

Features Of Mitochondrial Disease
Mitochondrial disease (MD) occurs when alteration of mitochondrial respiratory chain (RC) Complex function due to genetic mutation produces a detectable disease state. Activities of Complexes I – V can be altered (Figure 1), and physiological consequences of mitochondrial RC defects include reduced metabolic capacity, reduced ATP synthesis, and increased oxidative and nitrosative stress1, 2. Mutations in nuclear DNA (nDNA) or mitochondrial DNA (mtDNA) can lead to defects in the Complexes essential for RC function or for the transport and assembly of mitochondrial proteins (Box 1). Additionally, as mtDNA mutations can impair mitochondrial function, mutations that affect the complement of factors that facilitate the recycling, synthesis, and import of nucleotides to the mitochondrial genome can also produce MD.
Figure 1. Diagram of Mitochondrial RC Components.
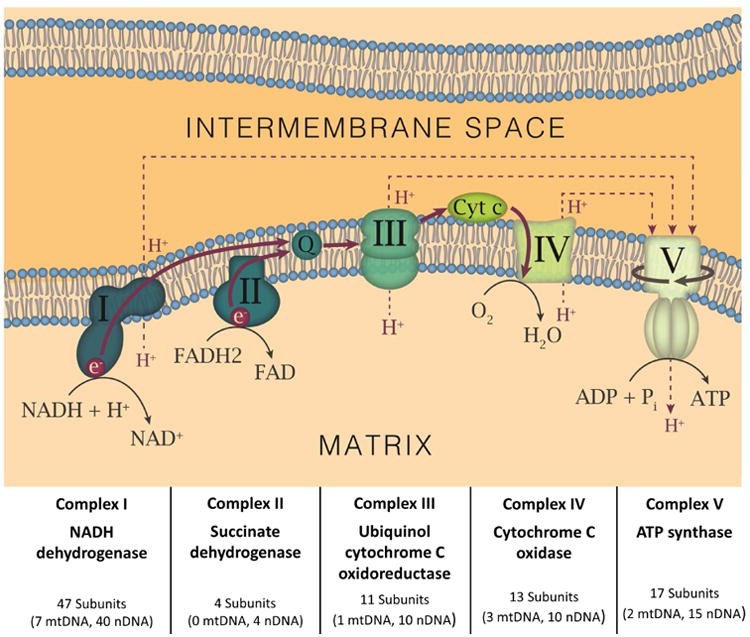
Each mitochondrial RC Complex is depicted and named, as are the critical cofactors, coenzyme Q (CoQ) and cytochrome c (Cyt C). For each Complex, the proportion of subunits encoded by the mtDNA and nDNA are shown. The flow of electrons (solid red lines/arrows) and protons (dashed red lines/arrows) in the RC are also displayed. The solid line in encircling Complex V depicts its rotation as it produces ATP.
Box 1. Nuclear and Mitochondrial Genomes in Mitochondrial Disease.
The mitochondrial genome encodes 37 genes, encoding 2 ribosomal RNAs, 22 transfer RNAs, and 13 subunits of RCs.
There are at least 1000 nuclear genes associated with mitochondrial function.
Some pathological patterns (COX negative/SDH overexpressing fibers) specifically suggest mutations in the mitochondrial genome.
Gene diagnostic panels are available for disease subsets, along with sequencing of the mitochondrial and nuclear genomes separately.
As MD can present with an extraordinary range of clinical symptoms and testing abnormalities, it is often in the clinical differential diagnosis of patients with diseases involving the brain, muscle, or liver. Symptoms of MD are manifold and include abnormalities of the motor, sensory, gastrointestinal, endocrine, and cardiovascular systems, as well as intolerance of some general anesthetics and anti-epileptic drugs, increased susceptibility to infection, and pregnancy loss 1, 2. The exceptional variation seen in the MD phenotype can be due to variations in the amount and distribution of dysfunctional mitochondria throughout the body. As discussed below, dramatic differences in clinical phenotype can be seen in different patients with the same mutation, particularly when that mutation is present in the mitochondrial genome. For MD due to mtDNA mutation, the clinical phenotype will depend on 1) the specific tissues that contain abnormal mitochondria, 2) the proportion of abnormal mitochondria within these tissues, and 3) the number of copies of mutant mtDNA within the tissue. For nuclear-encoded defects, the phenotype is driven by 1) the dependence of that tissue on mitochondrial respiration and 2) the tissue specific expression of the protein and of other potentially compensatory proteins. A number of distinct MD syndromes have been described (Box 2), but it should be noted that these constitute particularly striking clinical phenotypes rather than the most common presentations of MD. In clinical practice, the most common presentation of MD is nonspecific, and may include the following issues:
Unexplained combination of neuromuscular and non-neuromuscular symptoms
Progressive course
Involvement of an increasing number of seemingly unrelated organs
Fluctuating symptomatology
Exercise intolerance due to premature fatigue after even mild activities (usually disproportionately severe compared to weakness)
Muscle cramps, stiffness, or the “second wind phenomenon” that is often seen in glycogenoses
Elevated lactate levels at rest
Box 2. Examples of Well-Described Mitochondrial Syndromes.
Mitochondrial Encephalomyopathy, Lactic Acidosis Stroke-like episodes (MELAS): Presentation includes weakness, headaches, followed by episodes of seizures and transient hemiparesis and cortical blindness.
Myoclonic Epilepsy with Ragged Red Fibers (MERRF): Presentation includes myoclonus (often the first symptom) followed by generalized epilepsy, ataxia, weakness, and dementia.
Progressive External Ophthalmoplegia Plus: Presentation includes ptosis followed by ophthalmoplegia, and this may be associated with weakness of the upper limbs and exercise intolerance.
Alpers Disease: Presentation includes severe encephalopathy with intractable epilepsy and hepatic failure.
Navajo Neurohepatopathy: Presentation includes hepatopathy, peripheral neuropathy, corneal anesthesia and scarring, acral mutilation, cerebral leukoencephalopathy, failure to thrive, and recurrent metabolic acidosis with intercurrent infections
Leigh syndrome: Presentation includes clinical evidence of brainstem/basal ganglia disease (including stepwise psychomotor retardation or regression), elevated blood or CSF lactate levels, and imaging abnormalities in the brainstem and basal ganglia.
Diagnostic approaches for MD are currently non-standard and better diagnostic tools for MD are needed2. The clinical heterogeneity of MD complicates diagnosis because it can symptomatically overlap with a broad range of diseases, and testing abnormalities among the MDs are not uniform. MD is often suspected in early childhood, and traditional diagnostic methods for MD include clinical presentation, family history, pathology, metabolic profiling, enzyme activity levels, electrophysiology, magnetic resonance imaging (MRI), magnetic resonance spectroscopy (MRS), and mtDNA analysis. Despite this arsenal of methods, the diagnostic workup for the identification of MD is not standard across providers or institutions. Diagnostic algorithms have been employed to predict the likelihood of MD 2. Nevertheless, there is no clear consensus on when MD can be excluded, or where follow-up confirmatory testing should be performed2.
MD is likely vastly underdiagnosed at a level of 5 in 100,000 children1, whereas its suspected prevalence may be as high as 1:5000 adults3. Conversely, a subset of patients with other causes of their symptoms may be incorrectly identified as having an MD due to secondary metabolic effects of their disease state. The determination of whether MD is present or can be excluded in a given patient can be extremely complex, given that:
Mitochondrial function can be secondarily affected due to the disease processes in non-mitochondrial diseases.
There is no specific or sensitive biomarker that will identify all or even most individuals with MD.
There can be extensive variability in the distribution of abnormal mitochondria within an individual patient, allowing a “false negative” testing profile to occur when tissues used for diagnosis do not contain the abnormal mitochondria.
There are no uniform, clear-cut pathological abnormalities to distinguish all MD patients from patients with other disorders, to the extent that some biopsy specimens from MD look structurally normal.
Unfortunately, the lack of standard strategies to identify true MD cases (especially less severe cases that do not have specific pathology) poses a substantial limitation on improving MD diagnosis. In our center, we have adopted a “three strike” practice to identify whether genetic testing of the nuclear or mitochondrial genome is likely to be useful (Box 3), which has led to the identification of patients with pathogenic mutations despite underwhelming test abnormalities on some of the other testing modalities. The remainder of this article provides descriptions of the diagnostic testing modalities that can be useful in the diagnosis of MD.
Box 3. Integrating Test Abnormalities to Diagnose MD… A “3 Strike” Approach.
-
Follow-up genetic testing is performed in cases with significant abnormalities in any 3 of the following categories
Clinical history
Light microscopy
Electron microscopy
ETC (electron transport chain) Activity testing
mtDNA content
Recognizing there may be rare “false negatives”, we generally don't go forward with genetic testing for mitochondrial disease on cases without abnormalities in at least 3 of the categories listed above.
Diagnostic Test Interpretation In Md
Biochemical Screening Tests
Plasma Lactate
Elevated lactate levels have long been associated with MD, though the finding is not a prerequisite for diagnosis. Plasma lactate can be normal or only mildly increased in many conditions, including mtDNA depletion syndromes. “False positive” elevations in lactate may be seen in status of poor tissue perfusion/oxygenation or when samples are improperly collected or handled. Lactate levels are also often increased after feeding4.
Cerebrospinal Fluid (CSF) Lactate
In Leigh syndrome, lactate elevation in the CSF is more consistent than elevations in blood. CSF elevations in lactate, however, may also been seen after seizures of any cause.
Amino Acid Levels
Measurement of amino acids in plasma from MD patients may demonstrate elevated alanine. This is formed from the transamination of pyruvate and reflects a persistent elevation of lactate but may also be seen in other metabolic disorders.
Urine Organic Acids
Lactic aciduria is often seen in MD and urine analysis should be performed to exclude other organic acidurias. More specific biomarkers of mitochondrial dysfunction such as 3-Methylglutaconic acid may be useful (although other genetic disorders including glycogen storage disease may also cause this). Elevations in tricarboxylic acid cycle intermediates may suggest MD but may also be seen in hepatic disease and starvation. Another common finding in MD is elevated urine amino acids that are suggestive of renal Fanconi syndrome5.
Cardiac Testing
Electrocardiogram (ECG)
Cardiomyopathy, pre-excitation, or incomplete heart block are well described in MELAS. Atrioventricular conduction defects are a specific concern in cases of Kearns-Sayre syndrome.
Echocardiogram
Cardiomyopathy is more common in MDs that also cause skeletal muscle myopathy. Systolic dysfunction is a frequent finding in MD, but the structural phenotype may vary. In MELAS, concentric left ventricular (LV) hypertrophy is common, whereas dilated cardiomyopathy is typical of Barth syndrome. Historically, mitochondrial mutations were thought to be the typical cause of LV myocardial noncompaction (also known as LV hypertrabeculation (LVHT)), but many other genetic syndromes can also lead to this cardiac phenotype6.
MRI Imaging of the Brain
MDs produce variable imaging abnormalities, and some patterns can be very helpful in diagnosis (Figure 2). Cerebral and cerebellar atrophy are frequently observed across a broad range of MDs. Additionally, metabolic impairment may produce “stroke-like” lesions of ischemia or infarction that may not correspond to specific vascular territories. In MELAS, increased T2 signal can be present, most frequently in the occipital areas. Slow spreading of these lesions can be noted in the weeks following the initial event. The stroke-like lesions of MELAS show increased apparent diffusion coefficient (ADC), in contrast to the reduction seen in ischemic strokes. These stroke-like lesions may be confused with posterior reversible encephalopathy syndrome (PRES), and it is possible that both disease states have a similar etiology related to endothelial dysfunction, ischemia, and subsequent vasogenic edema. The imaging abnormalities seen in Leigh syndrome are typically different from those of MELAS, including bilateral symmetric hyperintense signal abnormalities in the brain stem and/or basal ganglia on T2-weighted MRI.
Figure 2. Brain Imaging Findings in MD.
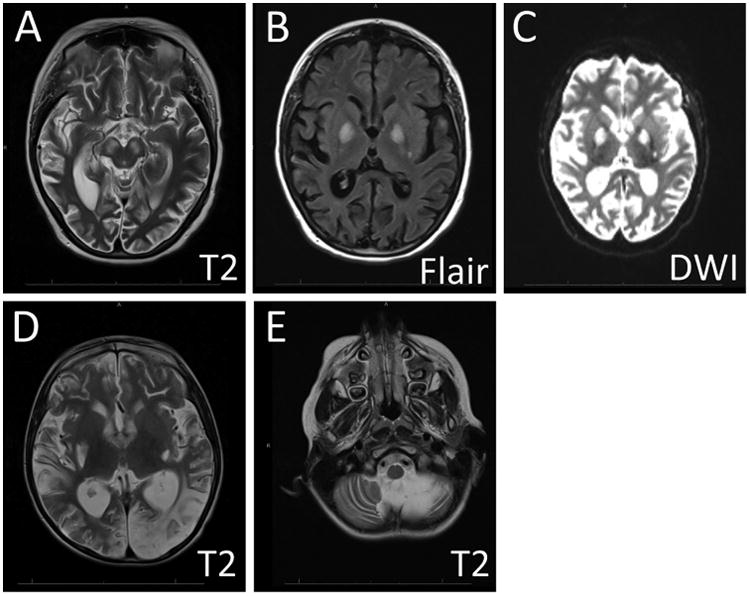
MRI images from two patients with MELAS are shown to illustrate the imaging findings in severe MD. Scans from the first patient (A-C) displayed patchy areas of restricted diffusion and long TR signal hyperintensity involving the parietal lobes, temporal lobes, and basal ganglia bilaterally. In the second patient (D-E), similar abnormalities were seen in the occipital lobes (D), and marked cerebellar atrophy was also noted (E). T2 = T2-weighted image sequence, FLAIR = Fluid-attenuated inversion recovery sequence, DWI = Diffusion weighted sequence.
Skeletal Muscle Biopsy
Light Microscopy
While muscle biopsy can be extremely useful in the diagnosis of muscle disease, the degree of histopathology observed in most cases of MD is underwhelming. Structural characteristics consistent with MD include abnormalities of mitochondrial size, shape, location, number, and internal architecture. In some instances, these abnormalities are sufficiently severe to produce “ragged red fibers” that show mitochondrial aggregates at the light microscopic level (Figure 3). In some cases, specific histochemical stains can be useful in identifying mitochondrial abnormalities. Stains for reduced nicotinamide adenine dinucleotide (NADH), cytochrome oxidase (COX), and succinate dehydrogenase (SDH) are commonly done on frozen muscle tissue to visualize the distribution of mitochondria and other cellular elements. While the NADH stain identifies both the mitochondria and sarcotubular elements, staining patterns on COX and SDH stains identify mitochondria more specifically. Cases with “ragged red fibers” on Gomori trichrome stain should have visible mitochondrial aggregates on these stains, and other abnormalities of mitochondrial distribution may also be noted. The COX stain can specifically be useful because mitochondrial dysfunction can produce “COX-negative fibers” that fail to stain on this preparation. While the number of COX-negative fibers increases even in normal muscle with increasing age, excessive numbers of COX-negative fibers may warrant further evaluation for MD. In cases where COX-negative fibers show overexpression of SDH, it may be useful to search for mtDNA mutations prior to testing the nuclear genome, as this staining pattern often corresponds to compensatory changes in SDH (which is predominantly nuclear-encoded) in response to significant alterations in COX (which is predominantly mitochondrial-encoded). In most cases of MD, however, it should be stressed once again that these very helpful pathological findings will likely be absent, and that the key value of muscle biopsy lies in its potential to assess mitochondrial structure and function through multiple assays.
Figure 3. Pathological Findings in Severe MD.
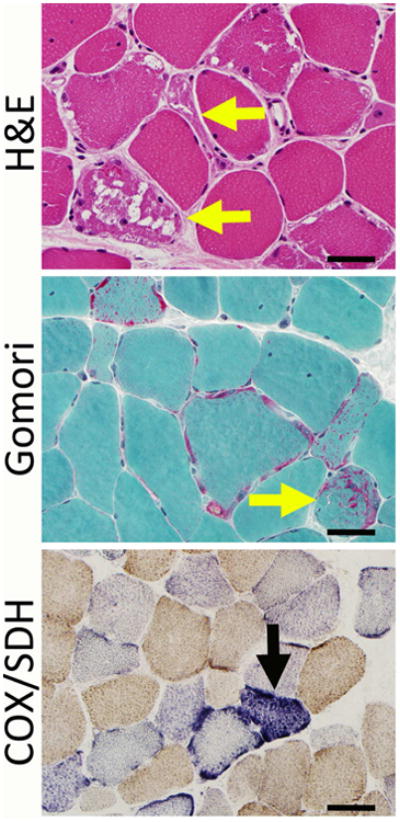
Severe mitochondrial disease can show alterations in fiber size, ragged red fibers (yellow arrows), and accumulations of mitochondria (here seen as red-stained subsarcolemmal areas on the Gomori trichrome stain). COX and SDH stains may show stains that are negative for COX, and some of these fibers may overexpress SDH (black arrow). Bar = 40 mm.
Electron Microscopy
Electron microscopy (EM) can be useful for the identification of structural abnormalities in mitochondria, as it can offer an excellent view of mitochondrial size, shape, internal complexity, and allows the identification for abnormal inclusions that may be present within mitochondria (Figure 4). EM studies should be evaluated by a diagnostician accustomed to working with human muscle biopsy tissue, as there is significant variation in “normal” mitochondrial morphology between species and even between different tissue types in humans. As noted above, the many MD cases have only mild abnormalities of mitochondrial ultrastructure, and some appear entirely normal. Even mild ultrastructural abnormalities, however, can be quite useful in determining the relevance of abnormal genetic or biochemical testing in these patients. EM can also be useful for the distinction of mitochondrial aggregates from other types of pathological aggregates that may be visible at the light microscopic level, including nemaline rods and myofibrillar inclusions.
Figure 4. Ultrastructural Findings in Severe MD.

(A) Ultrastructural appearance of normal mitochondria in a patient without MD. (B) Low-power image illustrating mitochondrial enlargement and excessively complex internal architecture. (C) Other cases show abnormally small or hypocomplex mitochondria. (D-F) Some mitochondrial abnormalities create inclusions within mitochondria. These inclusions can appear paracrystalline (D,E), or may be less defined (F). Excessive numbers of mitochondria may also be seen in MD (D). Bar = 500 nm.
Enzymatic Testing
As skeletal muscle biopsies harboring significant MD may not display structural abnormalities, the evaluation of mitochondrial RC enzymatic function can often be helpful in determining the likelihood of MD. These assays monitor the individual activity of the RC Complexes by following the oxidation/reduction of specific substrates or substrate analogues. The most commonly used assay involves physically and chemically disrupting the muscle sample to liberate intact mitochondrial complexes. Artificial substrates are then added to this suspension and the rate of production of the product is measured using spectrophotometry. These results are typically normalized to a tricarboxylic acid cycle control such as citrate synthase and/or mitochondrial Complex II to control for mitochondrial density. While these assays can be extremely helpful, they are also quite technically complex to perform and maintain results due to numerous technical factors 12, and they are typically performed as a send-out assay to one of several reference laboratories. The reports from these laboratories can be quite helpful in assessing the activity of the RC, but there are a few key issues to consider when ordering and interpreting these assays:
The biochemical activity of these enzymes will be highly dependent on the interval between tissue removal and freezing, and may also be significantly affected by freeze/thaw cycles. Optimal results are obtained by using samples that were frozen within 30 minutes of removal from the patient. Specimens with excessive removal-to-freezing delays will show poor function of both the RC components and the internal control enzymes.
The activity of RC Complexes may be abnormal due to secondary effects of a number of disease states, so it is important to evaluate the biopsy for other disorders before sending out tissue for this assay.
These assays evaluate the activity of mitochondrial RC Complexes I-IV, but do not routinely evaluate the activity of complex V. We would recommend contacting the reference laboratories performing this testing to determine the best way to evaluate complex V, if this is specifically suspected in a given patient.
Some cases with significant MD may yield entirely normal mitochondrial electron transport chain activity results. This may be because the ratio of components is normal but there is a reduction in mitochondrial mass.
Genetic Tests
Definitive genetic diagnosis of MD may require evaluations of both the mtDNA and the nDNA for mutations, either through targeted diagnostic panels or through mtDNA or nDNA sequencing. While the first described mutations in MD were found in the mtDNA, mtDNA-encoded defects only cause approximately 15% of all known MD and most mitochondrial proteins are actually coded in the nDNA. Thus, “next generation” nDNA sequencing can be a powerful tool for MD diagnosis7, 8. Unfortunately, the interpretation of next generation sequencing data for MD diagnosis can be challenging, given the large number of target genes for sequencing and the potentially poor correlation between the genetic data and the clinical/biochemical phenotype.9. Whole genome sequencing (WGS) is on the horizon, but much more work is required to assign mutations to aspects of mitochondrial dysfunction and to identify which mutations are truly disease-causing.
Experimental/Highly Specialized Approaches
Understanding global metabolism through analyses of the lipidome, metabolome, proteome, and transcriptome is progressing, and some disease-specific metabolites have been identified for some non-MD diseases10. The “omics” approach, however, is in need of substantial development before it approaches clinical utility. Improved bioinformatics tools will be required to integrate the enormous amount of data yielded by omics methods.
Mechanistic information on MD has largely arisen from mitochondrial RC activity assays on Complex components, isolated from their native matrix from fresh or frozen tissue, or from cultured cells. These assays are (like most clinical biochemical assays) performed under non-physiologic basal conditions and with very different substrate concentrations than are seen in-vivo. Complex interactions between the substrates of these assays and other cellular components can lead to erroneous results 11. Additionally, these assays cannot perform a functional assessment of processes that require intact mitochondrial structures (such as those involved with mitochondrial membrane potential or coupling). Also, some components of the RC (such as Complex V) cannot be tested using this method. Thus, additional specialized methods of mitochondrial assessment may be used, some of which involve direct measurements of viable, functioning mitochondria. While such assays offer the opportunity to measure phenotypes including oxygen consumption or ATP generation [reviewed in 13], they also are limited to certain clinical sites due to their requirement for freshly-isolated tissue. In all of the currently employed assays, mitochondrial function is not assessed in its native-organ context and the need persists for a functional measurement of mitochondria in tissue. Our research group and others are working on new techniques for such assessments (including electron paramagnetic resonance (EPR) spectroscopy 14), which will hopefully further improve the techniques available for understanding MD mechanisms and the relevance of abnormal genetic testing results in the near future.
Conclusion
As a disease class with exceptional phenotypic and genetic variability, MD presents a diagnostic challenge in many patients. However, it is often possible to identify clinically relevant mitochondrial dysfunction using complementary diagnostic testing methods, even in mild cases. While the development of improved diagnostic testing methods would be helpful, it may be possible to improve the diagnostic evaluation of MD in the short term through the employment of comprehensive, multidisciplinary diagnostic approaches such as the “three strike” approach that we have employed over the past few years.
Keypoints.
Mitochondrial diseases (MD) are a heterogeneous group of disorders with symptoms of organ dysfunction across multiple body systems.
The unifying feature in MD is the dysfunction of mitochondrial respiratory chain Complex function due to genetic mutations.
Diagnosis of MD can be complicated by the lack of “gold standard” diagnostic testing strategies and the potential for “false negative” test results due to sampling issues.
By integrating data obtained from clinical, imaging, pathological, molecular, and enzymatic assessments, it is often possible to identify MD despite these issues.
Synopsis.
Mitochondrial disease (MD) occurs when alteration of mitochondrial respiratory chain Complex function due to genetic mutation produces a detectable disease state. These mutations may be found in either the nuclear or mitochondrial genomes, and may only be present in a subset of cells or body tissues. Thus, the phenotype of MD is extremely variable and the definitive diagnosis of MD can be complex. This article provides a brief description of the strategy we employ in the diagnosis of MD, by integrating data from clinical, imaging, pathological, molecular, and enzymatic assessments. Additional information on characteristic findings seen in “classic” MD syndromes is also provided.
Acknowledgments
We would like to thank Ms. Maggie Beatka and Dr. Hui Meng for their artwork in the design of Figure 1. EM images were obtained at the Medical College of Wisconsin's Electron Microscopy Core Facility. This work was supported by grants from the Children's Hospital of Wisconsin Research Institute (CRI) and the Clinical and Translational Science Institute (CTSI) of Southeastern Wisconsin (through funding provided by NCATS 8UL1 TR000055).
Footnotes
Disclosure Statement: Dimmock Disclosures: Dr. Dimmock is a member of advisory boards for Audentes Therapeutics and Biomarin. He has been funded for work performed for Biomarin, Shire, Alexion, Genzyme/Sanofi, and Cytonet. He has worked as a paid consultant for Illumina and Complete Genomics. He has also participated in a sponsored research agreement with Demeter Therapeutics.
Lawlor Disclosures: Dr. Lawlor is a member of advisory boards for Audentes Therapeutics, and has been supported by sponsored research agreements by Audentes Therapeutics and Solid Biosciences. He is a scientific collaborator with Acceleron Pharma and Pfizer. He has recently been a consultant for Sarepta Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
David P. Dimmock, Human Molecular Genetics Center and Division of Genetics, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, 53226, USA
Michael W. Lawlor, Division of Pediatric Pathology and Neuroscience Research Center, Department of Pathology and Laboratory Medicine, Medical College of Wisconsin, Milwaukee, WI, 53226, USA
References
- 1.Chinnery PF. Mitochondrial Disorders Overview. In: Pagon RA, Bird TD, Dolan CR, editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993. updated 2010. [Google Scholar]
- 2.Parikh S, Goldstein A, Koenig MK, et al. Practice patterns of mitochondrial disease physicians in North America. Part 1: diagnostic and clinical challenges Mitochondrion. 2014;14(1):26–33. doi: 10.1016/j.mito.2013.07.116. [DOI] [PubMed] [Google Scholar]
- 3.Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of neurology. 2015;77(5):753–759. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorburn DR, Rahman S. In: Mitochondrial DNA-Associated Leigh Syndrome and NARP. Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R); Seattle (WA): 1993. [Google Scholar]
- 5.Wortmann SB, Kluijtmans LA, Rodenburg RJ, et al. 3-Methylglutaconic aciduria--lessons from 50 genes and 977 patients. Journal of inherited metabolic disease. 2013;36(6):913–921. doi: 10.1007/s10545-012-9579-6. [DOI] [PubMed] [Google Scholar]
- 6.Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Texas Heart Institute journal / from the Texas Heart Institute of St Luke's Episcopal Hospital, Texas Children's Hospital. 2013;40(4):385–394. [PMC free article] [PubMed] [Google Scholar]
- 7.Goh V, Helbling D, Biank V, et al. Next-generation sequencing facilitates the diagnosis in a child with twinkle mutations causing cholestatic liver failure. Journal of pediatric gastroenterology and nutrition. 2012;54(2):291–294. doi: 10.1097/MPG.0b013e318227e53c. [DOI] [PubMed] [Google Scholar]
- 8.Legati A, Reyes A, Nasca A, et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochimica et biophysica acta. 2016 doi: 10.1016/j.bbabio.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 9.Kemp JP, Smith PM, Pyle A, et al. Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain. 2011;134(Pt 1):183–195. doi: 10.1093/brain/awq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gloerich J, Wevers RA, Smeitink JA, et al. Proteomics approaches to study genetic and metabolic disorders. Journal of research. 2007;6(2):506–512. doi: 10.1021/pr060487w. [DOI] [PubMed] [Google Scholar]
- 11.Spinazzi M, Casarin A, Pertegato V, et al. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion. 2011;11(6):893–904. doi: 10.1016/j.mito.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Gellerich FN, Mayr JA, Reuter S, et al. The problem of interlab variation in methods for mitochondrial disease diagnosis: enzymatic measurement of respiratory chain complexes. Mitochondrion. 2004;4(5-6):427–439. doi: 10.1016/j.mito.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal. 2011;435(2):297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett B, Helbling D, Meng H, et al. Potentially diagnostic electron paramagnetic resonance spectra elucidate the underlying mechanism of mitochondrial dysfunction in the deoxyguanosine kinase deficient rat model of a genetic mitochondrial DNA depletion syndrome. Free radical biology & medicine. 2016;92:141–151. doi: 10.1016/j.freeradbiomed.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]