

Abstract Abstract
This review summarizes our current knowledge on lung vasculogenesis and angiogenesis during normal lung development and the regulation of fetal and postnatal pulmonary vascular tone. In comparison to that of the adult, the pulmonary circulation of the fetus and newborn displays many unique characteristics. Moreover, altered development of pulmonary vasculature plays a more prominent role in compromised pulmonary vasoreactivity than in the adult. Clinically, a better understanding of the developmental changes in pulmonary vasculature and vasomotor tone and the mechanisms that are disrupted in disease states can lead to the development of new therapies for lung diseases characterized by impaired alveolar structure and pulmonary hypertension.
Keywords: pulmonary circulation, fetal, newborn, endothelial, smooth muscle
Lung development has been classically divided into five overlapping stages in humans and rodents, on the basis of gross histological features. They are termed the embryonic (human: weeks 4–7 of pregnancy; mice: days E9.5–E12), pseudoglandular (humans: weeks 5–17; mice: E12–E16.5), canalicular (humans: weeks 16–26; mice: E16.5–E17.5), saccular (humans: weeks 24–38; mice: E17.5–P4), and alveolarization stages (humans: week 36 to infancy; mice: P4–P14).1 The development of the lung starts with the formation the primary left and right lung buds in the ventral foregut endoderm, at about week 4 of pregnancy in the human and at day E9.5 in the mouse. During the pseudoglandular stage, the major airway architecture, including the conducting airways and respiratory bronchiole of the lung, is established, resulting from recursive branching morphogenesis.2,3 This process is likely to be achieved by three geometric forms, termed domain branching, planar bifurcation, and orthogonal bifurcation.4 Continuity between the heart and the capillary plexus of the lung occurs as early as day 34 of pregnancy in the human, with the artery extending from the outflow tract of the heart and the vein connecting to the prospective left atrium. With the development of the circulation, a mesenchymal capillary plexus forms between the arteries and the veins.5,6 A similar phenomenon has been found at day E10.5 in the mouse.7 At the end of the pseudoglandular stage, all preacinar pulmonary and bronchial arteries are in place and correspond to the bronchial branching pattern in human lung.2,3
During the canalicular stage, a great increase in the number of lung capillaries occurs. These capillaries are arranged in close apposition to the epithelium to form the first air-blood barrier. The subsequent saccular stage is characterized by the formation of the last generation of airways, which end with clusters of thin-walled primitive alveoli (saccules). At this stage capillaries form a bilayer within the cellular intersaccular septa. Alveolarization is the final stage of lung development. During this period, alveoli are formed by the outgrowth of secondary septa that subdivide terminal saccules into anatomic alveoli. The interalveolar septa are thinned, the double capillary layer matures into a single-layer adult form, and the microvasculature undergoes marked growth and development.2,3 Studies by Abman and colleagues8 suggest that the disturbed alveolar growth and vasculogenesis may constitute an important factor for bronchopulmonary dysplasia (BPD).
The normal development of pulmonary vasculature is an essential part of lung development. The impairment of this process is involved in the pathogenesis of various pediatric pulmonary vascular diseases (PVDs), such as pulmonary artery hypertension and BPD. In comparison with adult PVDs, disruption of lung vascular growth and development plays an especially prominent role in the pathobiology of pediatric PVDs.3,8 In this review, we summarize our current knowledge on lung vasculogenesis and angiogenesis during normal lung development and the regulation of fetal and postnatal pulmonary vascular tone. Clearly, a better understanding of the mechanisms underlying the developing changes in pulmonary vasculature and pulmonary vasoreactivity would be of help to combat pediatric PVDs more effectively.
Vasculogenesis and angiogenesis
Formation of the pulmonary circulation is dependent on two basic processes: vasculogenesis and angiogenesis. Vasculogenesis is the de novo formation of blood vessels from angioblasts or endothelial precursor cells that migrate and differentiate in response to local cues (growth factors, extracellular matrix) to form vascular tubes. Angiogenesis is the formation of new blood vessels from preexisting ones.9 There are currently three different postulations regarding pulmonary vascular development: one proposes that central angiogenesis occurs concurrently with peripheral vasculogenesis,10-12 a second considers vasculogenesis as the primary process in the developing lung,7,13,14 and the third suggests that distal angiogenesis may act as the mechanism for lung vascular morphogenesis.5,15 These discrepant models may result, in part, from the lack of genetic tools for sophisticated cell lineage tracing as well as the lack of a time-based simultaneous detection of related signaling profiles.9
The development of pulmonary vasculature is closely correlated with and interacts with the airway growth. Studies demonstrate that lung vascularization initially originates in the mesenchyme, distal to the epithelium. In response to epithelial-derived vascular endothelial growth factor (VEGF), the endothelial cells move toward the epithelium, where they eventually form epithelium-capillary bilayers, which are essential for gas exchange.16 In mice, epithelial deletion of Wntless (Wls), a mediator of WNT ligand secretion, results in decreased distal pulmonary microvascular development. Epithelial Wls–mutant mice die at birth from respiratory failure due to lung hypoplasia and pulmonary hemorrhage.17,18 Conversely, impairment of pulmonary vasculature development by VEGF inhibitors in both fetal and newborn rats is accompanied by diminished alveolar development, histologically similar to pathologic findings in clinical BPD.19,20
A critical step of microvascular maturation occurs during the alveolar stage of lung development.21 Capillaries, which are initially organized as double capillary layers in the immature gas-exchange region, later fuse or remodel to form a single capillary layer. This process is completed in the rat by about the third postnatal week but can persist postnatally in infants with developmental lung disorders and Down syndrome (Thurlbeck). The alveolar wall thins, and its cellular composition changes. In the rat, the thickness of the alveolar wall and the air-gas barrier (the distance between alveolar gas and capillary blood) decreases by 20%–25%. Between birth and adulthood, the alveolar and capillary surface areas expand nearly 20-fold and the capillary volume by 35-fold. Further expansion of the capillary network subsequently occurs via two angiogenic mechanisms: sprouting angiogenesis from preexisting vessels and intussusceptive growth. Intussusceptive microvascular growth occurs by internal division of the preexisting capillary plexus (insertion of transcapillary tissue pillars) without sprouting, which may underlie alveolar growth and remodeling throughout adult life and thus be amenable to therapeutic modulation for lung regeneration.22,23 Much more must be learned about the anatomic events underlying lung vascular development and time-specific mechanisms that regulate growth and function at each stage. Nonetheless, current knowledge, gained mostly from developmental biology and cancer literature about key angiogenic growth factors, has been exploited to investigate the role of lung angiogenesis during normal alveolar development.
Role of angiogenic growth factors during lung vascular development
VEGF is pivotal for proper blood vessel formation
In mammals, 5 VEGF molecules have been identified: VEGF-A, placenta growth factor, VEGF-B, VEGF-C, and VEGF-D. Among them, VEGF-A (hereafter referred to as VEGF) is the one essential molecule for all aspects of vascular development, from the endothelial lineage specification to vessel maintenance. VEGF exerts its effect by binding to transmembrane tyrosine kinase receptors, VEGF receptors 1 (VEGFR-1, flt-1) and 2 (VEGFR-2, flk-1/KDR), and neuropilin 1, which are expressed on vascular endothelium. VEGF signaling is transduced via VEGFR-2, whereas VEGFR-1 modulates this process in a negative manner. Upon binding of VEGF, activated VEGFR-2 undergoes dimerization and autophosphorylation of specific intracellular tyrosine residues of the receptors. VEGFR-2 activation stimulates several signaling pathways, including the rapidly accelerated fibrosarcoma kinase (Raf)–mitogen-activated protein kinase kinase (MEK)–extracellular signal–regulated protein kinase 1 and 2 (ERK 1/2) pathway, which enhances cell proliferation; the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)–protein kinase B (PKB) pathway, which promotes cell survival; the cell division cycle 42 (Cdc42)–p38 mitogen-activated protein kinase (p38 MAPK) pathway; and tyrosine-protein kinase CSK (Src), which induces cell migration. Src activation and PI3K-PKB-mediated increase in nitric oxide (NO) production can increase cell permeability, which may facilitate angiogenesis by providing exposure to the proangiogenic extracellular environment (Fig. 1).24-26
Figure 1.
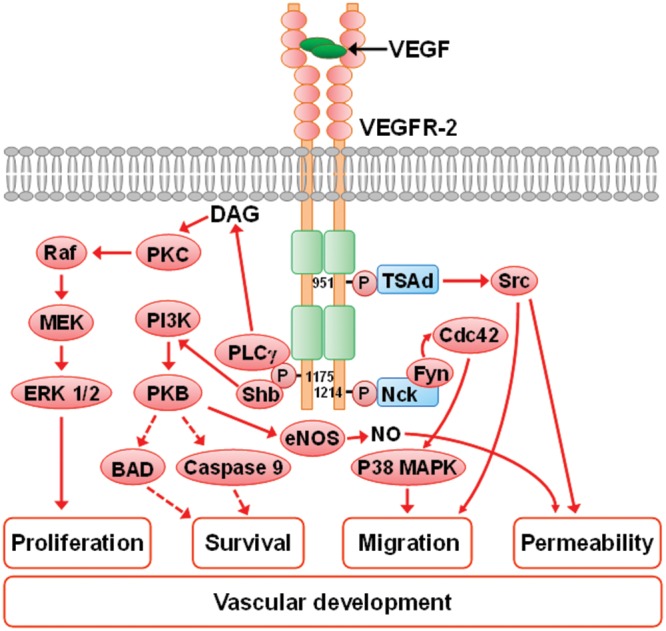
Scheme depicting the possible mechanism of VEGFR-2-mediated vascular effects (cell proliferation, survival, migration, and enhanced permeability) related to vascular formation. VEGFR-2 is composed of an extracellular domain containing seven immunoglobulin-like domains, a transmembrane domain, and an intracellular domain of two kinase domains separated by a kinase-insert domain. The binding of the VEGF dimer to the receptor leads to the dimerization and autophosphorylation of specific intracellular tyrosine residues of the receptors. The phosphorylation of Y1175 recruits PLCγ, resulting in DAG release and activation of PKC and the Raf-MEK-ERK 1/2 signaling pathway and thus in stimulated cell proliferation. The phosphorylated Y1175 may also recruit adaptor protein Shb, which leads to activation of PI3K-PKB signaling; consequently, BAD and caspase are suppressed, which results in increased cell survival. Increased PI3K-PKB activity increases eNOS-dependent NO production, resulting in increased cell permeability, which facilitates angiogenesis through providing a proangiogenic extracellular environment. The phosphorylation of Y951 provides a binding site for TSAd, which causes the activation of Src, leading to increased cell migration and permeability. Increased cell migration also results from activation of Cdc42-p38 MAPK as a result of the phosphorylation of Y1214 and the formation of the Nck-Fyn complex. Solid arrows indicate activation and dashed arrows inhibition. BAD: B-cell lymphoma 2-associated death promoter; Cdc42: cell division cycle 42; DAG: diacylglycerol; eNOS: endothelial nitric oxide synthase; ERK 1/2: extracellular signal–regulated protein kinase 1 and 2; Fyn: proto-oncogene tyrosine-protein kinase Fyn; MEK: mitogen-activated protein kinase; Nck: Nck adaptor protein 1; NO, nitric oxide; p38 MAPK: p38 mitogen-activated protein kinase; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; PKB: protein kinase B, also known as Akt; PKC: protein kinase C; PLCγ: phosphoinositide phospholipase C-γ; Raf: rapidly accelerated fibrosarcoma kinases; Shb: SH2-domain-containing adaptor protein B; Src: tyrosine-protein kinase CSK; TSAd: T cell-specific adaptor molecule; VEGF: vascular endothelial growth factor; VEGFR-2: VEGF receptor 2.
The absolute requirement of VEGF for development of the embryonic vasculature in mice has been demonstrated by inactivation studies of VEGF alleles27,28 and knockouts of VEGFR-129 and VEGFR-2.30 In each of these studies, VEGF-related gene inactivation causes lethal phenotypes that are characterized by deficient organization of endothelial cells. Inducible Cre-loxP-mediated gene targeting or administration of a soluble VEGF receptor chimeric protein (mFlt (1–3)-IgG) to inactivate VEGF in early postnatal life increased mortality, stunted body growth, and impaired organ development. The impact of VEGF inhibition lessened with postnatal maturation in this model.31
VEGF promotes normal lung growth
The spatial relationship between receptor and ligand suggests that VEGF plays a role in the development of the alveolar capillary bed. In addition, pharmacological and genetic inactivation of VEGF arrests alveolar development. VEGF messenger RNA (mRNA) and protein are localized to distal airway epithelial cells and the basement membrane subjacent to the airway epithelial cells.32 VEGFR-1 and VEGFR-2 mRNA expression increases during normal mouse lung development33,34 and is localized to the pulmonary endothelial cells closely apposed to the developing epithelium.35 VEGF120, VEGF164, and VEGF188 are present in alveolar type II cells in the developing mouse lung, and their expression peaks during the canalicular stage, when most of the vessel growth occurs in the lung, then decreases until postnatal day 10 (P10), when it increases to levels that are maintained through adulthood.35
Targeted exon deletion of the VEGF gene reveals that mice that lack the heparin-binding isoforms VEGF164 and VEGF188 display a variety of vascular defects, including a significant reduction in the formation of air spaces and capillaries, resulting in distended and underdeveloped alveoli.36 Likewise, pharmacological and genetic VEGF inhibition during alveolar development decreases alveolarization and pulmonary arterial density, features encountered in clinical BPD.19,20,36-39 Chronic treatment of adult rats with the VEGFR-1 and VEGFR-2 blocker SU5416 leads to enlargement of the air spaces, indicative of emphysema,40 suggesting that VEGF is required not only for the formation but also for the maintenance of the pulmonary vasculature and alveolar structures throughout adulthood. Conversely, lung overexpression of VEGF during normal lung development disrupts the lung architecture, suggesting a tight regulation of VEGF-driven angiogenesis to insure proper lung development.37,41
In a series of elegant experiments, Vu and colleagues used pharmacological VEGF inhibition in lung renal capsule grafts39 and genetic VEGF inactivation42 to show that selective inactivation of VEGF in respiratory epithelium results in an almost complete absence of pulmonary capillaries in the fetal mouse, demonstrating the dependence of pulmonary capillary development on epithelium-derived VEGF. Deficient capillary formation in VEGF-deficient lungs was associated with a defect in primary septum formation, coupled with suppression of epithelial cell proliferation and decreased hepatocyte growth factor (HGF) expression. Lung endothelial cells express HGF, and selective deletion of the HGF receptor gene in respiratory epithelium phenocopies the malformation of septa, confirming the requirement for epithelial HGF signaling in normal septum formation and suggesting that HGF serves as an endothelium-derived factor that signals to the epithelium. These observations suggest that inhibition of vascular growth in the fetus directly impairs distal airspace growth.42 Selective VEGF inactivation during the early postnatal period causes similar disruption of distal lung development, resulting in sustained reductions of alveolar and vascular growth in the infant mouse.26
Perturbation of NO is associated with arrested lung vascular growth
The role of the endothelium-derived relaxing factor NO in the regulation of the pulmonary vascular tone in the perinatal period is well established; however, little was known about its potential role in the structural development of the pulmonary vasculature.3 Lungs of late-fetal and neonatal endothelial NO synthase (eNOS)–deficient mice have a paucity of distal arteries and reduced alveolarization43 and are more susceptible to failed vascular and alveolar growth after exposure to mild hypoxia and hyperoxia.44 Recent studies suggest that VEGF-induced lung angiogenesis is in part mediated by NO. SU5416 (a VEGF inhibitor)-induced arrested alveolar and vascular growth in newborn rats is associated with decreased lung eNOS protein expression and NO production; treatment with inhaled NO improves vascular and alveolar growth in this model.45 Soluble guanylyl cyclase (sGC) is the primary effector enzyme for signaling by NO.3 In sGC-α1 knockout mouse pups, the number of small airway structures and the lung volume are decreased, compared with those in wild-type mice, indicating the involvement of NO-cGMP (cyclic guanosine monophosphate) signaling in lung development.46 However, recombinant human VEGF protein treatment restores lung structure after exposure to mild hyperoxia in eNOS−/− postnatal mice, suggesting that VEGF can enhance vascular and alveolar growth through non-eNOS-related mechanisms, such as HGF signaling or enhanced vitamin A production.26,47
Counterintuitively, NF-κB, a transcription factor traditionally associated with inflammation, plays a unique protective role in the neonatal lung. Constitutive NF-κB expression is higher in the neonatal lung than in the adult lung, and inhibition of constitutive NF-κB activity decreases pulmonary endothelial cell survival and proliferation and impairs in vitro angiogenesis. NF-κB inhibition during the alveolar stage of lung development in mice reduces pulmonary capillary density and leads to alveolar simplification reminiscent of BPD.48
The Forkhead Box (Fox) family of transcription factors plays important roles in regulating expression of genes involved in cellular proliferation and differentiation. Newborn foxf1+/− mice with low Foxf1 levels die with defects in alveolarization and lung capillaries, whereas those with increased pulmonary Foxf1 levels survive and display normal adult lung morphology.34 Endothelial-specific deletion of Foxf1 is embryonically lethal and results in fewer blood vessels in the lung.49 In humans, FOXF1 haploinsufficiency is associated with alveolar capillary dysplasia with misalignment of pulmonary veins.50
EphrinB2 and its receptor EphB4 belong to the Eph/ephrin family, which is critically involved in cardiovascular development.51,52 Expression of ephrinB2 and its receptor EphB4 peaks during the stage of intense lung vascular growth in the developing rat lung. EphrinB2 and EphB4 are expressed in cells that ultimately differentiate into arteries and veins, respectively, preceding the formation of morphologically distinct arteries and veins.53-55 Expression of ephrinB2 and EphB4 is not restricted to arterial versus venous vessels until the canalicular stage.16,52 In vivo ephrinB2 knockdown using intranasal small interfering RNA during the postnatal stage of alveolar development and vascular maturation arrests alveolar and vascular growth.56 Mice homozygous for the hypomorphic knock-in allele ephrinB2ΔV/ΔV die by 2 weeks of age with enlarged airspaces.57
Axonal guidance cues (AGCs) contribute to lung angiogenesis
AGCs are molecules that guide the outgrowth of axons to their targets. Among these AGCs, members of the ephrin family also promote angiogenesis, cell migration, and organogenesis outside the nervous system. Thus, AGCs are also appealing candidates in guiding the outgrowth of secondary crests during alveolar development and repair.58 Semaphorin 3, another molecule of the AGC family, may have an important role in the orchestration of pulmonary vascular development during alveolarization. The congenital loss of semaphorin 3–neuropilin 1 signaling in transgenic mice resulted in disrupted alveolar-capillary interface formation and high neonatal mortality.59-62
Role of lung endothelial progenitor cells during lung vascular development
Endothelial cell function is important for lung growth
Administration of an anti-PECAM-1 antibody that inhibits endothelial cell migration, but not proliferation or survival in vitro, also impairs alveolarization in neonatal rats, without reducing endothelial cell content.63 In the endothelium, platelet endothelial cell adhesion molecule (PECAM-1), in addition to being an adhesion molecule, affects the expression and activity of eNOS. Endoglin is an essential molecule for angiogenesis. Its expression and activity are compromised in the absence of PECAM-1.64 Pulmonary capillary endothelial cells may also initiate and sustain alveologenesis by increasing MMP14 (matrix metalloproteinase 14)-dependent bioavailability of epidermal growth factor receptor ligands on the epithelial cells.65,66
Lung endothelial progenitor cells (EPCs)
An observation by Schwartz and Benditt67 suggested that the presence of fast-growing endothelial niches within the aortic intima that were enriched with replication-competent cells was required to repopulate injured or senesced cells. More recently, large differences in proliferative potential have been documented between pulmonary macro- and microvascular endothelial cells,68 with rat pulmonary microvascular endothelial cells proliferating twice as fast as rat pulmonary artery endothelial cells (PAECs). Thus, the pulmonary microcirculation seems to be enriched with EPCs that display vasculogenic competence while maintaining functional endothelial microvascular specificity.69 These so-called resident microvascular endothelial progenitor cells (RMEPCs) are highly proliferative and express endothelial cell markers (CD31, CD144, eNOS, and von Willebrand factor) and progenitor cell antigens (CD34 and CD309) but not the leukocyte marker CD45. RMEPCs interact with Griffonia simplicifolia, display restrictive barrier properties, and possess vasculogenic capacity, as demonstrated in vitro and in vivo, forming ultrastructurally normal de novo vessels. Nucleosome assembly protein 1, incriminated in the control of cell growth, is preferentially expressed by RMEPCs and functions as an important regulator of the proliferative and vasculogenic endothelial cell phenotype.70 These RMEPCs share features of human cord blood–derived endothelial colony-forming cells (ECFCs). Developing human fetal and neonatal rat lungs contain ECFCs with robust proliferative potential, secondary colony formation, and de novo blood vessel formation in vivo when transplanted into a matrigel plug under the skin of immunodeficient mice.71 Furthermore, human fetal lung ECFCs exposed to hyperoxia in vitro and neonatal rat ECFCs isolated from hyperoxic alveolar growth–arrested lungs mimicking BPD proliferate less, show decreased clonogenic capacity, and form fewer capillary-like networks. These findings indicate that impaired ECFC function contributes to the lack of lung repair and the persistence of arrested alveolar growth in BPD. Consequently, exogenous administration of human cord blood–derived ECFCs restores alveolar and lung vascular growth in hyperoxic rodents.71 However, lung engraftment was low, suggesting that the effect of these exogenous ECFCs was through a paracrine effect. Accordingly, administration of cell-free ECFC-derived conditioned media exerted similar therapeutic benefits in the hyperoxia- and bleomycin-induced models of BPD in rodents.71,72 Overall, these findings further support the crucial role of angiocrine-derived factors in lung repair and offers promising new therapeutic strategies to prevent/restore lung damage.
Diversity of the smooth muscle cell (SMC) phenotype in development
During development, SMCs undergo rapid proliferation, during which time the vessel wall acquires its compliment of SMCs. Replication rates decrease as the animal matures, with quiescence of SMCs in the adult animal.73-75 However, after injury to an adult artery, SMC replication rates increase, at least transiently, to levels similar to those exhibited during embryonic life.76 Similarly, adult SMCs do not exhibit serum-independent growth and have lower growth responses to mitogen stimulation than fetal SMCs. In an observation that is consistent with high in vivo growth rates, several investigators have reported the isolation of unique fetal aortic SMCs that differ substantially from adult cells. These SMCs, isolated from rat pups, demonstrate a characteristic epitheloid morphology, secrete platelet-derived growth factor (PDGF), and express high levels of cytochrome P450 IA1, PDGF-b chain, and elastin mRNAs.77,78
An even more interesting cell has been isolated from earlier embryonic periods that exhibits an intrinsically activated growth phenotype, dependent on Akt and mTOR (protein kinase B and mechanistic target of rapamycin, respectively) signaling, apparently independent of autocrine secretion of growth factors. This cell is ultimately growth inhibited by the timed acquisition of the endogenously produced growth factor PTEN (phosphatase and tensin homolog).78 In contrast, SMCs isolated in culture from uninjured adult aortas or carotid arteries show a spindle-shaped morphology, low to undetectable PDGF secretion, and greatly reduced cytochrome P450 IA1 and elastin mRNA expression. During normal development, SMCs demonstrate compositional changes in cytoskeletal and contractile protein expression.79 Fetal and neonatal aortic SMCs express primarily nonmuscle β-actin, whereas adult SMCs express predominantly SMC-specific α-actin isoforms.80 A gradual increase in the number of desmin- and tropomyosin-positive aortic SMCs also occurs with increasing age.81 A reversion from the adult to the fetal or neonatal phenotype is often observed for each of these parameters in SMCs in experimental intimal thickening, atherosclerotic plaques, and pulmonary hypertension.75,82,83 These observations strongly suggest that SMCs undergo maturation from a fetal to an adult phenotype during normal development and that reexpression of “fetal-like” markers of cell phenotype occurs upon arterial injury or entry of SMCs into a growth phase. It is also clear that expression of fetal or less differentiated cell phenotypic characteristics may have a significant impact on functional responses of the injured vessel wall.
An obvious question raised by these observations is, what is the response of the fetal and neonatal circulation (still undifferentiated) to injury? With regard to the pulmonary circulation, infants dying with persistent pulmonary hypertension often exhibit a striking distal extension of SMCs, thickening of the media and adventitia, and excessive accumulation of the matrix proteins in vessel walls that appear to be acquired over very short periods of time. The SMC and adventitial changes appear particularly malignant when compared to adult forms of pulmonary hypertension. In fact, numerous observations demonstrate that the proliferative and matrix-producing potential of the neonatal pulmonary circulation in response to injury exceeds that of the adult. For example, when exposed to hypoxic, hyperoxic, or toxic stimuli, neonatal animals develop more severe, more rapidly progressive, and potentially less reversible pulmonary hypertension and right ventricular hypertrophy than adult animals. Further, structural changes, including cell proliferation and matrix-protein accumulation, are more dramatic than those observed in adults.84-86
The observation that neonatal cells normally exist in a “synthetic” or “primed” state provides support for the notion that in neonates the potential for vascular cell growth is heightened.75 Alterations in receptor expression, signal transduction (including protein kinase C [PKC]), proto-oncogene expression (including c-fos and c-myc), and growth factor production have been demonstrated during periods of rapid cell growth and differentiation and are consistent with the rapid proliferative response of the neonate to growth-promoting stimuli.77,87,88 Matrix protein–producing genes for elastin and collagen demonstrate their greatest activity in late fetal and early neonatal life.89,90 Tropoelastin mRNA expression, for example, is nearly completely suppressed in the adult.91 Thus, the hypothesis has been raised that the capacity of the SMCs to respond to matrix-producing stimuli depends not only on local hormonal and cell-cell interactions but also on the developmental state of the cell. The possibility that the normal fetal-to-adult transition in vascular cell phenotype may fail to occur under certain conditions in the neonatal period has also been raised. The persistence of fetal-like characteristics of vascular wall cells could therefore contribute to the excessive growth and matrix-protein production observed under conditions of severe neonatal pulmonary hypertension.
Site-specific heterogeneity of SMC phenotype in vascular wall
In addition to the developmentally regulated differences in SMC phenotype, there is a growing body of literature documenting the existence of phenotypic heterogeneity within the resident SMC population of a given tissue at a specified developmental stage. It is becoming clear that SMCs in the pulmonary artery (and aorta) form a phenotypically heterogeneous, yet highly organized, cell population.92,93 Distinct phenotypic differences have been documented in SMCs at specific locations and along the longitudinal axis of the pulmonary artery and aorta.93 Marked differences in the production of elastin and collagen have been demonstrated in aortic SMCs, with elastin production being greatest near the heart.94 An inverse gradient for the expression of desmin has also been demonstrated.95 These differences in the biosynthetic phenotype are stable in culture, suggesting the existence of multiple medial SMC phenotypes.
Regulation of SMC proliferation during development
Much of the available literature suggests that the bulk of vascular SMC replication during development occurs before birth. With regard to the SMCs, the active replicative state slows considerably in the perinatal period and then virtually ceases under normal conditions by adulthood. Therefore, an understanding of the factors and mechanisms responsible for cell proliferation in fetal and neonatal life and the mechanisms that suppress cell replication once morphogenesis is complete is crucial if we are to understand the proliferative response of the vascular SMC to injury.
Cook et al.74 defined the distinct phases of SMC replication that occur during early vascular development. At the embryonic stage, the rat aorta is characterized by a very high in vivo replication rate, significant autonomous capabilities in vitro, and a generalized lack of responsiveness to exogenously added SMC mitogens. The capacity for autocrine growth in vitro appears to be lost between gestational days 18–20 in the fetal rat and appears to be correlated with a drop in in vivo replication rates just after the embryonic-fetal transition. Autocrine growth was due not to hyperresponsiveness of the cells to growth stimuli but rather to intrinsically controlled developmental timing mechanisms operating independently of exogenous growth factors.
In contrast, mechanisms involving growth factors are likely involved in the regulation of vascular cell growth in the neonatal period. While the autonomous growth capability observed in fetal cells has been lost, neonatal cells demonstrate enhanced growth capacity, compared to adult cells. Neonatal bovine pulmonary artery SMCs are more responsive than adult cells to several growth-promoting stimuli. Specifically, SMCs harvested from the lobar pulmonary artery of neonatal calves grow faster in serum-containing media than matched SMCs obtained from an adult. Although the neonatal cells lack autocrine growth capacity, they are more resistant to the induction of a quiescent state than their adult counterparts. This has been demonstrated by measuring basal [3H]-thymidine incorporation after 72 hours of serum deprivation.96 In addition, these neonatal vascular cells also have enhanced mitogenic responses to growth factors such as insulin-like growth factor 1 (IGF-1) and phorbol myristate acetate, factors that activate different but potentially synergistic growth-promoting pathways.97,98 The mechanisms responsible for these enhanced growth responses remain unknown but may involve expression of increased numbers of cell surface receptors specific for a particular ligand or increased ability to generate second messengers in response to growth factor binding. For instance, IGF-1 binding levels have been found to be much higher in near-term fetal and neonatal aortic SMCs than in the adult. IGF-2 binding activity is increased over IGF-1 levels in fetal cells but then falls below them in the postnatal period.99 Thus, receptor expression for each relevant growth factor may be uniquely regulated during development and could contribute to enhance neonatal responsiveness to growth stimuli.
Increased responsiveness of neonatal cells to phorbol esters, cell-permeable activators of PKC, is also intriguing, because in this case the cell surface receptors have been bypassed altogether. This signal transduction pathway has been shown to be important in the pulmonary artery SMC (PASMC) proliferative response to hypoxia, as well as in the hypertrophic response to mechanical loading, and is susceptible to development change.97,98,100,101 In addition, increased expression of certain isotypes of PKC have been shown to cause increased responsiveness to growth factors and to promote cell differentiation.102 A modest increase in whole-cellular PKC catalytic activity in neonatal versus adult bovine PASMCs has been shown.103 More work on intrinsic differences in signaling pathways leading to enhanced growth responses in neonatal versus adult cells may lead to explanations of exaggerated growth responses to pathologic stimuli.
There may also be more subtle developmental differences in the growth responses of vascular cells to mitogenic signals. For instance, it has been demonstrated that neonatal rat aortic SMCs exhibit altered responses to transforming growth factor beta (TGF-β), a family of peptides capable of stimulating or inhibiting replication in adult SMCs. In neonatal SMCs, TGF-β1 acts exclusively as a growth inhibitor. In addition, TGF-β1 acts to significantly increase the size of these cultured neonatal rat cells. Adult cells do not demonstrate this increase in size in response to TGF-β1. Here, then, is an instance where the type, rather than the magnitude, of the response is uniquely altered in neonatal vascular cells.104,105
Differences in growth properties of neonatal bovine vascular cells, as compared to those of adult cells, can be further enhanced by application of hypoxic stress to experimental animals before harvesting. The vascular-wall SMCs subsequently obtained from hypoxia-exposed animals grow even faster in culture than their normoxia-exposed counterparts. The mechanisms of these “acquired” growth properties are unknown. They could represent a regression to the autocrine (exogenous growth factor–independent) growth properties of fetal vascular cells or a further increase in the as-yet-unidentified mechanisms that underlie enhanced neonatal cell responsiveness to mitogens. Here there is great interest in the possibility of epigenetic changes in these cells leading to this growth phenotype.
Age-specific regulation of extracellular matrix expression by SMC
There is little question that a major function of the SMCs in the developing vessel is the production of connective tissue, particularly elastin and collagen. In large arteries, such as the aorta, elastin and collagen make up more than 70% of the dry weight of the vessel wall.106 Thus, study of the mechanisms controlling elastin synthesis during normal vascular development, as well as the elastogenic response to injury in both the neonate and the adult, is crucial for a complete understanding of vascular growth in health and disease. In most mammalian tissues, the bulk of elastin production occurs during fetal and neonatal periods and is essentially complete by the first decade of life. For instance, the levels of tropoelastin mRNA parallel the deposition of elastin in the developing bovine nuchal ligament.107 In addition, the steady-state levels of tropoelastin mRNA correlate with the functional levels of the mRNA as determined by cell-free translation and immunoprecipitation, indicating pretranslational regulation. The temporal sequence of elastogenesis is unique. Elastic fibers are deposited during a defined developmental window and persist for the life of the organism. In contrast, many other extracellular matrix proteins are continually made and turned over.
Although factors that influence the initiation, maintenance, and cessation of elastogenesis in vivo are unknown, results obtained from in vitro models have demonstrated that various mediators affect tropoelastin expression, and these may have regulatory functions in vivo. Several mediators, including TGF-β, IGF-1, and glucocorticoids, coordinately increase elastin synthesis and tropoelastin mRNA. In general, the stimulation effected by these mediators is about 2–4-fold above control levels.89,108 Repressors of tropoelastin synthesis include epidermal growth factor,109 recombinant interleukin 1,110 1,25-dihydroxyvitamin D3 (1,25(OH)2D3),111 and phorbol esters.112 Unlike mediators that stimulate production, factors and conditions that downregulate tropoelastin synthesis profoundly influence production and, with sufficient exposure, can completely repress tropoelastin expression. Because elastogenesis proceeds through defined stages, diverse regulatory mechanisms may be active during different periods of expression. Indeed, tropoelastin pre-mRNA undergoes age-specific alternative splicing, resulting in the translation of multiple distinct protein isoforms.113,114 This may have particular importance in the early lung, since tropoelastin expression is also noted in alveolar-tip cells and abrogated expression is associated with decreased alveolarization and vascular growth.115
The SMC response in lobar pulmonary arteries to hypertension reveals yet another example that elastin production is controlled by different means at various stages of development. Increased deposition of elastin is characteristic of the vascular remodeling seen in pulmonary hypertension in both newborns and adults, but the site of this increased production differs between the two groups. In neonates, when elastogenesis is ongoing, increased deposition is detected in the medial wall and is apparently a consequence of sustained fetal expression.116 By maturity, tropoelastin expression in medial SMCs has ceased, but renewed production is evident in neointimal cells.117 It is not clear why some SMCs in the adult arteries do not contribute to matrix remodeling associated with pulmonary hypertension, but this age-specific response suggests that not all adult SMCs have the capacity to reexpress tropoelastin.
Regulation of vasomotor tone
During fetal life, the lung is fluid filled, oxygen tension is low, and pulmonary blood flow is limited, while pulmonary vascular resistance (PVR) exceeds systemic vascular resistance. Blood is shunted away from the lungs at the level of the ductus arteriosus (DA).3 At birth, pulmonary blood flow increases 8–10-fold,118 and pulmonary arterial pressure decreases to 50% of systemic levels within 24 hours after birth,119 concomitant with an increase in oxygen tension. The critical physiologic stimuli that account for perinatal pulmonary vasodilatation include rhythmic distention of the lung120 and an increase in oxygen tension121-124 and shear stress.125 Data accumulated over the past several decades implicate distinct and complementary roles for both PAECs and PASMCs in promoting fetal lung growth and development and in mediating the postnatal adaptation of the pulmonary circulation.
In the human fetus at weeks 20 and 38 of gestation, blood flow to the lungs is 13% and 21% of biventricular output, respectively. Maternal hyperoxygenation has no effect on human fetal pulmonary blood flow between weeks 20 and 26 of gestation, but significant increases occur in the fetus from week 31 to week 36 of gestation, suggesting that the capacity of the pulmonary circulation to sense and respond to an increase in oxygen tension is developmentally regulated.122 In fetal lambs at 0.4 and at 1.0 gestation, pulmonary blood flow is 3.7% and 7.0% of the combined cardiac output, respectively.123 In near-term and term fetal lambs, pulmonary arteries and veins display vigorous responses to various vasoactive agents and changes in oxygen tension.121,124 However, the acute pulmonary vascular dilator response to increased oxygen tension is not sustained during more prolonged exposure, suggesting the presence of developmental vasoregulatory mechanisms that oppose vasodilation in the fetus.121,125
With the onset of breathing in the human infant, pulmonary blood flow increases from 21% to 100% of cardiac output to enable exchange of gas in the lungs.122 There are several vasoactive molecules that play a key role in modulating pulmonary vascular tone, including endothelium-derived nitric oxide (EDNO), prostaglandin I2 (PGI2), endothelin 1 (ET-1), and platelet-activating factor (PAF). The role of each of these molecules in the perinatal pulmonary circulation is addressed below.3,126
Pulmonary artery endothelial cell (PAEC)
The PAEC plays a critically important role in the developing pulmonary circulation, both during and after fetal life. Vasoactive products produced by the pulmonary endothelial cells simultaneously preserve and constrain fetal pulmonary blood flow. During fetal life, inhibition of ET-1, a vasoconstrictive protein produced by the endothelium, causes marked pulmonary vasodilation.127 Conversely, pharmacologic inhibition of NO decreases pulmonary blood flow and increases pulmonary artery blood pressure.128 These two observations suggest that carefully circumscribed pulmonary blood flow is of critical importance in normal pulmonary vascular development.
In response to key perinatal physiologic stimuli, the pulmonary endothelium elaborates vasoactive mediators such as NO, endothelin, and prostaglandins.127-130 PAEC NO production is necessary131 for the normal transition of the pulmonary circulation from a high- to a low-resistance circuit. Among the most important properties of the pulmonary circulation is the ability to sense and respond to alterations in oxygen tension. During air-breathing life, the pulmonary circulation adapts to respond to decreases in oxygen tension in such a manner as to match ventilation and perfusion and thereby mitigate hypoxemia. At birth, it is biologically imperative for the pulmonary circulation to respond to an acute increase in oxygen tension. Interestingly, the capacity of the pulmonary circulation to sense and respond to an acute increase in oxygen tension is developmentally regulated. Not until gestation is approximately 85% complete is the fetal pulmonary circulation able to respond to an increase in oxygen tension.124 Vasodilator agents that act through endothelial cell–mediated mechanisms produce only transient pulmonary vasodilation.121,132 The relatively narrow developmental window wherein the fetal pulmonary circulation is able to respond to an increase in oxygen tension may serve a protective purpose, as either too much or too little pulmonary blood flow compromises pulmonary vascular development.133,134
The capacity of PAECs to produce NO significantly enhances the ability of the pulmonary circulation to dilate in response to increasing oxygen tension with maturation.135 By the end of gestation, the pulmonary circulation is exquisitely sensitive to alterations in oxygen tension, as an increase of only 8 Torr results in a 3-fold increase in pulmonary blood flow.136 Several investigators have demonstrated that production of NO by the endothelial cells mediates oxygen-induced pulmonary vasodilatation.128,137 In a confluent monolayer of fetal PAECs, an acute increase in oxygen tension directly increases cytosolic calcium. In the PAEC, an acute increase in oxygen tension results in membrane depolarization and an increased rate of calcium entry. The sustained and progressive increase in PAEC intracellular calcium concentration ([Ca2+]i) entails calcium release from intracellular stores. The observation that acetylcholine causes a greater increase in PAEC cytosolic calcium under hypoxic than under normoxic conditions provides inferential evidence that the low-oxygen-tension environment of the normal fetus may ready the perinatal pulmonary circulation to respond to an acute increase in oxygen tension by augmented loading of intracellular stores. In PAECs and lung tissue derived from fetal lambs with chronic uterine pulmonary hypertension, the response of the pulmonary circulation to vasodilator stimuli is attenuated. Expression of eNOS and NO production are decreased, characterized by impaired pulmonary vasodilation at delivery.138 PAECs derived from this ovine model of perinatal pulmonary hypertension of the newborn (PPHN) do not respond to an acute increase in oxygen tension with either an increase in [Ca2+]i or NO production, and they grow and form tubes poorly in vitro.139 These findings suggest that intrauterine events, including sustained hemodynamic stress, cause sustained endothelial cell dysfunction in the early perinatal period.
The response of the PAEC to birth-related physiologic stimuli is critically important for postnatal adaptation of the pulmonary circulation. PAECs must contract and increase production of NO131,140 and prostacyclin,141,142 as well as decrease endothelin production,143 in a remarkably short time frame to facilitate the transition of the neonatal pulmonary circulation from high to low tone and thereby enable effective gas exchange to occur in the lungs. Shear stress is an essential physiologic stimulus that prompts PAECs to increase NO production. In the chronically instrumented fetal lamb model, pharmacologic inhibition of NO production prevents shear-stress-induced pulmonary vasodilatation.128 The increase in fluid shear stress after birth resulting from increased pulmonary blood flow also increases NO production by altered phosphorylation of eNOS and increased eNOS expression. In primary cultures of ovine fetal PAECs, shear stress activates eNOS through reduction in phosphorylation of eNOS at Thr495 and increased phosphorylation at Ser1177.143,144 A complex consisting of PECAM-1, vascular endothelial cadherin, and VEGFR-2 is localized at the gap junction of endothelial cells, serving as a mechanotransducer for shear stress. PECAM-1 is activated by shear stress, resulting in a Src family kinase–mediated, ligand-independent transactivation of VEGFR-2. The activation of VEGFR-2 may activate eNOS through Akt-dependent phosphorylation of Ser1177. Activation of VEGFR-2 can also cause Ser1177 phosphorylation via PKA.145
In fetal PAECs, acute shear stress initially upregulates eNOS expression that may be due to increased phosphorylation of c-Jun146 and reduced binding of STAT3 to the eNOS promoter.147 Heat shock protein 90 (Hsp90), a constitutively expressed molecular chaperone, provides an additional level of control for endothelial cell NO production. Hsp90 associates with eNOS to facilitate eNOS activation. Hsp90 also recruits activated Akt in the eNOS complex and maintains Akt activity, both by preventing proteosomal degradation of upstream PI3K and by inhibiting dephosphorylation of Akt by phosphatase 2A, thereby enhancing Akt-mediated activation of eNOS (Fig. 2).148 In resistance pulmonary arteries of the piglet, the physical interactions between Hsp90 and eNOS mature over the first weeks of life.149 In isolated perfused lungs of newborn piglets, Hsp90 inhibition reduces eNOS-Hsp90 interaction and NO production and thereby attenuates acetylcholine-induced pulmonary vasodilatation.150 Moreover, decreased association of Hsp90 with eNOS occurs in pulmonary hypertension induced by prenatal ductal ligation in fetal lambs. As a result of the diminished interaction, NO release is impaired, as is the vasodilator response to adenosine triphosphate (ATP), an important mediator of perinatal pulmonary vasodilatation.151
Figure 2.
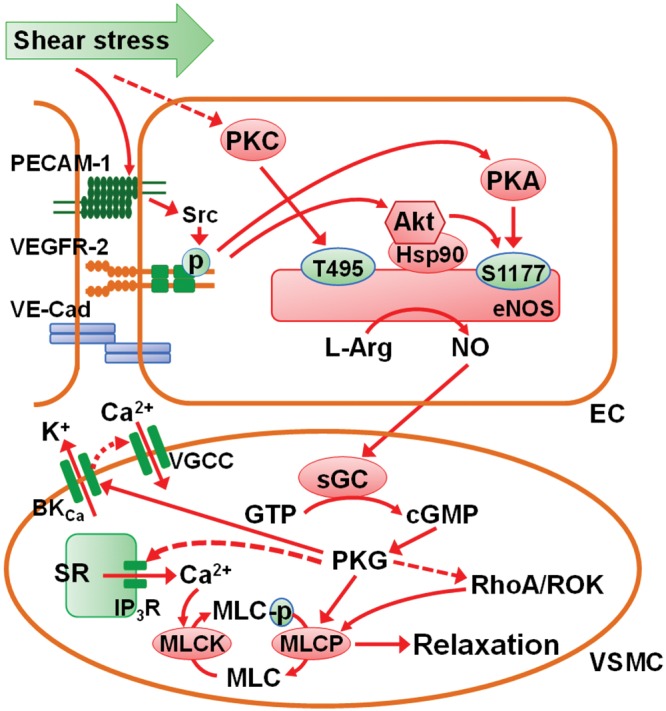
Mechanism for shear stress–induced activation of eNOS- and NO-mediated relaxation of vascular smooth muscle cells (VSMCs). A complex consisting of PECAM-1, VE-cadherin, and VEGFR-2 localized at cell-cell junctions serves as a mechanotransducer for shear stress. The tension exerted by shear stress on PECAM-1 activates a Src family kinase, resulting in ligand-independent transactivation of VEGFR-2, followed by activation of Akt (also known as protein kinase B), phosphorylation of serine 1177 (S1177) of eNOS, and production of NO. Activation of VEGFR-2 can also cause S1177 phosphorylation via PKA. Heat shock protein 90 (Hsp90), when associated with eNOS, can recruit Akt into the eNOS complex and maintains Akt activity. In contrast to S1177, the phosphorylated threonine 495 (T495) of eNOS exerts inhibitory effect on the enzyme. T495 can also be phosphorylated by PKC. Shear stress may enhance eNOS activity by inhibiting PKC activity. NO released from the endothelial cells (ECs) can readily diffuse into the underlying VSMCs, where it activates sGC, resulting in the conversion of guanosine triphosphate (GTP) to cGMP (cyclic guanosine monophosphate). The elevated cGMP causes vasodilatation, primarily via PKG, by reducing the intracellular Ca2+ concentration and decreasing the sensitivity of myofilaments to Ca2+. PKG stimulates Ca2+-activated K+ (BKCa) channels, resulting in membrane hyperpolarization and suppression of Ca2+ entry. PKG also reduces the release of Ca2+ from sarcoplasmic reticulum (SR) into cytosol by inhibiting IP3 receptor (IP3R) activity. The decreased cytosolic Ca2+ concentration leads to diminished activity of myosin light-chain kinase (MLCK), decreased phosphorylation of myosin light chain (MLC), and reduced contractility. PKG causes reduced Ca2+ sensitivity of myofilaments by directly stimulating the activity of myosin light-chain phosphatase (MLCP) as well as interfering with the inhibitory action of RhoA/Rho kinase (ROK) signaling on MLCP. These actions leads to increased dephosphorylation of MLC and thus relaxation of VSMCs. Solid arrows indicate activation and dashed arrows inhibition. eNOS: endothelial nitric oxide synthase; IP3R: inositol 1,4,5-trisphosphate receptor; L-Arg: l-arginine; NO: nitric oxide; PECAM-1: platelet endothelial cell adhesion molecule; PKA: protein kinase A; PKC: protein kinase C; PKG: cGMP-dependent protein kinase; sGC: soluble guanylyl cyclase; Src: tyrosine-protein kinase CSK; VGCC: voltage-gated Ca2+ channel; VE-cad: vascular endothelial cadherin; VEGFR-2: vascular endothelial growth factor receptor 2.
Pulmonary endothelial cells produce the prostaglandin molecules PGI2 and PGE2, both of which are potent vasodilators. Substantial evidence suggests that these molecules play a meaningful role in modulating fetal and neonatal pulmonary vascular tone. In isolated resistance pulmonary arteries and veins of term fetal lambs, the cyclooxygenase inhibitor indomethacin has no effect on arteries under low oxygen tension but causes contraction of arteries under higher oxygen tensions, suggesting that oxygen may regulate the synthesis of the dilator prostanoids.152 PGI2 production has been found to be more sensitive to the inhibitory effect of hypoxia in fetal ovine pulmonary arteries and veins than in those of adult sheep.153 In fetal goats and lambs, ventilation-induced pulmonary vasodilatation is associated with increased production of PGI2. Despite pharmacologic inhibition of cyclooxygenase, PVR decreased with the onset of air-breathing life, indicating that while PGI2 is involved in the decrease in PVR at birth, it is not absolutely required.129,130,141 The decrease in PVR caused by PGI2 at birth is modest in comparison to that induced by EDNO.154
Vasodilatation induced by PGI2 and PGE2 is mainly mediated by cyclic adenosine monophosphate (cAMP), which is elevated after the activation of adenylyl cyclase (AC). Forskolin, a direct AC stimulator, caused a greater relaxation of pulmonary veins in newborn than in fetal lambs, whereas 8-Br-cAMP induced similar responses in vessels of both ages. Furthermore, the stimulated AC activity is greater in newborn than in fetal veins. Hence, the greater response of pulmonary veins of the newborns may in part be attributable to AC activity greater than that in the fetal vessels.155
Recent data demonstrate that RhoA, a member of the Rho family of small GTPases,156 plays a significant role in determining vascular tone,157 cell contraction, migration, cell permeability,158 gene expression, and differentiation.159,160 RhoA activation occurs with GTP (guanosine triphosphate) association and translocation of the molecule to the plasma membrane where Rho kinase (ROK) is stimulated.161 RhoA activation can result via a G-protein-coupled receptor or via tyrosine kinases. In the pulmonary vasculature, hypoxia activates RhoA.157,162 RhoA activation increases Ca2+ sensitivity of the contractile myofilaments in vascular SMCs, in part, through inhibition of light-chain phosphatase. Inhibition of myosin light-chain phosphatase results from phosphorylation of the myosin-binding subunit of myosin light chain phosphatase (MYPT-1) or the myosin light chain phosphatase inhibitor protein CPI-17.163 Phosphorylation of CPI-17 by ROK may be more significant in sustained vasoconstriction than is phosphorylation of MYPT-1.164,165
During fetal life, ROK activity maintains high PVR in the fetal lung.166 Moreover, ROK inhibition causes NO-independent perinatal pulmonary vasodilatation. Because compromised NO production plays a role in PPHN, ROK inhibition might provide an additional therapeutic tool. ROK inhibition increases PAEC NO production and barrier function of endothelial cell monolayers and restores growth and tube formation of PAECs from experimental PPHN in vitro.139,167 These findings suggest that, in addition to its role in SMCs, ROK activity further modulates endothelial cell growth and functions in the normal developing lung and PPHN.
The consequences of an attenuated response of endothelial cells to an acute increase in oxygen tension are significant as blood shunts away from the lungs, resulting in severe central hypoxemia that is unresponsive to high concentrations of inspired oxygen.168 Recent data provide clear evidence that inadequate production of NO and relatively increased production of endothelin play an etiologic role in PPHN.169 The observation that endothelial cells from an animal model of PPHN demonstrate no increase in either [Ca2+]i or only a minimal increase in NO production in response to an acute increase in oxygen tension provides the first direct evidence that chronic intrauterine pulmonary hypertension compromises endothelial cell oxygen sensing and NO production.138 Moreover, the observation that acetylcholine caused a greater increase in cytosolic calcium in PAECs from hypoxic than in those from normoxic lambs suggests that the normally low oxygen tension environment of the developing pulmonary circulation prepares the lung for extrauterine, air-breathing life, providing a mechanistic link between endothelial cell cytosolic calcium and an acute increase in oxygen tension, one of the central perinatal physiologic stimuli at birth.118
The observation that fetal PAECs maintained in culture respond to an acute increase in oxygen tension with an increase in cytosolic calcium, in direct contrast to the response of PAECs derived from the adult pulmonary vasculature,170,171 lends weight to the proposition that maturation-related differences in the pulmonary circulation extend not only to the molecular expression but also to the physiology of individual cellular constituents. Further, the observation that PAECs derived from fetal lambs with chronic intrauterine pulmonary hypertension do not respond to an acute increase in oxygen tension with either an increase in cytosolic calcium or increased NO production138 provides still further evidence of a link, albeit an inferential one, between an acute increase in oxygen tension and NO production in fetal PAECs.
Thus, in the perinatal pulmonary circulation, PAECs are able to sense and respond to an acute increase in oxygen tension with an increase in cytosolic calcium. The acute increase in oxygen tension leads to membrane depolarization and an increase in the rate of extracellular calcium entry. Augmented entry of extracellular calcium leads to calcium release from inositol triphosphate (IP3)–sensitive intracellular calcium stores. The sustained elevation of PAEC calcium results, in part, from release of calcium from ryanodine-sensitive stores. From a teleologic perspective, the response of the PAECs to an acute increase in oxygen tension is a biologic imperative. Compromised PAEC oxygen sensing may underlie the attenuated response of the pulmonary circulation to perinatal vasodilator stimuli, thereby leading to PPHN.
CYCLIC GMP signaling
NO causes pulmonary vasodilatation, primarily via activation of soluble guanylate cyclase (sGC). NO binds to the heme moiety of sGC, thereby inducing a conformational change that increases cGMP production from GTP by approximately 200-fold.172,173 In pulmonary arteries of piglets, the molecular expression of the sGC β1 subunit increases with age in a manner that parallels the increasing response to NO.174 Moreover, pulmonary sGC activity stimulated by sodium nitroprusside, a NO donor, is approximately 7-fold greater in 1- and 8-day-old rats than in adult rats.175
PKG (cGMP-dependent protein kinase) is the primary enzyme responsible for vasodilatation caused by NO-cGMP activity. In vascular SMCs, relaxation is contingent on activation of the type I isoform (PKG-I).176,177 Calcium-sensitive potassium (BKCa) channels are an important phosphorylation target of PKG,178,179 and the activation of PKG by NO-cGMP signaling seems to be sufficient to activate BKCa channels.180 Reduced PKG-I expression may contribute to the development of nitrate tolerance, a condition wherein the vascular sensitivity to nitrovasodilators is diminished.181,182 The capacity to induce cGMP-mediated relaxation in pulmonary arteries increases with maturation.183,184
In response to sustained exposure to normoxia, the expression and activity of PKG that favor diminished tone in PASMCs normally increase.185 Experimental evidence indicates that PKG has three distinct effects that mediate diminished SMC tone. First, receptor phosphorylation by PKG enables ryanodine-sensitive calcium stores to produce a local and quantal release of calcium, resulting in BKCa channel opening, membrane hyperpolarization, and subsequent closure of calcium channels to decrease cytosolic calcium.178,186 Second, PKG directly activates the BKCa channel through phosphorylation at serine 1072 of the alpha chain.187,188 Third, PKG may inhibit the voltage-operated calcium channel (VOCC) by direct phosphorylation of the channel or by PKG-induced activation of a phosphatase.189 In addition to the effects on PKG-I expression, sustained normoxia decreases expression of PASMC BKCa and VOCCs.190
Evidence from studies in fetal and newborn ovine pulmonary vessels indicates that cGMP-mediated relaxation is primarily mediated by PKG in the perinatal period.191-193 Furthermore, PKG activity is regulated by oxygen tension. Relaxation of pulmonary arteries and veins of term fetal lambs to 8-Br-cGMP, a cell-permeable cGMP analog, is greater after exposure for 4 hours to normoxia (partial pressure of oxygen [Po2]: 140 mmHg) than after exposure to hypoxia (Po2: 30 mmHg). The decreased relaxation of pulmonary veins to cGMP in hypoxia may result from reduced expression of PKG protein and mRNA as well as posttranscriptional modification of PKG by peroxynitrite and other reactive oxygen species, specifically in veins.193,194 Acute hypoxia also induces an accumulation of ubiquitinated PKG-I in ovine fetal and newborn PASMCs. Such a modification was not evident in ovine fetal systemic (cerebral) artery SMCs. Ubiquitinated PKG-Iα was unable to bind the cGMP analog 8-(2-aminoethyl)thioguanosine-3ʹ,5ʹ (AET)-cGMP, a ligand for the unmodified protein, and reoxygenation reversed the acute hypoxia-induced accumulation of ubiquitinated PKG-I. PKG-I ubiquitination induced by acute hypoxia appears to play a unique role in the regulation of the pulmonary vascular SMC vasoreactivity and relaxation mediated by the NO-cGMP-PKG-I pathway.195
Role of the calcium-sensitive K+ channel in the perinatal lung
While physical factors and vasoactive products elaborated by the pulmonary vascular endothelium are involved in the regulation of perinatal pulmonary vascular tone,3,131 sustained and progressive perinatal pulmonary vasodilatation requires pulmonary vascular K+ channel activation. PASMC K+ activation causes membrane hyperpolarization, closure of VOCCs, a decrease in cytosolic calcium, and vasodilatation.196 Data demonstrate that activation of BKCa channels in the pulmonary vasculature plays a critical role in perinatal pulmonary vasodilatation. The first definitive evidence that BKCa channel activation mediates O2-induced fetal pulmonary vasodilatation came from studies in acutely instrumented late-gestation ovine fetuses.197 O2-induced fetal pulmonary vasodilatation was blocked by either tetraethylammonium (TEA), a K+ channel blocker, or iberiotoxin, a specific BKCa channel antagonist, but was unaffected by glibenclamide, a blocker of the KATP channel, suggesting that O2 causes fetal pulmonary vasodilatation through BKCa channel activation.196,197 Inhibitors of either guanylate cyclase– or cyclic nucleotide–dependent kinases also attenuated O2-dependent fetal pulmonary vasodilation, implying that elevated fetal O2 acts to increase guanylate cyclase activity and cGMP concentration and to activate cyclic nucleotide–dependent kinases, causing BKCa channel activation and vasodilatation.197 Vasodilatation of perinatal pulmonary arteries caused by ventilation was inhibited by TEA.198 This observation implies that ventilation causes sustained and progressive perinatal pulmonary vasodilatation through activation of TEA-sensitive K+ channels. NO causes perinatal pulmonary vasodilatation, in part, through activation of the BKCa channel.199 Even shear stress–induced fetal pulmonary vasodilatation, induced by compression of the DA during fetal life, requires BKCa and voltage-dependent K+ channel activation.125
Ontogeny of the pulmonary vascular K+ channel
While the BKCa channel is crucially important in the perinatal pulmonary circulation, its importance seems to decrease with maturation. The observation that the K+ channel setting resting membrane potential (RMP) in the pulmonary circulation changes after birth from a BKCa to a voltage-gated K+ (Kv) channel suggests developmental regulation of K+ channels in the pulmonary circulation.200 In coordination with a maturation-related decrease in BKCa channel expression and activation, the Kv channel expression increases with maturation. There is relatively more Kv2.1 channel protein and message in the adult than in the fetal and neonatal pulmonary circulation. The increase in the Kv2.1 channel parallels the increasing capacity of PASMCs to sense and respond to acute decreases in oxygen tension as, in response to acute hypoxia, [Ca2+]i increases more rapidly and to a greater degree in adult than in fetal PASMCs.201 The relatively greater abundance of the Kv2.1 channel in the adult may represent an adaptation that allows the pulmonary circulation to respond to a specific physiologic signal that is relevant to a particular developmental stage. Reports that the O2 sensor in the adult PASMC is a Kv channel are consistent with this view,202 since the Kv channel is inactivated by acute hypoxia, causing PASMC depolarization, opening of VOCCs, an increase in [Ca2+]i, and vasoconstriction. Thus, the maturation-related increase in hypoxic pulmonary vasoconstriction that has been previously reported might derive from the parallel increases in Kv2.1 channel activity, protein, and message with maturation.203,204
In contrast to pulmonary vascular Kv channel expression and activity, pulmonary vascular BKCa activity and expression decrease with maturation. Data from both pulmonary arteries and PASMCs in primary culture indicate that BKCa channel expression is greatest in the fetal lung and decreases with maturation.205 Indeed, the physiology of pulmonary vascular SMCs is consistent with the molecular biology. In fetal PASMCs, RMP is determined by the BKCa channel, while in adult PASMCs, Kv channel activity determines RMP.200 In fetal PASMCs, an acute increase in oxygen tension results in membrane hyperpolarization and a decrease in PASMC [Ca2+]i and has no effect on adult pulmonary arterial membrane potential or cytosolic calcium.205 Taken together, these observations indicate that the developmental regulation of BKCa channel expression allows for the fetal PASMCs to be uniquely well adapted to respond to an acute increase in oxygen tension with a decrease in [Ca2+]i and vasorelaxation.
Hypoxia-inducible factor-1 (HIF-1) modulates BKCa channel expression
HIF-1, a key transcription factor that is central to oxygen homeostasis, enables the cell to respond to changes in oxygen availability, an essential response in many developmental, physiological, and pathological processes. Semenza206 and others have shown that HIF-1 regulates the transcription of many genes involved in the cellular and systemic response to oxygen availability, including genes involved in angiogenesis (i.e., VEGF), oxygen transport (i.e., erythropoietin), and energy metabolism (i.e., glycolytic enzymes). HIF-1 is also a key player in pathophysiological processes such as cancer, because hypoxic microenvironments in a tumor trigger expression of angiogenic genes that promote the growth of the newly vascularized tumor.206,207
From a developmental perspective, the transition from the relatively low-oxygen-tension intrauterine environment to air-breathing life entails an unprecedented and absolutely required response to an increase in oxygen availability. The physiologically low-O2 environment of the fetus might be essential for fetal vascular growth and lung morphogenesis206 as well as other early embryo developmental programs.208 Because HIF-1 is a master regulator of the oxygen response, it follows that the molecular mechanisms responsible for fetal life and its transition to newborn life might involve it. Under normoxic conditions, the α chain of HIF-1 (HIF-1α) is eliminated by degradation, rendering HIF-1 inactive. In low-O2 environments, HIF-1 forms a dimer, is stabilized, and can translocate to the cell nucleus, thereby promoting transcription of hypoxia-sensitive genes.209
Deferoxamine mesylate (DFX) is an iron chelator that prevents HIF-1α degradation. Normoxic fetal PASMCs treated with DFX increased expression of the α subunit of the BKCa channel, implying that hypoxia may upregulate BKCa channel expression via HIF-1.210 In human PASMCs, knockdown of HIF-1α, a master transcriptional factor for the hypoxic response, using a short hairpin RNA plasmid blocked the hypoxic induction of BKCa β1-subunit expression.211 Considering that HIF-1α is more stable and less prone to destruction in fetal and neonatal PASMCs than in adult PASMCs,212 it is possible that the greater activity of BKCa channels in perinatal pulmonary vasculature could be partially ascribed to a high level of HIF-1α. In contrast to BKCa channels, the expression of Kv channels is downregulated by HIF-1α in PASMCs of the rat and mice. This effect is indirect, resulting from HIF-1-mediated upregulation of ET-1 and the subsequent ET-1-mediated suppression of its expression (Fig. 3).213 Changes in oxygen tension at birth might signal an important transcriptional adaptation of the fetus to the air-breathing life of the newborn infant. Resnik et al.212 investigated the role of HIF-1 in that transition and found that expression of HIF-1α is developmentally regulated, as is the expression of the prolyl and asparagyl hydroxylases and other competing factors that control HIF-1α expression and activity. The well-coordinated developmental regulation of the components of the HIF-1 pathway in the fetus suggests that the fetus represents a particular transcriptional paradigm that, in response to differential oxygen availability, produces a specific physiological and developmental response.
Figure 3.
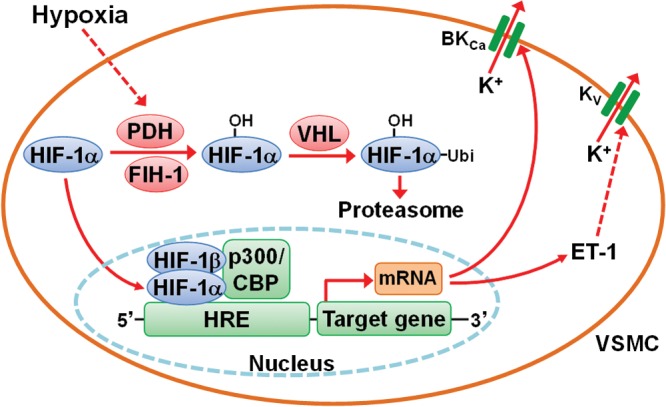
Role of hypoxia-inducible factor-1 (HIF-1) on the expression of Ca2+-activated K+ (BKCa) and voltage gated K+ (Kv) channels in pulmonary arterial smooth muscle cells (PASMCs). In mammalian cells, including PASMCs, prolyl hydroxylase domain–containing enzyme (PHD) and factor inhibiting HIF-1 (FIH-1) act as cellular oxygen sensors and control the abundance of the α isoform of HIF-1 (HIF-1α). Under normoxia, HIF-1α is hydroxylated by PHD and FIH-1 and ubiquitinated by the von Hippel–Lindau protein (VHL), resulting in proteasomal degradation. The activities of PHD and FIH-1 depend on the presence of oxygen, and hence these enzymes are inhibited under hypoxic conditions, leading to reduced degradation of HIF-1α. Consequently, HIF-1α is translocated into the nucleus, dimerizes with the β isoform of HIF-1 (HIF-1β), forms a complex with the transcriptional coactivator p300/CBP and binds to hypoxia-responsive elements (HRE) of HIF target genes to induce the expression of BKCa. HIF also promotes the expression of ET-1, while increased ET-1 suppresses the expression of Kv channels. Solid arrows indicate activation and dashed arrows inhibition. CBP: cAMP-response element-binding protein (CREB)–binding protein; ET-1: endothelin 1; Ubi: ubiquitin.
Platelet-activating factor (PAF)
PAF, a molecule that modulates vascular tone, was originally described as a soluble factor involved in leukocyte-dependent histamine and serotonin release from platelets.214 In 1972, Benveniste et al.215 demonstrated that this soluble factor was also released from rabbit basophils after immunoglobulin E stimulation and coined the term “PAF.” Multiple cell types release PAF in response to inflammatory stimuli such as angiotensin II, thrombin, histamine, leukotrienes, or tumor necrosis factor α.216 Most cells that produce PAF also have PAF receptors and are, therefore, PAF targets.217 PAF acts via specific receptors on responsive cells.218 PAF receptor binding leads to activation of phospholipase C and consequently increased production of IP3 and diacylglycerol. The elevated IP3 stimulates Ca2+ release from the intracellular calcium stores while the increased diacylglycerol level causes PKC activation and enhanced sensitivity of myofilaments to Ca2+. These events evoke or augment vasoconstriction.219
The hemodynamic effects of PAF are contingent on both anatomical site and developmental stage. In fetal lambs, PAF receptor blockade reduces PVR more than systemic vascular resistance, suggesting that PAF plays a role in the maintenance of the high-tone fetal pulmonary vasculature.220 PAF levels are higher in fetal than in adult lung and plasma.221 Moreover, hypoxia augments PAF receptor expression and PAF levels decrease after birth.222 Hypoxia serves to potentiate the release of calcium from IP3-sensitive stores.223
Within 90 minutes after birth, plasma PAF levels decrease in newborn lambs, perhaps because of upregulation of PAF-acetylhydrolase (PAF-Ah) activity and mRNA expression.221 PAF-Ah inactivates PAF and thus reduces its vasoconstrictive action. In PASMCs and in the whole lung, PAF-Ah activity is augmented by oxygenation.221 Elevated cGMP and cAMP levels resulting from oxygen-induced release of EDNO and PGI2 may further decrease PAF receptor binding and PAF-stimulated IP3 production in a PKG-dependent manner.224 These developmental changes likely facilitate the postnatal adaptation of the pulmonary circulation.
Role of veins in regulation of pulmonary circulation
In the past, pulmonary veins have been viewed as conduit vessels similar to those in the systemic circulation; however, over the past 2 decades, a large amount of evidence has accumulated to indicate that pulmonary veins can exhibit substantial vasoactivity and contribute to the regulation of total pulmonary vasomotor tone and resistance.3,225 In the fetus, pulmonary veins contribute a significant fraction to total PVR. Studies show that the veins may play a more important role in modulating the pulmonary circulation in the fetus and the newborn than in the adult.226-229 It is not clear why, teleologically, there is a need for an increased venous resistance and venous reactivity in the lungs during the perinatal period; however, venous reactivity does appear to be at a maximum in the fetal lung. In perinatal sheep, not only acetylcholine (an EDNO-dependent vasodilator) but also exogenous NO causes a greater increase in the intracellular content of cGMP and relaxation in pulmonary veins than in arteries.3 At birth, the veins, as well as the arteries, relax in response to EDNO and dilator prostaglandins, thereby assisting in the fall in PVR. These effects are oxygen dependent and modulated by PKG. In a number of species, including the human, pulmonary veins are also the primary sites of action of certain vasoconstrictors, such as endothelin230 and thromboxane.231 In various pathological conditions, there is an increased synthesis of these vasoactive agents that may lead to pulmonary venous constriction, increased microvascular pressures for fluid filtration, and formation of pulmonary edema. The significant role of veins in regulation of the pulmonary circulation must be appreciated to better prevent, diagnose, and treat lung disease.
Ductus arteriosus (DA)
Closure of the DA at birth is a necessary step for the normal transition from fetal to air-breathing life.232 Previous studies have demonstrated that an acute increase in Po2 causes an increase in tension of DA rings. In rabbit DA SMCs, acute normoxia inhibits the K+ current, leading to membrane depolarization.233-235 Pharmacologic blockade of the Kv channels mimics the effect of acute normoxia. Patent DA is a common complication of extreme prematurity. Preterm rabbit DAs display reduced O2 constriction. This is associated with decreased O2-sensitive K+ current and decreased expression of Kv channels.236 Accordingly, overexpression of the O2-sensitive K+ channels Kv1.5 or Kv2.1 via gene transfer confers O2 responsiveness to preterm rabbit DAs. Consistent with such data, 4-aminopyridine mimics an acute increase in oxygen tension in DA SMCs.237 Moreover, preventing extracellular Ca2+ entry accentuates the initial increase in [Ca2+]i in DA SMCs exposed to acute normoxia.
An acute increase in oxygen tension directly increases [Ca2+]i in a subconfluent monolayer of DA SMCs. Pharmacologic inactivation of voltage-sensitive, but not ATP- or calcium-sensitive, K+ channels mimics the effect of an acute increase in oxygen tension, providing support for the proposition that Kv channel inactivation mediates the O2-induced increase in DA SMC [Ca2+]i.237 Alternative reports demonstrate that an acute increase in oxygen tension initially decreases the rate of calcium entry into DA SMCs.238 Data demonstrate that entry of extracellular calcium is not necessary for the initial increase in DA SMC [Ca2+]I. Rather, release of calcium from IP3-sensitive stores accounts for the initial increase in DA SMC [Ca2+]i. The sustained and progressive increase in DA SMC [Ca2+]i entails subsequent extracellular calcium entry.237 The initial release of Ca2+ from intracellular stores is necessary to trigger the second phase of extracellular Ca2+ entry, in a process termed calcium-induced calcium entry. The observation that blockade of calcium release from ryanodine-sensitive stores has no effect on the normoxia-induced increase in DA SMCs offers further support for the notion that release of calcium from an IP3-sensitive store plays a pivotal role in the normoxia-induced increase in DA SMC [Ca2+]i.238 Thus, pathological patency of the DA may result from defects in oxygen sensing.
Concluding remarks
This review provides information surrounding the complexity of the developmental regulation of pulmonary vascular growth, structure, and physiology. Both the vascular endothelium and the SMCs are adapted to modulate vascular growth and tone in the low-oxygen-tension environment of the developing lung. Cellular constituents of the pulmonary circulation possess developmentally regulated properties that ensure well-coordinated cell proliferation and growth in the fetal and neonatal pulmonary circulation, in conjunction with the distal airspace. Maturation of molecular mechanisms underlie the ability of the lung circulation to adapt to postnatal life with an exponential increase in pulmonary blood flow at birth. Should the pulmonary circulation be subject to stress or injury during fetal life, alterations in the production of critical growth factors, vasoactive molecules, or ion channel expression or increases in cell and matrix proliferation might ensue, resulting in the failure of transition at birth due to abnormal pulmonary vascular growth, structural remodeling, and vascular tone and vasoreactivity. Finally, we speculate that insights into mechanisms that preserve endothelial maturation and function may further enhance lung development and long-term outcomes in diverse forms of neonatal pulmonary hypertension associated with preterm birth, developmental lung diseases, and congenital heart disease.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Roth-Kleiner M, Post M. Similarities and dissimilarities of branching and septation during lung development. Pediatr Pulmonol 2005;40(2):113–134. [DOI] [PubMed]
- 2.Herriges M, Morrisey EE. Lung development: orchestrating the generation and regeneration of a complex organ. Development 2014;141(3):502–513. [DOI] [PMC free article] [PubMed]
- 3.Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev 2010;90(4):1291–1335. [DOI] [PubMed]
- 4.Metzger RJ, Klein OD, Martin GR, Krasnow MA. The branching programme of mouse lung development. Nature 2008;453(7196):745–750. [DOI] [PMC free article] [PubMed]
- 5.Pereda J, Sulz L, San Martin S, Godoy-Guzmán C. The human lung during the embryonic period: vasculogenesis and primitive erythroblasts circulation. J Anat 2013;222(5):487–494. [DOI] [PMC free article] [PubMed]
- 6.Hall SM, Hislop AA, Haworth SG. Origin, differentiation, and maturation of human pulmonary veins. Am J Respir Cell Mol Biol 2002;26(3):333–340. [DOI] [PubMed]
- 7.Schachtner SK, Wang Y, Baldwin HS. Qualitative and quantitative analysis of embryonic pulmonary vessel formation. Am J Respir Cell Mol Biol 2000;22(2):157–165. [DOI] [PubMed]
- 8.Abman SH, Baker C, Gien J, Mourani P, Galambos C. The Robyn Barst Memorial Lecture: Differences between the fetal, newborn, and adult pulmonary circulations: relevance for age-specific therapies (2013 Grover Conference series). Pulm Circ 2014;4(3):424–440. [DOI] [PMC free article] [PubMed]
- 9.Peng T, Morrisey EE. Development of the pulmonary vasculature: current understanding and concepts for the future. Pulm Circ 2013;3(1):176–178. [DOI] [PMC free article] [PubMed]
- 10.deMello DE, Sawyer D, Galvin N, Reid LM. Early fetal development of lung vasculature. Am J Respir Cell Mol Biol 1997;16(5):568–581. [DOI] [PubMed]
- 11.Maeda S, Suzuki S, Suzuki T, Endo M, Moriya T, Chida M, Kondo T, Sasano H. Analysis of intrapulmonary vessels and epithelial-endothelial interactions in the human developing lung. Lab Investig 2002;82(3):293–301. [DOI] [PubMed]
- 12.Papamatheakis DG, Blood AB, Kim JH, Wilson SM. Antenatal hypoxia and pulmonary vascular function and remodeling. Curr Vasc Pharmacol 2013;11(5):616–640. [DOI] [PMC free article] [PubMed]
- 13.Hall SM, Hislop AA, Pierce CM, Haworth SG. Prenatal origins of human intrapulmonary arteries: formation and smooth muscle maturation. Am J Respir Cell Mol Biol 2000;23(2):194–203. [DOI] [PubMed]
- 14.Anderson-Berry A, O’Brien EA, Bleyl SB, Lawson A, Gundersen N, Ryssman D, Sweeley J, et al. Vasculogenesis drives pulmonary vascular growth in the developing chick embryo. Dev Dyn 2005;233(1):145–153. [DOI] [PubMed]
- 15.Parera MC, van Dooren M, van Kempen M, de Krijger R, Grosveld F, Tibboel D, Rottier R. Distal angiogenesis: a new concept for lung vascular morphogenesis. Am J Physiol Lung Cell Mol Physiol 2005;288(1):L141–L149. [DOI] [PubMed]
- 16.Schwarz MA, Caldwell L, Cafasso D, Zheng H. Emerging pulmonary vasculature lacks fate specification. Am J Physiol Lung Cell Mol Physiol 2009;296(1):L71–L81. [DOI] [PMC free article] [PubMed]
- 17.Cornett B, Snowball J, Varisco BM, Lang R, Whitsett J, Sinner D. Wntless is required for peripheral lung differentiation and pulmonary vascular development. Dev Biol 2013;379(1):38–52. [DOI] [PMC free article] [PubMed]
- 18.Jiang M, Ku WY, Fu J, Offermanns S, Hsu W, Que J. Gpr177 regulates pulmonary vasculature development. Development 2013;140(17):3589–3594. [DOI] [PMC free article] [PubMed]
- 19.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 2000;279(3):L600–L607. [DOI] [PubMed]
- 20.Tang JR, Karumanchi SA, Seedorf G, Markham N, Abman SH. Excess soluble vascular endothelial growth factor receptor-1 in amniotic fluid impairs lung growth in rats: linking preeclampsia with bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2012;302(1):L36–L46. [DOI] [PMC free article] [PubMed]
- 21.Chao CM, El Agha E, Tiozzo C, Minoo P, Bellusci S. A breath of fresh air on the mesenchyme: impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia. Front Med 2015;2:27. doi:10.3389/fmed.2015.00027. [DOI] [PMC free article] [PubMed]
- 22.De Spiegelaere W, Casteleyn C, Van den Broeck W, Plendl J, Bahramsoltani M, Simoens P, Djonov V, Cornillie P. Intussusceptive angiogenesis: a biologically relevant form of angiogenesis. J Vasc Res 2012;49(5):390–404. [DOI] [PubMed]
- 23.Ackermann M, Houdek JP, Gibney BC, Ysasi A, Wagner W, Belle J, Schittny JC, et al. Sprouting and intussusceptive angiogenesis in postpneumonectomy lung growth: mechanisms of alveolar neovascularization. Angiogenesis 2014;17(3):541–551. [DOI] [PMC free article] [PubMed]
- 24.Nagy JA, Dvorak AM, Dvorak HF. Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harbor Perspect Med 2012;2(2):a006544. doi:10.1101/cshperspect.a006544. [DOI] [PMC free article] [PubMed]
- 25.Voelkel NF, Gomez-Arroyo J. The role of vascular endothelial growth factor in pulmonary arterial hypertension: the angiogenesis paradox. Am J Respir Cell Mol Biol 2014;51(4):474–484. [DOI] [PubMed]
- 26.Yun EJ, Lorizio W, Seedorf G, Abman SH, Vu TH. VEGF and endothelium-derived retinoic acid regulate lung vascular and alveolar development. Am J Physiol Lung Cell Mol Physiol 2016;310(4):L287–L298. [DOI] [PMC free article] [PubMed]
- 27.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996;380(6573):439–442. [DOI] [PubMed]
- 28.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996;380(6573):435–439. [DOI] [PubMed]
- 29.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995;376(6535):66–70. [DOI] [PubMed]
- 30.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995;376(6535):62–66. [DOI] [PubMed]
- 31.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development 1999;126(6):1149–1159. [DOI] [PubMed]
- 32.Healy AM, Morgenthau L, Zhu X, Farber HW, Cardoso WV. VEGF is deposited in the subepithelial matrix at the leading edge of branching airways and stimulates neovascularization in the murine embryonic lung. Dev Dyn 2000;219(3):341–352. [DOI] [PubMed]
- 33.Gebb SA, Shannon JM. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev Dyn 2000;217(2):159–169. [DOI] [PubMed]
- 34.Kalinichenko VV, Lim L, Stolz DB, Shin B, Rausa FM, Clark J, Whitsett JA, Watkins SC, Costa RH. Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous null for the Forkhead Box f1 transcription factor. Dev Biol 2001;235(2):489–506. [DOI] [PubMed]
- 35.Ng YS, Rohan R, Sunday ME, deMello DE, D’Amore PA. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn 2001;220(2):112–121. [DOI] [PubMed]
- 36.Galambos C, Ng YS, Ali A, Noguchi A, Lovejoy S, D’Amore PA, deMello DE. Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am J Respir Cell Mol Biol 2002;27(2):194–203. [DOI] [PubMed]
- 37.Le Cras TD, Spitzmiller RE, Albertine KH, Greenberg JM, Whitsett JA, Akeson AL. VEGF causes pulmonary hemorrhage, hemosiderosis, and air space enlargement in neonatal mice. Am J Physiol Lung Cell Mol Physiol 2004;287(1):L134–L142. [DOI] [PubMed]
- 38.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation 2005;112(16):2477–2486. [DOI] [PubMed]
- 39.Zhao L, Wang K, Ferrara N, Vu TH. Vascular endothelial growth factor co-ordinates proper development of lung epithelium and vasculature. Mech Dev 2005;122(7–8):877–886. [DOI] [PubMed]
- 40.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Investig 2000;106(11):1311–1319. [DOI] [PMC free article] [PubMed]
- 41.Akeson AL, Cameron JE, Le Cras TD, Whitsett JA, Greenberg JM. Vascular endothelial growth factor-A induces prenatal neovascularization and alters bronchial development in mice. Pediatr Res 2005;57(1):82–88. [DOI] [PubMed]
- 42.Yamamoto H, Yun EJ, Gerber HP, Ferrara N, Whitsett JA, Vu TH. Epithelial-vascular cross talk mediated by VEGF-A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev Biol 2007;308(1):44–53. [DOI] [PubMed]
- 43.Han RN, Babaei S, Robb M, Lee T, Ridsdale R, Ackerley C, Post M, Stewart DJ. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ Res 2004;94(8):1115–1123. [DOI] [PubMed]
- 44.Balasubramaniam V, Tang JR, Maxey A, Plopper CG, Abman SH. Mild hypoxia impairs alveolarization in the endothelial nitric oxide synthase (eNOS) deficient mouse. Am J Physiol Lung Cell Mol Physiol 2003;284(6):L964–L971. [DOI] [PubMed]
- 45.Balasubramaniam V, Maxey AM, Morgan DB, Markham NE, Abman SH. Inhaled NO restores lung structure in eNOS-deficient mice recovering from neonatal hypoxia. Am J Physiol Lung Cell Mol Physiol 2006;291(1):L119–L127. [DOI] [PubMed]
- 46.Bachiller PR, Cornog KH, Kato R, Buys ES, Roberts JD Jr. Soluble guanylate cyclase modulates alveolarization in the newborn lung. Am J Physiol Lung Cell Mol Physiol 2013;305(8):L569–L581. [DOI] [PMC free article] [PubMed]
- 47.Seedorf G, Metoxen AJ, Rock R, Markham N, Ryan S, Vu T, Abman SH. Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1098–L1110. [DOI] [PMC free article] [PubMed]
- 48.Iosef C, Alastalo TP, Hou Y, Chen C, Adams ES, Lyu SC, Cornfield DN, Alvira CM. Inhibiting NF-κB in the developing lung disrupts angiogenesis and alveolarization. Am J Physiol Lung Cell Mol Physiol 2012;302(10):L1023–L1036. [DOI] [PMC free article] [PubMed]
- 49.Ren X, Ustiyan V, Pradhan A, Cai Y, Havrilak JA, Bolte CS, Shannon JM, Kalin TV, Kalinichenko VV. FOXF1 transcription factor is required for formation of embryonic vasculature by regulating VEGF signaling in endothelial cells. Circ Res 2014;115(8):709–720. [DOI] [PMC free article] [PubMed]
- 50.Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 2009;84(6):780–791. [DOI] [PMC free article] [PubMed]
- 51.Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harbor Perspect Biol 2013;5(9):a009159. doi:10.1101/cshperspect.a009159. [DOI] [PMC free article] [PubMed]
- 52.Barquilla A, Pasquale EB. Eph receptors and ephrins: therapeutic opportunities. Annu Rev Pharmacol Toxicol 2015;55:465–487. [DOI] [PMC free article] [PubMed]
- 53.Kume T. Specification of arterial, venous, and lymphatic endothelial cells during embryonic development. Histol Histopathol 2010;25(5):637–646. [DOI] [PMC free article] [PubMed]
- 54.Bautch VL, Caron KM. Blood and lymphatic vessel formation. Cold Spring Harbor Perspect Biol 2015;7(3):a008268. doi:10.1101/cshperspect.a008268. [DOI] [PMC free article] [PubMed]
- 55.Gale NW, Baluk P, Pan L, Kwan M, Holash J, DeChiara TM, McDonald DM, Yancopoulos GD. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev Biol 2001;230(2):151–160. [DOI] [PubMed]
- 56.Vadivel A, van Haaften T, Alphonse RS, Rey-Parra GJ, Ionescu L, Haromy A, Eaton F, Michelakis E, Thébaud B. Critical role of the axonal guidance cue ephrinB2 in lung growth, angiogenesis, and repair. Am J Respir Crit Care Med 2012;185(5):564–574. [DOI] [PubMed]
- 57.Wilkinson GA, Schittny JC, Reinhardt DP, Klein R. Role for ephrinB2 in postnatal lung alveolar development and elastic matrix integrity. Dev Dyn 2008;237(8):2220–2234. [DOI] [PubMed]
- 58.Prodhan P, Kinane TB. Developmental paradigms in terminal lung development. Bioessays 2002;24(11):1052–1059. [DOI] [PubMed]
- 59.Vadivel A, Alphonse RS, Collins JJ, van Haaften T, O’Reilly M, Eaton F, Thébaud B. The axonal guidance cue semaphorin 3C contributes to alveolar growth and repair. PLoS ONE 2013;8(6):e67225. doi:10.1371/journal.pone.0067225. [DOI] [PMC free article] [PubMed]
- 60.Becker PM, Tran TS, Delannoy MJ, He C, Shannon JM, McGrath-Morrow S. Semaphorin 3A contributes to distal pulmonary epithelial cell differentiation and lung morphogenesis. PLoS ONE 2011;6(11):e27449. doi:10.1371/journal.pone.0027449. [DOI] [PMC free article] [PubMed]
- 61.Joza S, Wang J, Fox E, Hillman V, Ackerley C, Post M. Loss of semaphorin-neuropilin-1 signaling causes dysmorphic vascularization reminiscent of alveolar capillary dysplasia. Am J Pathol 2012;181(6):2003–2017. [DOI] [PubMed]
- 62.Joza S, Wang J, Tseu I, Ackerley C, Post M. Fetal, but not postnatal, deletion of semaphorin-neuropilin-1 signaling affects murine alveolar development. Am J Respir Cell Mol Biol 2013;49(4):627–636. [DOI] [PubMed]
- 63.DeLisser HM, Helmke BP, Cao G, Egan PM, Taichman D, Fehrenbach M, Zaman A, et al. Loss of PECAM-1 function impairs alveolarization. J Biol Chem 2006;281(13):8724–8731. [DOI] [PubMed]
- 64.Park S, Sorenson CM, Sheibani N. PECAM-1 isoforms, eNOS and endoglin axis in regulation of angiogenesis. Clin Sci 2015;129(3):217–234. [DOI] [PMC free article] [PubMed]
- 65.Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 2011;147(3):539–553. [DOI] [PMC free article] [PubMed]
- 66.Rafii S, Cao Z, Lis R, Siempos II, Chavez D, Shido K, Rabbany SY, Ding BS. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol 2015;17(2):123–136. [DOI] [PMC free article] [PubMed]
- 67.Schwartz SM, Benditt EP. Clustering of replicating cells in aortic endothelium. Proc Natl Acad Sci USA 1976;73(2):651–653. [DOI] [PMC free article] [PubMed]
- 68.Solodushko V, Fouty B. Proproliferative phenotype of pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 2007;292(3):L671–L677. [DOI] [PubMed]
- 69.Alvarez DF, Huang L, King JA, ElZarrad MK, Yoder MC, Stevens T. Lung microvascular endothelium is enriched with progenitor cells that exhibit vasculogenic capacity. Am J Physiol Lung Cell Mol Physiol 2008;294(3):L419–L430. [DOI] [PubMed]
- 70.Clark J, Alvarez DF, Alexeyev M, King JA, Huang L, Yoder MC, Stevens T. Regulatory role for nucleosome assembly protein-1 in the proliferative and vasculogenic phenotype of pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol 2008;294(3):L431–L439. [DOI] [PubMed]
- 71.Alphonse RS, Vadivel A, Fung M, Shelley WC, Critser PJ, Ionescu L, O’Reilly M, et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 2014;129(21):2144–2157. [DOI] [PMC free article] [PubMed]
- 72.Baker CD, Seedorf GJ, Wisniewski BL, Black CP, Ryan SL, Balasubramaniam V, Abman SH. Endothelial colony-forming cell conditioned media promote angiogenesis in vitro and prevent pulmonary hypertension in experimental bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2013;305(1):L73–L81. [DOI] [PMC free article] [PubMed]
- 73.Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of endothelium. Lab Investig 1983;49(3):327–333. [PubMed]
- 74.Cook CL, Weiser MC, Schwartz PE, Jones CL, Majack RA. Developmentally timed expression of an embryonic growth phenotype in vascular smooth muscle cells. Circ Res 1994;74(2):189–196. [DOI] [PubMed]
- 75.Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp 2007;283:174–191. [DOI] [PubMed]
- 76.Weiser-Evans MC, Quinn BE, Burkard MR, Stenmark KR. Transient reexpression of an embryonic autonomous growth phenotype by adult carotid artery smooth muscle cells after vascular injury. J Cell Physiol 2000;182(1):12–23. [DOI] [PubMed]
- 77.Seifert RA, Schwartz SM, Bowen-Pope DF. Developmentally regulated production of platelet-derived growth factor-like molecules. Nature 1984;311(5987):669–671. [DOI] [PubMed]
- 78.Mourani PM, Garl PJ, Wenzlau JM, Carpenter TC, Stenmark KR, Weiser-Evans MC. Unique, highly proliferative growth phenotype expressed by embryonic and neointimal smooth muscle cells is driven by constitutive Akt, mTOR, and p70S6K signaling and is actively repressed by PTEN. Circulation 2004;109(10):1299–1306. [DOI] [PubMed]
- 79.Glukhova MA, Frid MG, Koteliansky VE. Developmental changes in expression of contractile and cytoskeletal proteins in human aortic smooth muscle. J Biol Chem 1990;265(22):13042–13046. [PubMed]
- 80.Owens GK, Thompson MM. Developmental changes in isoactin expression in rat aortic smooth muscle cells in vivo: relationship between growth and cytodifferentiation. J Biol Chem 1986;261(28):13373–13380. [PubMed]
- 81.Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles: dependence on tissue type. J Biol Chem 1984;259(23):14383–14388. [PubMed]
- 82.Gabbiani G, Kocher O, Bloom WS, Vandekerckhove J, Weber K. Actin expression in smooth muscle cells of rat aortic intimal thickening, human atheromatous plaque, and cultured rat aortic media. J Clin Investig 1984;73(1):148–152. [DOI] [PMC free article] [PubMed]
- 83.Tajsic T, Morrell NW. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr Physiol 2011;1(1):295–317. [DOI] [PubMed]
- 84.Haworth SG. Development of the normal and hypertensive pulmonary vasculature. Exp Physiol 1995;80(5):843–853. [DOI] [PubMed]
- 85.Gao Y, Raj JU. Hypoxic pulmonary hypertension of the newborn. Compr Physiol 2011;1(1):61–79. [DOI] [PubMed]
- 86.Ivy DD, Abman SH, Barst RJ, Berger RM, Bonnet D, Fleming TR, Haworth SG, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D117–D126. [DOI] [PubMed]
- 87.Müller R, Bravo R, Burckhardt J, Curran T. Induction of c-fos gene and protein by growth factors precedes activation of c-myc. Nature 1984;312(5996):716–720. [DOI] [PubMed]
- 88.Rozengurt E. Early signals in the mitogenic response. Science 1986;234(4773):161–166. [DOI] [PubMed]
- 89.Mariani TJ, Sandefur S, Pierce RA. Elastin in lung development. Exp Lung Res 1997;23(2):131–145. [DOI] [PubMed]
- 90.Watson WH, Ritzenthaler JD, Roman J. Lung extracellular matrix and redox regulation. Redox Biol 2016;8:305–315. [DOI] [PMC free article] [PubMed]
- 91.Noguchi A, Samaha H, deMello DE. Tropoelastin gene expression in the rat pulmonary vasculature: a developmental study. Pediatr Res 1992;31(3):280–285. [DOI] [PubMed]
- 92.Frid MG, Aldashev AA, Dempsey EC, Stenmark KR. Smooth muscle cells isolated from discrete compartments of the mature vascular media exhibit unique phenotypes and distinct growth capabilities. Circ Res 1997;81(6):940–952. [DOI] [PubMed]
- 93.Frid MG, Dempsey EC, Durmowicz AG, Stenmark KR. Smooth muscle cell heterogeneity in pulmonary and systemic vessels: importance in vascular disease. Arterioscler Thromb Vasc Biol 1997;17(7):1203–1209. [DOI] [PubMed]
- 94.Davidson JM, Hill KE, Alford JL. Developmental changes in collagen and elastin biosynthesis in the porcine aorta. Dev Biol 1986;118(1):103–111. [DOI] [PubMed]
- 95.Osborn M, Caselitz J, Weber K. Heterogeneity of intermediate filament expression in vascular smooth muscle: a gradient in desmin positive cells from the rat aortic arch to the level of the arteria iliaca communis. Differentiation 1981;20(3):196–202. [DOI] [PubMed]
- 96.Badesch DB, Lee PD, Parks WC, Stenmark KR. Insulin-like growth factor I stimulates elastin synthesis by bovine pulmonary arterial smooth muscle cells. Biochem Biophys Res Commun 1989;160(1):382–387. [DOI] [PubMed]
- 97.Dempsey EC, McMurtry IF, O’Brien RF. Protein kinase C activation allows pulmonary artery smooth muscle cells to proliferate to hypoxia. Am J Physiol Lung Cell Mol Physiol 1991;260(2):L136–L145. [DOI] [PubMed]
- 98.Dempsey EC, Stenmark KR, McMurtry IF, O’Brien RF, Voelkel NF, Badesch DB. Insulin-like growth factor I and protein kinase C activation stimulate pulmonary artery smooth muscle cell proliferation through separate but synergistic pathways. J Cell Physiol 1990;144(1):159–165. [DOI] [PubMed]
- 99.Lee PD, Hintz RL, Rosenfeld RG, Benitz WE. Presence of insulinlike growth factor receptors and lack of insulin receptors on fetal bovine smooth muscle cells. In Vitro Cell Dev Biol 1988;24(9):921–926. [DOI] [PubMed]
- 100.Malkinson AM, Girard PR, Kuo JF. Strain-specific postnatal changes in the activity and tissue levels of protein kinase C. Biochem Biophys Res Commun 1987;145(2):733–739. [DOI] [PubMed]
- 101.Komuro I, Katoh Y, Kaida T, Shibazaki Y, Kurabayashi M, Hoh E, Takaku F, Yazaki Y. Mechanical loading stimulates cell hypertrophy and specific gene expression in cultured rat cardiac myocytes: possible role of protein kinase C activation. J Biol Chem 1991;266(2):1265–1268. [PubMed]
- 102.Cuadrado A, Molloy CJ, Pech M. Expression of protein kinase C I in NIH 3T3 cells increases its growth response to specific activators. FEBS Lett 1990;260(2):281–284. [DOI] [PubMed]
- 103.Dempsey EC, Badesch DB, Dobyns EL, Stenmark KR. Enhanced growth capacity of neonatal pulmonary artery smooth muscle cells in vitro: dependence on cell size, time from birth, insulin-like growth factor I, and auto-activation of protein kinase C. J Cell Physiol 1994;160(3):469–481. [DOI] [PubMed]
- 104.Majesky MW, Lindner V, Twardzik DR, Schwartz SM, Reidy MA. Production of transforming growth factor β1 during repair of arterial injury. J Clin Investig 1991;88(3):904–910. [DOI] [PMC free article] [PubMed]
- 105.Majesky MW, Giachelli CM, Reidy MA, Schwartz SM. Rat carotid neointimal smooth muscle cells reexpress a developmentally regulated mRNA phenotype during repair of arterial injury. Circ Res 1992;71(4):759–768. [DOI] [PubMed]
- 106.Rhodin JAG. Architecture of the vessel wall. In: Bohr DH, Somlyo AP, Sparks HV Jr., eds. The cardiovascular system: vascular smooth muscle. Handbook of physiology. Compr Physiol 2011;Suppl 7:1–31. doi:10.1002/cphy.cp020201.
- 107.Parks WC, Secrist H, Wu LC, Mecham RP. Developmental regulation of tropoelastin isoforms. J Biol Chem 1988;263(9):4416–4423. [PubMed]
- 108.Pierce RA, Mariencheck WI, Sandefur S, Crouch EC, Parks WC. Glucocorticoids upregulate tropoelastin expression during late stages of fetal lung development. Am J Physiol Lung Cell Mol Physiol 1995;268(3):L491–L500. [DOI] [PubMed]
- 109.Ichiro T, Tajima S, Nishikawa T. Preferential inhibition of elastin synthesis by epidermal growth factor in chick aortic smooth muscle cells. Biochem Biophys Res Commun 1990;168(2):850–856. [DOI] [PubMed]
- 110.Berk JL, Franzblau C, Goldstein RH. Recombinant interleukin-1β inhibits elastin formation by a neonatal rat lung fibroblast subtype. J Biol Chem 1991;266(5):3192–3197. [PubMed]
- 111.Pierce RA, Kolodziej ME, Parks WC. 1,25-Dihydroxyvitamin D3 represses tropoelastin expression by a posttranscriptional mechanism. J Biol Chem 1992;267(16):11593–11599. [PubMed]
- 112.Parks WC, Kolodziej ME, Pierce RA. Phorbol ester-mediated downregulation of tropoelastin expression is controlled by a posttranscriptional mechanism. Biochemistry 1992;31(29):6639–6645. [DOI] [PubMed]
- 113.Yeh H, Ornstein-Goldstein N, Indik Z, Sheppard P, Anderson N, Rosenbloom JC, Cicila G, Yoon K, Rosenbloom J. Sequence variation of bovine elastin mRNA due to alternative splicing. Collagen Relat Res 1987;7(4):235–247. [DOI] [PubMed]
- 114.Rosenbloom J, Abrams WR, Indik Z, Yeh H, Ornstein-Goldstein N, Bashir MM. Structure of the elastin gene. Ciba Found Symp 1995;192:59–74. [DOI] [PubMed]
- 115.Bland RD, Xu L, Ertsey R, Rabinovitch M, Albertine KH, Wynn KA, Kumar VH, et al. Dysregulation of pulmonary elastin synthesis and assembly in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 2007;292(6):L1370–L1384. [DOI] [PubMed]
- 116.Prosser IW, Stenmark KR, Suthar M, Crouch EC, Mecham RP, Parks WC. Regional heterogeneity of elastin and collagen gene expression in intralobar arteries in response to hypoxic pulmonary hypertension as demonstrated by in situ hybridization. Am J Pathol 1989;135(6):1073–1088. [PMC free article] [PubMed]
- 117.Botney MD, Kaiser LR, Cooper JD, Mecham RP, Parghi D, Roby J, Parks WC. Extracellular matrix protein gene expression in atherosclerotic hypertensive pulmonary arteries. Am J Pathol 1992;140(2):357–364. [PMC free article] [PubMed]
- 118.Dawes GS, Mott JC, Widdicombe JG, Wyatt DG. Changes in the lungs of the newborn lamb. J Physiol 1953;121(1):141–162. [DOI] [PMC free article] [PubMed]
- 119.Emmanouilides GC, Moss AJ, Duffie ER Jr., Adams FH. Pulmonary arterial pressure changes in human newborn infants from birth to 3 days of age. J Pediatr 1964;65(3):327–333. [DOI] [PubMed]
- 120.Cassin S, Dawes GS, Mott JC, Ross BB, Strang LB. The vascular resistance of the fetal and newly ventilated lung of the lamb. J Physiol 1964;171(1):61–79. [DOI] [PMC free article] [PubMed]
- 121.Accurso FJ, Alpert B, Wilkening RB, Petersen RG, Meschia G. Time-dependent response of fetal pulmonary blood flow to an increase in fetal oxygen tension. Respir Physiol 1986;63(1):43–52. [DOI] [PubMed]
- 122.Rasanen J, Wood DC, Debbs RH, Cohen J, Weiner S, Huhta JC. Reactivity of the human fetal pulmonary circulation to maternal hyperoxygenation increases during the second half of pregnancy: a randomized study. Circulation 1998;97(3):257–262. [DOI] [PubMed]
- 123.Rudolph A. Distribution and regulation of blood flow in the fetal and neonatal lamb. Circ Res 1985;57(6):811–821. [DOI] [PubMed]
- 124.Morin FC III, Egan EA, Ferguson W, Lundgren CE. Development of pulmonary vascular response to oxygen. Am J Physiol Heart Circ Physiol 1988;254(3):H542–H546. [DOI] [PubMed]
- 125.Storme L, Rairigh RL, Parker TA, Cornfield DN, Kinsella JP, Abman SH. K+-channel blockade inhibits shear stress-induced pulmonary vasodilation in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 1999;276(2):L220–L228. [DOI] [PubMed]
- 126.Heymann MA. Control of the pulmonary circulation in the fetus and during the transitional period to air breathing. Eur J Obstet Gynecol Reprod Biol 1999;84(2):127–132. [DOI] [PubMed]
- 127.Ivy DD, Kinsella JP, Abman SH. Physiologic characterization of endothelin A and B receptor activity in the ovine fetal pulmonary circulation. J Clin Investig 1994;93(5):2141–2148. [DOI] [PMC free article] [PubMed]
- 128.Cornfield DN, Chatfield BA, McQueston JA, McMurtry IF, Abman SH. Effects of birth-related stimuli on l-arginine-dependent pulmonary vasodilation in ovine fetus. Am J Physiol Heart Circ Physiol 1992;262(5):H1474–H1481. [DOI] [PubMed]
- 129.Leffler CW, Tyler TL, Cassin S. Effect of indomethacin on pulmonary vascular response to ventilation of fetal goats. Am J Physiol Heart Circ Physiol 1978;235(4):H346–H351. [DOI] [PubMed]
- 130.Leffler CW, Hessler JR, Green RS. The onset of breathing at birth stimulates pulmonary vascular prostacyclin synthesis. Pediatr Res 1984;18(10):938–942. [DOI] [PubMed]
- 131.Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol Heart Circ Physiol 1990;259(6):H1921–H1927. [DOI] [PubMed]
- 132.Accurso F, Wilkening R. Temporal response of the fetal pulmonary circulation to pharmacologic vasodilators. Proc Soc Exp Biol Med 1988;187(1):89–98. [DOI] [PubMed]
- 133.Abman S, Accurso F. Acute effects of partial compression of ductus arteriosus on fetal pulmonary circulation. Am J Physiol Heart Circ Physiol 1989;257(2):H626–H634. [DOI] [PubMed]
- 134.Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts: a model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 1995;92(3):606–613. [DOI] [PubMed]
- 135.Shaul PW, Wells LB. Oxygen modulates nitric oxide production selectively in fetal pulmonary endothelial cells. Am J Respir Cell Mol Biol 1994;11(4):432–438. [DOI] [PubMed]
- 136.Assali NS, Kirchbaum TH, Dilts PV. Effects of hyperbaric oxygen on utero placental and fetal circulation. Circ Res 1968;22(5):573–588. [DOI] [PubMed]
- 137.Tiktinsky MH, Morin FC III. Increasing oxygen tension dilates fetal pulmonary circulation via endothelium-derived relaxing factor. Am J Physiol Heart Circ Physiol 1993;265(1):H376–H380. [DOI] [PubMed]
- 138.Tirosh R, Resnik ER, Herron J, Sukovich DJ, Hong Z, Weir EK, Cornfield DN. Acute normoxia increases fetal pulmonary artery endothelial cell cytosolic Ca2+ via Ca2+-induced Ca2+ release. Pediatr Res 2006;60(3):258–263. [DOI] [PubMed]
- 139.Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signaling. Am J Respir Crit Care Med 2007;176(11):1146–1153. [DOI] [PMC free article] [PubMed]
- 140.Fineman JR, Wong J, Morin FC III, Wild LM, Soifer SJ. Chronic nitric oxide inhibition in utero produces persistent pulmonary hypertension in newborn lambs. J Clin Investig 1994;93(6):2675–2683. [DOI] [PMC free article] [PubMed]
- 141.Leffler CW, Hessler JR, Green RS. Mechanism of stimulation of pulmonary prostacyclin synthesis at birth. Prostaglandins 1984;28(6):877–887. [DOI] [PubMed]
- 142.Shaul PW, Farrar MA, Magness RR. Oxygen modulation of pulmonary arterial prostacyclin synthesis is developmentally regulated. Am J Physiol Heart Circ Physiol 1993;265(2):H621–H628. [DOI] [PubMed]
- 143.Rosenberg AA, Kennaugh J, Koppenhafer SL, Loomis M, Chatfield BA, Abman SH. Elevated immunoreactive endothelin-1 levels in newborn infants with persistent pulmonary hypertension. J Pediatr 1993;123(1):109–114. [DOI] [PubMed]
- 144.Kumar S, Sud N, Fonseca FV, Hou Y, Black SM. Shear stress stimulates nitric oxide signaling in pulmonary arterial endothelial cells via a reduction in catalase activity: role of protein kinase Cδ. Am J Physiol Lung Cell Mol Physiol 2010;298(1):L105–L116. [DOI] [PMC free article] [PubMed]
- 145.Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Investig 2016;126(3):821–828. [DOI] [PMC free article] [PubMed]
- 146.Wedgwood S, Mitchell CJ, Fineman JR, Black SM. Developmental differences in the shear stress-induced expression of endothelial NO synthase: changing role of AP-1. Am J Physiol Lung Cell Mol Physiol 2003;284(4):L650–L662. [DOI] [PubMed]
- 147.Sud N, Kumar S, Wedgwood S, Black SM. Modulation of PKCδ signaling alters the shear stress-mediated increases in endothelial nitric oxide synthase transcription: role of STAT3. Am J Physiol Lung Cell Mol Physiol 2009;296(3):L519–L526. [DOI] [PMC free article] [PubMed]
- 148.Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev 2009;89(2):481–534. [DOI] [PubMed]
- 149.Fike CD, Pfister SL, Slaughter JC, Kaplowitz MR, Zhang Y, Zeng H, Frye NR, Aschner JL. Protein complex formation with heat shock protein 90 in chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Heart Circ Physiol 2010;299(4):H1190–H1204. [DOI] [PMC free article] [PubMed]
- 150.Aschner JL, Zeng H, Kaplowitz MR, Zhang Y, Slaughter JC, Fike CD. Heat shock protein 90-eNOS interactions mature with postnatal age in the pulmonary circulation of the piglet. Am J Physiol Lung Cell Mol Physiol 2009;296(3):L555–L564. [DOI] [PMC free article] [PubMed]
- 151.Konduri GG, Ou J, Shi Y, Pritchard KA Jr. Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 2003;285(1):H204–H211. [DOI] [PubMed]
- 152.Wang Y, Coceani F. Isolated pulmonary resistance vessels from fetal lambs: contractile behavior and responses to indomethacin and endothelin-1. Circ Res 1992;71(2):320–330. [DOI] [PubMed]
- 153.Ibe BO, Hillyard RM, Raj JU. Heterogeneity in prostacyclin and thromboxane synthesis in ovine pulmonary vascular tree: effect of age and oxygen tension. Exp Lung Res 1996;22(3):351–374. [DOI] [PubMed]
- 154.Zenge JP, Rairigh RL, Grover TR, Storme L, Parker TA, Kinsella JP, Abman SH. NO and prostaglandin interactions during hemodynamic stress in the fetal ovine pulmonary circulation. Am J Physiol Lung Cell Mol Physiol 2001;281(5):L1157–L1163. [DOI] [PubMed]
- 155.Gao Y, Zhou H, Ibe BO, Raj JU. Prostaglandins E2 and I2 cause greater relaxations in pulmonary veins than in arteries of newborn lambs. J Appl Physiol 1996;81(6):2534–2539. [DOI] [PubMed]
- 156.Mita M, Yanagihara H, Hishinuma S, Saito M, Walsh MP. Membrane depolarization-induced contraction of rat caudal arterial smooth muscle involves Rho-associated kinase. Biochem J 2002;364(2):431–440. [DOI] [PMC free article] [PubMed]
- 157.Robertson TP, Dipp M, Ward JP, Aaronson PI, Evans AM. Inhibition of sustained hypoxic vasoconstriction by Y-27632 in isolated intrapulmonary arteries and perfused lung of the rat. Br J Pharmacol 2000;131(1):5–9. [DOI] [PMC free article] [PubMed]
- 158.Wójciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc2 regulate endothelial cell permeability. J Cell Sci 2001;114(7):1343–1355. [DOI] [PubMed]
- 159.McMurtry IF, Bauer NR, Fagan KA, Nagaoka T, Gebb SA, Oka M. Hypoxia and Rho/Rho-kinase signaling: lung development versus hypoxic pulmonary hypertension. Adv Exp Med Biol 2003;543:127–137. [PubMed]
- 160.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 2005;97(2):185–191. [DOI] [PubMed]
- 161.Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-kinase in the cardiovascular system. Circ Res 2016;118(2):352–366. [DOI] [PubMed]
- 162.Wang Z, Jin N, Ganguli S, Swartz DR, Li L, Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol 2001;25(5):628–635. [DOI] [PubMed]
- 163.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 2003;83(4):1325–1358. [DOI] [PubMed]
- 164.Kitazawa, T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem 2000;275(14):9897–9900. [DOI] [PubMed]
- 165.Niiro N, Koga Y, Ikebe M. Agonist-induced changes in the phosphorylation of the myosin-binding subunit of myosin light chain phosphatase and CPI17, two regulatory factors of myosin light chain phosphatase, in smooth muscle. Biochem J 2003;369(1):117–128. [DOI] [PMC free article] [PubMed]
- 166.Parker TA, Roe G, Grover TR, Abman SH. Rho kinase activation maintains high pulmonary vascular resistance in the ovine fetal lung. Am J Physiol Lung Cell Mol Physiol 2006;291(5):L976–L982. [DOI] [PubMed]
- 167.Alvira CM, Sukovich DJ, Lyu SC, Cornfield DN. Rho kinase modulates postnatal adaptation of the pulmonary circulation through separate effects on pulmonary artery endothelial and smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2010;299(6):L872–L878. [DOI] [PMC free article] [PubMed]
- 168.Kinsella JP, Abman SH. Recent developments in the pathophysiology and treatment of persistent pulmonary hypertension of the newborn. J Pediatr 1995;126(6):853–864. [DOI] [PubMed]
- 169.Christou H, Adatia I, Van Marter LJ, Kane JW, Thompson JE, Stark AR, Wessel DL, Kourembanas S. Effect of inhaled nitric oxide on endothelin-1 and cyclic guanosine 5ʹ-monophosphate plasma concentrations in newborn infants with persistent pulmonary hypertension. J Pediatr 1997;130(4):603–611. [DOI] [PubMed]
- 170.Hampl V, Cornfield DN, Cowan NJ, Archer SL. Hypoxia potentiates nitric oxide synthesis and transiently increases cytosolic calcium levels in pulmonary artery endothelial cells. Eur Respir J 1995;88(4):815–822. [PubMed]
- 171.Stevens T, Cornfield DN, McMurtry IF, Rodman DM. Acute reductions in Po2 depolarize pulmonary artery endothelial cells and decrease [Ca2+]i. Am J Physiol Heart Circ Physiol 1994;266(4):H1416–H1421. [DOI] [PubMed]
- 172.Follmann M, Griebenow N, Hahn MG, Hartung I, Mais FJ, Mittendorf J, Schäfer M, et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew Chem Int Ed Engl 2013;52(36):9442–9462. [DOI] [PubMed]
- 173.Gao Y. Conventional and unconventional mechanisms for soluble guanylyl cyclase signaling. J Cardiovasc Pharmacol 2016;67(5):367–372. [DOI] [PubMed]
- 174.Moreno L, Gonzalez-Luis G, Cogolludo A, Lodi F, Lopez-Farre A, Tamargo J, Villamor E, Perez-Vizcaino F. Soluble guanylyl cyclase during postnatal porcine pulmonary maturation. Am J Physiol Lung Cell Mol Physiol 2005;288(1):L125–L130. [DOI] [PubMed]
- 175.Bloch KD, Filippov G, Sanchez LS, Nakane M, de la Monte SM. Pulmonary soluble guanylate cyclase, a nitric oxide receptor, is increased during the perinatal period. Am J Physiol Lung Cell Mol Physiol 1997;272(3):L400–L406. [DOI] [PubMed]
- 176.Morgado M, Cairrão E, Santos-Silva AJ, Verde I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci 2012;69(2):247–266. [DOI] [PMC free article] [PubMed]
- 177.Hofmann F, Wegener JW. cGMP-dependent protein kinases (cGK). Methods Mol Biol 2013;1020:17–50. [DOI] [PubMed]
- 178.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 1993;265(1):C299–C303. [DOI] [PubMed]
- 179.Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci USA 1994;91(16):7583–7587. [DOI] [PMC free article] [PubMed]
- 180.Swayze RD, Braun AP. A catalytically inactive mutant of type I cGMP-dependent protein kinase prevents enhancement of large conductance, calcium-sensitive K+ channels by sodium nitroprusside and cGMP. J Biol Chem 2001;276(23):19729–19737. [DOI] [PubMed]
- 181.Soff GA, Cornwell TL, Cundiff DL, Gately S, Lincoln TM. Smooth muscle cell expression of type I cyclic GMP-dependent protein kinase is suppressed by continuous exposure to nitrovasodilators, theophylline, cyclic GMP, and cyclic AMP. J Clin Investig 1997;100(10):2580–2587. [DOI] [PMC free article] [PubMed]
- 182.Dou D, Zheng X, Qin X, Qi H, Liu L, Raj JU, Gao Y. Role of PKG in development of nitroglycerin tolerance of porcine coronary arteries. Br J Pharmacol 2008;153(3):497–507. [DOI] [PMC free article] [PubMed]
- 183.Abman SH, Chatfield BA, Rodman DM, Hall SL, McMurtry IF. Maturational changes in endothelium-derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Physiol Lung Cell Mol Physiol 1991;260(41):L280–L285. [DOI] [PubMed]
- 184.Kolber KA, Gao Y, Raj JU. Maturational changes in endothelium-derived nitric oxide-mediated relaxation of ovine pulmonary arteries. Biol Neonate 2000;77(2):123–130. [DOI] [PubMed]
- 185.Gao Y, Dhanakoti S, Trevino EM, Sander FC, Portuga AM, Raj JU. Effect of oxygen on cyclic GMP-dependent protein kinase-mediated relaxation in ovine fetal pulmonary arteries and veins. Am J Physiol Lung Cell Mol Physiol 2003;285(3):L611–L618. [DOI] [PubMed]
- 186.Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, et al. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand 1998;164(4):577–587. [DOI] [PubMed]
- 187.Alioua A, Tanaka Y, Wallner M, Hofmann F, Ruth P, Meera P, Toro L. The large conductance, voltage-dependent, and calcium-sensitive K+ channel, Hslo, is a target of cGMP-dependent protein kinase phosphorylation in vivo. J Biol Chem 1998;273(49):32950–32956. [DOI] [PubMed]
- 188.Fukao M, Mason HS, Britton FC, Kenyon JL, Horowitz B, Keef KD. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J Biol Chem 1999;274(16):10927–10935. [DOI] [PubMed]
- 189.Keef KD, Hume JR, Zhong J. Regulation of cardiac and smooth muscle Ca2+ channels (CaV1.2a,b) by protein kinases. Am J Physiol Cell Physiol 2001;281(6):C1743–C1756. [DOI] [PubMed]
- 190.Resnik E, Herron J, Keck M, Sukovich D, Linden B, Cornfield DN. Chronic intrauterine pulmonary hypertension selectively modifies pulmonary artery smooth muscle cell gene expression. Am J Physiol Lung Cell Mol Physiol 2006;290(3):L426–L433. [DOI] [PubMed]
- 191.Gao Y, Dhanakoti S, Tolsa JF, Raj JU. Role of protein kinase G in nitric oxide- and cGMP-induced relaxation of newborn ovine pulmonary veins. J Appl Physiol 1999;87(3):993–998. [DOI] [PubMed]
- 192.Dhanakoti SN, Gao Y, Nguyen MQ, Raj JU. Involvement of cGMP-dependent protein kinase in the relaxation of ovine pulmonary arteries to cGMP and cAMP. J Appl Physiol 2000;88(5):1637–1642. [DOI] [PubMed]
- 193.Negash S, Gao Y, Zhou W, Liu J, Chinta S, Raj JU. Regulation of cGMP-dependent protein kinase-mediated vasodilation by hypoxia-induced reactive species in ovine fetal pulmonary veins. Am J Physiol Lung Cell Mol Physiol 2007;293(4):L1012–L1020. [DOI] [PubMed]
- 194.Sander F, Gao Y, Raj JU. Hypoxia down-regulates cyclic guanidine monophosphate-dependent protein kinase in fetal pulmonary vascular smooth muscle cell through generation of reactive oxygen species and promotes development of pulmonary hypertension. Chest 2005;128(6 suppl):577S–5788S. [DOI] [PubMed]
- 195.Ramchandran R, Pilipenko E, Bach L, Raghavan A, Reddy SP, Raj JU. Hypoxic regulation of pulmonary vascular smooth muscle cyclic guanosine monophosphate-dependent kinase by the ubiquitin conjugating system. Am J Respir Cell Mol Biol 2012;46(3):323–330. [DOI] [PMC free article] [PubMed]
- 196.Cornfield DN. Developmental regulation of oxygen sensing and ion channels in the pulmonary vasculature. Adv Exp Med Biol 2010;661:201–220. [DOI] [PubMed]
- 197.Cornfield DN, Reeve HL, Tolarova S, Weir EK, Archer S. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proc Natl Acad Sci USA 1996;93(15):8089–8094. [DOI] [PMC free article] [PubMed]
- 198.Tristani-Firouzi M, Martin EB, Tolarova S, Weir EK, Archer SL, Cornfield DN. Ventilation-induced pulmonary vasodilation at birth is modulated by potassium channel activity. Am J Physiol Heart Circ Physiol 1996;271(40):H2353–H2359. [DOI] [PubMed]
- 199.Saqueton CB, Miller RB, Porter VA, Milla CE, Cornfield DN. NO causes perinatal pulmonary vasodilation through K+-channel activation and intracellular Ca2+ release. Am J Physiol Lung Cell Mol Physiol 1999;276(6):L925–L932. [DOI] [PubMed]
- 200.Reeve HL, Weir EK, Archer SL, Cornfield DN. A maturational shift in pulmonary K+ channels, from Ca2+ sensitive to voltage dependent. Am J Physiol Lung Cell Mol Physiol 1998;275(6):L1019–L1025. [DOI] [PubMed]
- 201.Cornfield DN, Saqueton CB, Porter VA, Herron J, Resnik E, Haddad IY, Reeve HL. Voltage-gated K+-channel activity in ovine pulmonary vasculature is developmentally regulated. Am J Physiol Lung Cell Mol Physiol 2000;278(6):L1297–L1304. [DOI] [PubMed]
- 202.Moudgil R, Michelakis ED, Archer SL. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006;13(8):615–632. [DOI] [PubMed]
- 203.Rendas A, Reid L. Response of the pulmonary circulation to acute hypoxia in the growing pig. J Appl Physiol 1982;52(4):811–814. [DOI] [PubMed]
- 204.Gordon JB, Hartup J, Hakim A. Developmental effects of hypoxia and indomethacin on distribution of vascular responses in lamb lungs. Pediatr Res 1989;26(4):325–329. [DOI] [PubMed]
- 205.Rhodes MT, Porter VA, Saqueton CB, Herron JM, Resnik ER, Cornfield DN. Pulmonary vascular response to normoxia and KCa channel activity is developmentally regulated. Am J Physiol Lung Cell Mol Physiol 2001;280(6):L1250–L1257. [DOI] [PubMed]
- 206.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol 2014;9:47–71. [DOI] [PubMed]
- 207.Shimoda LA, Laurie SS. HIF and pulmonary vascular responses to hypoxia. J Appl Physiol 2014;116(7):867–874. [DOI] [PMC free article] [PubMed]
- 208.Sainson RC, Harris AL. Regulation of angiogenesis by homotypic and heterotypic notch signalling in endothelial cells and pericytes: from basic research to potential therapies. Angiogenesis 2008;11(1):41–51. [DOI] [PubMed]
- 209.Bishop T, Ratcliffe PJ. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ Res 2015;117(1):65–79. [DOI] [PMC free article] [PubMed]
- 210.Resnik E, Herron J, Fu R, Ivy DD, Cornfield DN. Oxygen tension modulates the expression of pulmonary vascular BKCa channel α- and β-subunits. Am J Physiol Lung Cell Mol Physiol 2006;290(4):L761–L768. [DOI] [PubMed]
- 211.Ahn YT, Kim YM, Adams E, Lyu SC, Alvira CM, Cornfield DN. Hypoxia-inducible factor-1α regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2012;302(3):L352–L359. [DOI] [PMC free article] [PubMed]
- 212.Resnik ER, Herron JM, Lyu SC, Cornfield DN. Developmental regulation of hypoxia-inducible factor 1 and prolyl-hydroxylases in pulmonary vascular smooth muscle cells. Proc Natl Acad Sci USA 2007;104(47):18789–18794. [DOI] [PMC free article] [PubMed]
- 213.Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2008;294(2):L309–L318. [DOI] [PubMed]
- 214.Henson PM. Release of vasoactive amines from rabbit platelets induced by sensitized mononuclear leukocytes and antigen. J Exp Med 1970;131(2):287–306. [DOI] [PMC free article] [PubMed]
- 215.Benveniste J, Henson PM, Cochrane CG. Leukocyte-dependent histamine release from rabbit platelets: the role of IgE, basophils, and platelet-activating factor. J Exp Med 1972;136(6):1356–1377. [DOI] [PMC free article] [PubMed]
- 216.Camussi G, Aglietta M, Malavasi F, Tetta C, Piacibello W, Sanavio F, Bussolino F. The release of platelet activating factor from human endothelial cells in culture. J Immunol 1983;131(5):2397–2403. [PubMed]
- 217.Honda Z, Nakamura M, Miki I, Minami M, Watanabe T, Seyama Y, Okado H, et al. Cloning by functional expression of platelet-activating factor receptor from guinea-pig lung. Nature 1991;349(6307):342–346. [DOI] [PubMed]
- 218.Bito H, Shimizu T. Molecular characterization and physiological functions of PAF receptor. Adv Exp Biol 1997;400A:215–221. [DOI] [PubMed]
- 219.Shimizu T, Honda Z, Nakamura M, Bito H, Izumi T. Platelet-activating factor receptor and signal transduction. Biochem Pharmacol 1992;44(6):1001–1008. [DOI] [PubMed]
- 220.Ibe BO, Hibler S, Raj JU. Platelet activating factor modulates pulmonary vasomotor tone in the perinatal lamb. J Appl Physiol 1998;85(3):1079–1085. [DOI] [PubMed]
- 221.Ibe BO, Pham HH, Kääpä P, Raj JU. Maturational changes in ovine pulmonary metabolism of platelet-activating factor: implications for postnatal adaptation. Mol Genet Metab 2001;74(3):385–395. [DOI] [PubMed]
- 222.Ibe BO, Sander FC, Raj JU. Platelet-activating factor receptors in lamb lungs are down-regulated immediately after birth. Am J Physiol Heart Circ Physiol 2000;278(4):H1168–H1176. [DOI] [PubMed]
- 223.Ibe BO, Portugal AM, Chaturvedi S, Raj JU. Oxygen-dependent PAF receptor binding and intracellular signaling in ovine fetal pulmonary vascular smooth muscle. Am J Physiol Lung Cell Mol Physiol 2005;288(5):L879–L886. [DOI] [PubMed]
- 224.Ibe BO, Ameer A, Portugal AM, Renteria L, Raj JU. Platelet-activating factor modulates activity of cyclic nucleotides in fetal ovine pulmonary vascular smooth muscle. J Pharmacol Exp Ther 2007;320(2):728–737. [DOI] [PubMed]
- 225.Gao Y, Raj JU. Role of veins in regulation of pulmonary circulation. Am J Physiol Lung Cell Mol Physiol 2005;288(2):L213–L226. [DOI] [PubMed]
- 226.Steinhorn RH, Morin FC III, Van Wylen DG, Gugino SF, Giese EC, Russell JA. Endothelium-dependent relaxations to adenosine in juvenile rabbit pulmonary arteries and veins. Am J Physiol Heart Circ Physiol 1994;266(5):H2001–H2006. [DOI] [PubMed]
- 227.Toga H, Bansal V, Raj JU. Differential responses of ovine intrapulmonary arteries and veins to acetylcholine. Respir Physiol 1996;104(2–3):197–204. [DOI] [PubMed]
- 228.Stephenson AH, Sprague RS, Lonigro AJ. 5,6-Epoxyeicosatrienoic acid reduces increases in pulmonary vascular resistance in the dog. Am J Physiol Heart Circ Physiol 1998;275(1):H100–H109. [DOI] [PubMed]
- 229.Arrigoni FI, Hislop AA, Haworth SG, Mitchell JA. Newborn intrapulmonary veins are more reactive than arteries in normal and hypertensive piglets. Am J Physiol Lung Cell Mol Physiol 1999;277(5):L887–L892. [DOI] [PubMed]
- 230.Walch L, de Montpreville V, Brink C, Norel X. Prostanoid EP1- and TP-receptors involved in the contraction of human pulmonary veins. Br J Pharmacol 2001;134(8):1671–1678. [DOI] [PMC free article] [PubMed]
- 231.Yoshimura K, Tod ML, Pier KG, Rubin LJ. Role of venoconstriction in thromboxane-induced pulmonary hypertension and edema in lambs. J Appl Physiol 1989;66(2):929–935. [DOI] [PubMed]
- 232.Gournay V. The ductus arteriosus: physiology, regulation, and functional and congenital anomalies. Arch Cardiovasc Dis 2011;104(11):578–585. [DOI] [PubMed]
- 233.Tristani-Firouzi M, Reeve HL, Tolarova S, Weir EK, Archer SL. Oxygen-induced constriction of rabbit ductus arteriosus occurs via inhibition of a 4-aminopyridine-voltage-sensitive potassium channel. J Clin Investig 1996;98(9):1959–1965. [DOI] [PMC free article] [PubMed]
- 234.Michelakis ED, Rebeyka I, Wu X, Nsair A, Thébaud B, Hashimoto K, Dyck JR, et al. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res 2002;91(6):478–486. [DOI] [PubMed]
- 235.Olschewski A, Hong Z, Linden BC, Porter VA, Weir EK, Cornfield DN. The contribution of the KCa channel to membrane potential and O2 sensitivity is decreased in an ovine model of PPHN. Am J Physiol Lung Cell Mol Physiol 2002;283(5):L1103–L1109. [DOI] [PubMed]
- 236.Thébaud B, Michelakis ED, Wu XC, Moudgil R, Kuzyk M, Dyck JR, Harry G, et al. Oxygen-sensitive Kv channel gene transfer confers oxygen responsiveness to preterm rabbit and remodeled human ductus arteriosus: implications for infants with patent ductus arteriosus. Circulation 2004;110(11):1372–1379. [DOI] [PubMed]
- 237.Keck M, Resnik E, Linden B, Anderson F, Sukovich DJ, Herron J, Cornfield DN. Oxygen increases ductus arteriosus smooth muscle cytosolic calcium via release of calcium from inositol triphosphate-sensitive stores. Am J Physiol Lung Cell Mol Physiol 2005;288(5):L917–L923. [DOI] [PubMed]
- 238.Thébaud B, Wu XC, Kajimoto H, Bonnet S, Hashimoto K, Michelakis ED, Archer SL. Developmental absence of the O2 sensitivity of L-type calcium channels in preterm ductus arteriosus smooth muscle cells impairs O2 constriction contributing to patent ductus arteriosus. Pediatr Res 2008;63(2):176–181. [DOI] [PubMed]