

Hereditary hemochromatosis is a rare iron overload disease caused by mutations in the genes involved in iron homeostasis, the most frequent being HFE-hemochromatosis. The classic phenotype of hemochromatosis is elevated transferrin saturation and serum ferritin, and progressive iron deposits mainly in parenchymal cells of the liver. The non-HFE hemochromatosis is caused by mutations in other genes: transferrin receptor 2 gene (TFR2), hemojuvelin and hepcidin genes (HJV and HAMP), and ferroportin gene (SLC40A1) causing ferroportin disease (FD), the most common form of non-HFE hemochromatosis (1,2) (Table 1).
TABLE 1.
Characteristics of hemochromatosis
| Subtypes of hemochromatosis | Gen (inheritance) | Age of onset | Biochemical features | Clinical expression | Iron overload/iron deposition | Pathogenesis |
| HFE hemochromatosis (type 1) | HFE (AR) | Adulthood | Increased plasma iron and ferritin concentrations, increased transferrin saturation | Fatigue, skin pigmentation, arthropathy, liver damage/cirrhosis, endocrine dysfunctions, cardiomyopathy | Variable/parenchymal | Decreased hepcidin expression |
| Juvenile hemochromatosis (type 2A) | HJV (AR) | Childhood to youth | The same as HFE hemochromatosis | Cardiomyopathy and hypogonadism are more prevalent, liver damage | Severe/parenchymal | Inhibition of hepcidin expression |
| Juvenile hemochromatosis (type 2B) | HAMP (AR) | Decreased hepcidin expression | ||||
| TfR2 hemochromatosis (type 3) | TFR2 (AR) | Youth adulthood | The same as HFE hemochromatosis | The same as HFE hemochromatosis | Variable-severe/parenchymal | Decreased hepcidin expression |
| Ferroportin disease (classical FD or type 4A) | SLC40A1 (AD) | Any age | Increased plasma iron and ferritin concentrations, normal to low transferrin saturation | Fatigue, arthropathy, endocrine dysfunctions | Mild/macrophagic | Decreased iron export from macrophages |
| Ferroportin disease (nonclassical FD or type 4B) | The same as HFE hemochromatosis | Fatigue, arthropathy, endocrine dysfunctions, liver damage | Mild/parenchymal | Resistance of ferroportin to hepcidin |
Patients in this study have the classical form of FD. AD = autosomal dominant; AR = autosomal recessive; FD = ferroportin disease.
A 9-year-old girl was referred by the pediatrician because of elevated ferritin and transaminases. She was clinically asymptomatic and the first evaluation revealed the following: alanine transaminase 72 U/L (0–53), aspartate aminotransferase 44 U/L (10–37), serum iron 78 μg/dL (25–120), serum transferrin 263 mg/dL (176–386); transferrin saturation 21.2% (7.5–45), serum ferritin 815 ng/dL (7–140), total IgE 616 IU/mL (0–87), CPK 95 U/L (21–215); normal values are given in parenthesis. Other parameters such as complete blood cell count, liver function, renal biochemistry, and immunoglobulins were normal. Screening laboratory tests to viral hepatitis, autoimmune hepatitis, Wilson disease, and α-1 antitrypsin deficiency were within the normal values.
The patient's 38-year-old father has been diagnosed as having hemochromatosis but without the mutations H63D, S65C, or C282Y in the HFE. He had hyperferritinemia, 4000 ng/dL (15–150) and iron overload in liver, spleen, and bone marrow but not in pancreas (Fig. 1A and B). He has been receiving treatment by apheresis for 8 years. The patient's mother, 39 years old, and 2 siblings, 5 and 6 years old, have no relevant clinical data.
FIGURE 1.
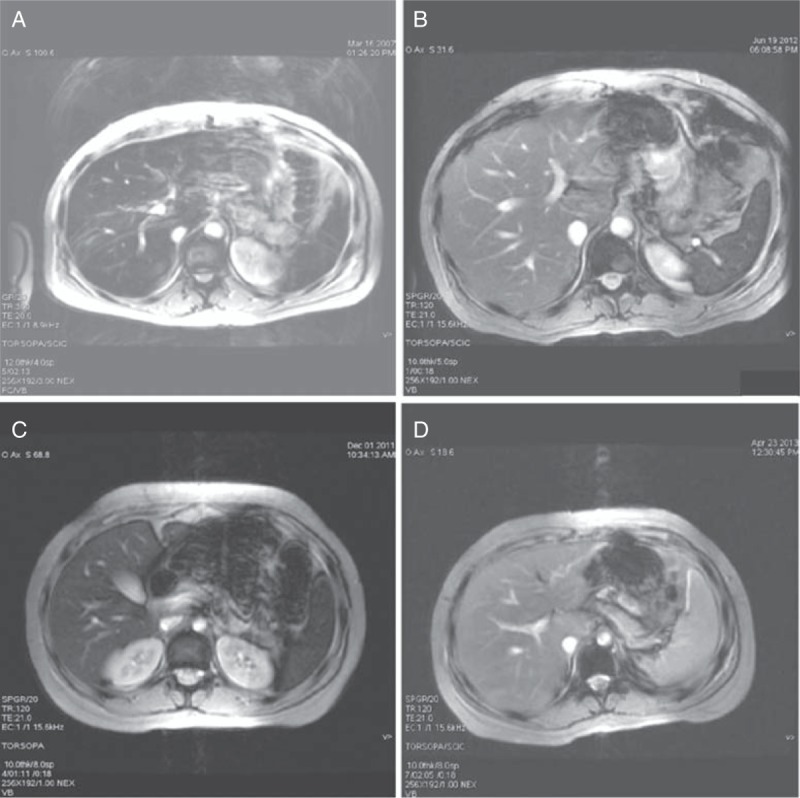
MRI T2∗∗ (GRE TR120, TE 21, flip angle 20°) of the upper abdomen of the patient's father (A and B) and patient (C and D). (A) MRI in March 2007, before apheresis treatment, revealed iron overload (not standardized criteria for iron quantifying at that moment); (B) MRI in June 2012, after apheresis treatment, liver iron content 60 μmol/g, calculated according to the protocol from the University of Rennes (normal value <36); (C) MRI before apheresis treatment, liver iron content 250 μmol/g and spleen and bone marrow signals are low because of iron deposition; (D) MRI after 12 sessions of apheresis treatment, liver and spleen signals are normal, liver iron content 60 μmol/g, and bone marrow hypointense signal persists. MRI = magnetic resonance imaging.
After the initial assessment of the 9-year-old girl, the expected diagnosis was FD given the exclusive elevation of ferritin, but not affecting transferrin saturation. Genetic analysis was performed in the HJV, HAMP, HFE, TFR2, and SLC40A1 genes. Written informed consent was obtained according to the guidelines by Instituto de Investigación Hospital 12 de Octubre, and the genetic study was in accordance with the ethical guidelines of the Declaration of Helsinki for Human Research.
The magnetic resonance imaging (MRI) study carried out on the patient in December 2011 revealed iron overloads in liver, spleen, and bone marrow, the liver iron concentration being 250 ± 50 μmol/g and iron accumulation mainly in the Kupffer cells (Fig. 1C).
The therapeutic protocol was venesections of 5 to 8 mL/kg and replacement with a saline solution with a 3-step key at intervals of 3 weeks, besides restricting the diet with iron-rich foods and limitation of citrus. The serum ferritin concentration decreased progressively, allowing the performance of venesections to be more spaced, and the serum transaminases concentrations were also normalized (Fig. 2A). MRI performed in April 2013 to monitor the iron burden showed a reduction in the liver iron content value (60 ± 30 μmol/g) (Fig. 1D) and slight hepatic fatty infiltration, but a low signal intensity persisted in the bone marrow.
FIGURE 2.
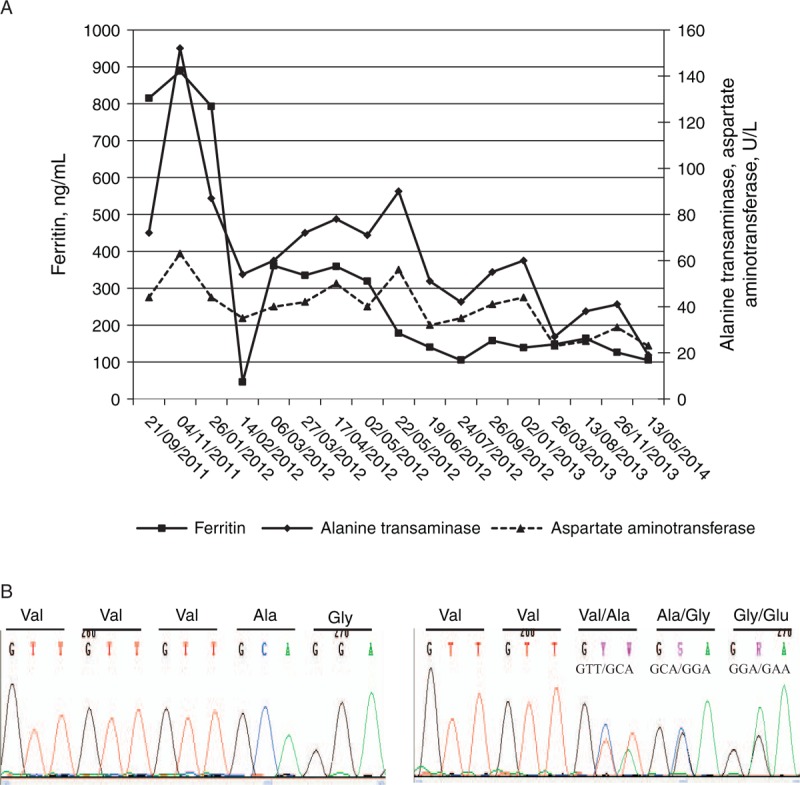
A venesection treatment was performed on the patient on all the dates indicated in the chart except for the first 2. The follow-ups of serum alanine transaminase, aspartate aminotransferase, and ferritin are shown. (B) Electropherogram showing control sequence of exon 5 of the SLC40A1 gene (left panel) and the mutation c.484_486delGTT in heterozygous state (right panel).
The patient had the mutation c.484_486delGTT (p.Val162del) in the exon 5 of SLC40A1 gene in heterozygosis, confirming the diagnosis of FD (Fig. 2B). This mutation was also found in heterozygosis in her father and her 6-year-old brother. No other pathogenic mutations were found in the genes studied.
The hepatic and iron parameters of the patient's brother were studied in December 2011 and no alterations were found except for elevated serum ferritin, 202 ng/mL (7–140), which increased to 293 ng/mL in August 2013. An MRI in January 2014 revealed a mild iron overload in the liver (55 ± 30 μmol/g). Serum ferritin was 359 ng/mL in May 2014 and therapy of venesections was begun (5 mL/kg body weight).
DISCUSSION
FD is an autosomal dominant disorder caused by mutations in the gene coding for ferroportin, the only known exporter of iron. Mutations in the SLC40A1 gene are heterogeneous and cause loss of function or classical FD (type 4A) and gain of function or nonclassical FD (subtype 4B). The loss of function is a result of impaired iron trafficking, caused by altered cell-surface expression of ferroportin or altered iron egress. The gain of function is a result of resistance of ferroportin to hepcidin, caused by alteration of the hepcidin-binding site. Hepcidin is a hormone regulator of iron metabolism, which modulates iron export by binding to ferroportin, inducing its internalization and subsequent degradation (3).
The 2 subtypes of FD are characterized by hyperferritinemia and iron overload; the most prevalent, the classical form with normal or low plasma iron and transferrin saturation, and accumulation of iron mainly in macrophages (type 4A), and the nonclassical form with classic HFE-hemochromatosis phenotype, high transferrin saturation, and parenchymal cell iron accumulation (type 4B) (Table 1) (1,2). MRI is a noninvasive tool to diagnose, classify, and follow up these 2 forms of FD without performing liver biopsy (4).
The phenotypic expression of FD is variable, even in patients harboring the same mutation. It is suggested that pathological, acquired, or genetic factors (such as metabolic syndrome and excessive alcohol consumption that probably decrease hepcidin expression) may affect clinical expression. The clinical expression of FD is less severe than other types of hemochromatosis, concerning iron overload and clinical features (Table 1). A review of studies on large cohorts of patients and families with FD reports a high penetrance of FD, rarely associated with fibrosis, except for nonclassical form with a higher risk of fibrosis and more severe hepatic iron overload (5).
FD has been reported worldwide in individual cases and in families, normally in middle-aged patients and also in children between 2 and 17 years, carrying a mutation in heterozygous state in the SLC40A1 gene. The allelic variant described in the present family, c.484_486delGTT (p.V162del) in the SLC40A1 gene, causes the classical form of FD. This mutation has been previously reported in adults in families from Italy, Greece, United Kingdom, Sri Lanka, and France, the youngest being 18 years old (5–7).
This document emphasizes that besides the importance of diagnosis and treatment of iron overload disease, it is also valuable to detect genetic defects particularly in cases that do not have classical HFE mutations. A family genetic study should be made and the carriers of the pathological mutation monitored, especially children.
Acknowledgments
The genetic study was supported by grants from the “Fundación Mutua Madrileña de Investigación Biomédica” (FMM 2012–0087) and the “Fundación Eugenio Rodríguez Pascual” (FERP 2013–0014).
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Bardou-Jacquet E, Ben Ali Z, Beaumont-Epinette MP, et al. Non-HFE hemochromatosis: pathophysiological and diagnostic aspects. Clin Res Hepatol Gastroenterol 2014; 38:143–154. [DOI] [PubMed] [Google Scholar]
- 2.Bardou-Jacquet E, Brissot P. Diagnostic evaluation of hereditary hemochromatosis (HFE and non-HFE). Hematol Oncol Clin North Am 2014; 28:625–635. [DOI] [PubMed] [Google Scholar]
- 3.Ganz T. Systemic iron homeostasis. Physiol Rev 2014; 93:1721–1741. [DOI] [PubMed] [Google Scholar]
- 4.Pietrangelo A, Corradini E, Ferrara F, et al. Magnetic resonance imaging to identify classic and nonclassic forms of ferroportin disease. Blood Cells Mol Dis 2006; 37:192–196. [DOI] [PubMed] [Google Scholar]
- 5.Mayr R, Janecke AR, Schranz M, et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 2010; 53:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callebaut I, Joubrel R, Pissard S, et al. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum Mol Genet 2014; 23:4479–4490. [DOI] [PubMed] [Google Scholar]
- 7.Le Lan C, Mosser A, Ropert M, et al. Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology 2011; 140:1199–1207.e1–2. [DOI] [PubMed] [Google Scholar]