosphatase conjugated antibody (Roche, Penzberg, Germany), the reaction product was visualized using BM purple (Roche, Penzberg, Germany).
Image acquisition and signal quantification
Confocal (Nikon A1R microscope, Nikon Imaging Center, University of Heidelberg) and widefield microscopy (Leica DM1000 microscope, and for fluorescent samples: Axio Observer equipped with ApoTome, Zeiss, Oberkochen, Germany) as well as the quantification of Lmx1a-, Foxa2-positive domains and the number of TH/Fox2-positive neurons of Kif3a cko and control animals was performed as described previously.26 Briefly, the perimeter of Lmx1a- and Foxa2-positive domains was measured and normalized to the inner perimeter of the ventricle in each embryo (n ≥ 3) using the ImageJ software package 1.48v (http://rsb.info.nih.gov/ij/<http://rsb.info.nih.gov/ij/>). Counting of TH/Foxa2-positive cells was performed using the cell counter plugin in ImageJ. For E18.5, cell numbers were normalized to controls (set at 100 %).
Statistical analysis
Statistical analysis of histological data was performed using an unpaired 2-sided Student‘s t-test (Prism 6, Graphpad). Statistical significance levels were set at p < 0.05. The values are shown as mean ± SD (standard deviation).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Joachim Kirsch and Oliver Brüstle for generous scientific support and Tamara Caspary for the anti-Arl13b antibodies.
Funding
The authors were supported by the German Research Society (KLT: DFG SFB 488, Teilprojekt B9, SB: Heisenberg-Fellowship (BL 767/2-1) and Research Grant (BL 767/3-1)); the University of New England (to KLT); the North-Rhine-Westphalia Repatriation Program of the Ministry for Innovation, Science and Research of North Rhine Westphalia (to SB) and the Maria von Linden-Program of the University of Bonn (to SB).
References
- [1]. Björklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci 2007; 30:194–202; PMID:17408759; http://dx.doi.org/10.1016/j.tins.2007.03.006 [DOI] [PubMed] [Google Scholar]
- [2]. Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, Kim SY, Adhikari A, Thompson KR, Andalman AS, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 2013; 493:537–41; PMID:23235822; http://dx.doi.org/10.1038/nature11740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. Winterer G, Weinberger DR. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci 2004; 27:683–90; PMID:15474169; http://dx.doi.org/10.1016/j.tins.2004.08.002 [DOI] [PubMed] [Google Scholar]
- [4]. Luo SX, Huang EJ. Dopaminergic Neurons and Brain Reward Pathways: From Neurogenesis to Circuit Assembly. Am J Pathol 2016; 186:478–88; PMID:26724386; http://dx.doi.org/10.1016/j.ajpath.2015.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 1989; 12:366–75; PMID:2479133; http://dx.doi.org/10.1016/0166-2236(89)90074-X [DOI] [PubMed] [Google Scholar]
- [6]. Studer L. Derivation of dopaminergic neurons from pluripotent stem cells. Prog Brain Res 2012; 200:243–63; PMID:23195422; http://dx.doi.org/10.1016/B978-0-444-59575-1.00011-9 [DOI] [PubMed] [Google Scholar]
- [7]. Blaess S, Ang SL. Genetic control of midbrain dopaminergic neuron development. Wiley Interdiscip Rev Dev Biol 2015; 4:113–34; PMID:25565353; http://dx.doi.org/10.1002/wdev.169 [DOI] [PubMed] [Google Scholar]
- [8]. Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996; 383:407–13; PMID:8837770; http://dx.doi.org/10.1038/383407a0 [DOI] [PubMed] [Google Scholar]
- [9]. Blaess S, Corrales JD, Joyner AL. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development 2006; 133:1799–809; PMID:16571630; http://dx.doi.org/10.1242/dev.02339 [DOI] [PubMed] [Google Scholar]
- [10]. Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 2013; 14:416–29; PMID:23719536; http://dx.doi.org/10.1038/nrm3598 [DOI] [PubMed] [Google Scholar]
- [11]. Sasai N, Briscoe J. Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip Rev Dev Biol 2012; 1:753–72; PMID:23799571; http://dx.doi.org/10.1002/wdev.43 [DOI] [PubMed] [Google Scholar]
- [12]. Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson K V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003; 426:83–7; PMID:14603322; http://dx.doi.org/10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- [13]. Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol 2003; 19:423–43; PMID:14570576; http://dx.doi.org/10.1146/annurev.cellbio.19.111401.091318 [DOI] [PubMed] [Google Scholar]
- [14]. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature 2005; 437:1018–21; PMID:16136078; http://dx.doi.org/10.1038/nature04117 [DOI] [PubMed] [Google Scholar]
- [15]. Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 2005; 1:e53; PMID:16254602; http://dx.doi.org/10.1371/journal.pgen.0010053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007; 317:372–6; PMID:17641202; http://dx.doi.org/10.1126/science.1139740 [DOI] [PubMed] [Google Scholar]
- [17]. Besse L, Neti M, Anselme I, Gerhardt C, Ruther U, Laclef C, Schneider-Maunoury S. Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 2011; 138:2079–88; PMID:21490064; http://dx.doi.org/10.1242/dev.059808 [DOI] [PubMed] [Google Scholar]
- [18]. Prakash N, Brodski C, Naserke T, Puelles E, Gogoi R, Hall A, Panhuysen M, Echevarria D, Sussel L, Weisenhorn DMV, et al. A Wnt1-regulated genetic network controls the identity and fate of midbrain-dopaminergic progenitors in vivo. Development 2006; 133:89–98; PMID:16339193; http://dx.doi.org/10.1242/dev.02181 [DOI] [PubMed] [Google Scholar]
- [19]. Tang M, Miyamoto Y, Huang EJ. Multiple roles of β-catenin in controlling the neurogenic niche for midbrain dopamine neurons. Development 2009; 136:2027–38; PMID:19439492; http://dx.doi.org/10.1242/dev.034330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Yang J, Brown A, Ellisor D, Paul E, Hagan N, Zervas M. Dynamic temporal requirement of Wnt1 in midbrain dopamine neuron development. Development 2013; 140:1342–52; PMID:23444360; http://dx.doi.org/10.1242/dev.080630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Oh EC, Katsanis N. Context-dependent regulation of Wnt signaling through the primary cilium. J Am Soc Nephrol 2013; 24:10–8; PMID:23123400; http://dx.doi.org/10.1681/ASN.2012050526 [DOI] [PubMed] [Google Scholar]
- [22]. Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol 2009; 111:p39-53; PMID:19276629; http://dx.doi.org/10.1159/000208212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF. Kif3a constrains β-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 2008; 10:70–6; PMID:18084282; http://dx.doi.org/10.1038/ncb1670 [DOI] [PubMed] [Google Scholar]
- [24]. Willaredt MA, Hasenpusch-Theil K, Gardner HAR, Kitanovic I, Hirschfeld-Warneken VC, Gojak CP, Gorgas K, Bradford CL, Spatz J, Wolfl S, et al. A crucial role for primary cilia in cortical morphogenesis. J Neurosci 2008; 28:12887–900; PMID:19036983; http://dx.doi.org/10.1523/JNEUROSCI.2084-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Ocbina PJR, Tuson M, Anderson K V. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS One 2009; 4:e6839; PMID:19718259; http://dx.doi.org/10.1371/journal.pone.0006839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Gazea M, Tasouri E, Tolve M, Bosch V, Kabanova A, Gojak C, Kurtulmus B, Novikov O, Spatz J, Pereira G, et al. Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev Biol 2016; 409:55–71; PMID:26542012; http://dx.doi.org/10.1016/j.ydbio.2015.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Bodea GO, Blaess S. Establishing diversity in the dopaminergic system. FEBS Lett 2015; 589:3773–85; PMID:26431946; http://dx.doi.org/10.1016/j.febslet.2015.09.016 [DOI] [PubMed] [Google Scholar]
- [28]. Kimmel RA, Turnbull DH, Blanquet V, Wurst W, Loomis CA, Joyner AL. Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev 2000; 14:1377–89; PMID:10837030 [PMC free article] [PubMed] [Google Scholar]
- [29]. Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 1998; 125:2759–70; PMID:9655799 [DOI] [PubMed] [Google Scholar]
- [30]. Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development 2006; 133:1811–21; PMID:16571625; http://dx.doi.org/10.1242/dev.02351 [DOI] [PubMed] [Google Scholar]
- [31]. Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet 1993; 3:241–6; PMID:8387379; http://dx.doi.org/10.1038/ng0393-241 [DOI] [PubMed] [Google Scholar]
- [32]. Blaess S, Stephen D, Joyner AL. Gli3 coordinates three-dimensional patterning and growth of the tectum and cerebellum by integrating Shh and Fgf8 signaling. Development 2008; 135:2093–103; PMID:18480159; http://dx.doi.org/10.1242/dev.015990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Blaess S, Bodea GO, Kabanova A, Chanet S, Mugniery E, Derouiche A, Stephen D, Joyner AL. Temporal-spatial changes in Sonic Hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Dev 2011; 6:29; PMID:21689430; http://dx.doi.org/10.1186/1749-8104-6-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Hayes L, Zhang Z, Albert P, Zervas M, Ahn S. Timing of Sonic hedgehog and Gli1 expression segregates midbrain dopamine neurons. J Comp Neurol 2011; 519:3001–18; PMID:21713771; http://dx.doi.org/10.1002/cne.22711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 2008; 11:277–84; PMID:18297065; http://dx.doi.org/10.1038/nn2059 [DOI] [PubMed] [Google Scholar]
- [36]. Jung B, Messias AC, Schorpp K, Geerlof A, Schneider G, Saur D, Hadian K, Sattler M, Wanker EE, Hasenoder S, et al. Novel small molecules targeting ciliary transport of Smoothened and oncogenic Hedgehog pathway activation. Sci Rep 2016; 6:22540; PMID:26931153; http://dx.doi.org/10.1038/srep22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell 2000; 102:175–87; PMID:10943838; http://dx.doi.org/10.1016/S0092-8674(00)00023-4 [DOI] [PubMed] [Google Scholar]
- [38]. Schiebel E. gamma-tubulin complexes: binding to the centrosome, regulation and microtubule nucleation. Curr Opin Cell Biol 2000; 12:113–8; PMID:10679351; http://dx.doi.org/10.1016/S0955-0674(99)00064-2 [DOI] [PubMed] [Google Scholar]
- [39]. Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell 2007; 12:767–78; PMID:17488627; http://dx.doi.org/10.1016/j.devcel.2007.03.004 [DOI] [PubMed] [Google Scholar]
- [40]. Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, Ros MR, Garcia-Verdugo JM, Alvarez-Buylla A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol 2008; 317:246–59; PMID:18353302; http://dx.doi.org/10.1016/j.ydbio.2008.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci 2007; 27:9780–9; PMID:17804638; http://dx.doi.org/10.1523/JNEUROSCI.5586-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]. Tang M, Luo SX, Tang V, Huang EJ. Temporal and spatial requirements of Smoothened in ventral midbrain neuronal development. Neural Dev 2013; 8:8; PMID:23618354; http://dx.doi.org/10.1186/1749-8104-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]. Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res 1991; 19:4293; PMID:1870982; http://dx.doi.org/10.1093/nar/19.15.4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Theiler K. The House Mouse. Development and normal stages from fertilization to 4 weeks of age. 2nd ed. Springer Verlag; 1989. [Google Scholar]