

Abstract
α-Olefins are the most abundant petrochemical feedstock beyond alkanes, yet their use in commodity chemical manufacture is largely focused on polymerization and hydroformylation. The development of byproduct-free catalytic C-C bond forming reactions that convert olefins to value-added products remains an important objective. Here, we review catalytic intermolecular reductive couplings of unactivated and activated olefin-derived nucleophiles with carbonyl partners. These processes represent an alternative to the longstanding use of stoichiometric organometallic reagents in carbonyl addition.
Graphical abstract
One Sentence Summary: The development of catalytic intermolecular olefin-carbonyl reductive coupling is reviewed.
Byproduct-free catalytic C-C bond forming reactions of α-olefins are of great commercial interest. Hydroformylation (1,2) and single-site alkene polymerization (3) are now among the largest volume applications of homogenous metal catalysis. Carbonyl compounds represent another abundant class of chemical feedstock that are derived from α-olefins via hydroformylation (oxo-products) (1,2) or Wacker oxidation (4). Despite the availability of α-olefins and carbonyl compounds, there is a striking paucity of catalytic processes for the coupling of these orthogonal feedstocks. Here, we review direct methods for the metal catalyzed reductive coupling of olefins with carbonyl compounds. Transformations are catalogued based on their use of (i) unactivated or less activated olefins (α-olefins, styrenes), (ii) conjugated olefins (1,3-dienes, 1,3-enynes) and (iii) highly activated olefins (enones, acrylates, vinyl azines) (Figure 1). Multicomponent reactions, reductive couplings of allenes, reductive carboxylation and carbonyl additions wherein olefins are reduced in situ to form stoichiometric quantities of premetalated reagent, for example through hydroboration or hydrozirconation, are not covered.
Figure 1.
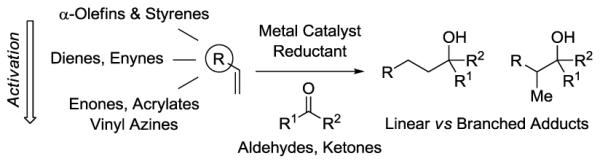
Metal catalyzed reductive coupling of unactivated and activated olefin-derived nucleophiles with carbonyl compounds.
α-Olefins and Styrenes
Intermolecular catalytic reductive coupling of simple linear α-olefins with unactivated carbonyl partners remains an unmet challenge. Titanocene-catalyzed silane-mediated reductive cyclizations of 1,5-enones and enals were reported by Buchwald (5,6) and Crowe (7) in 1995; however, intermolecular variants remain elusive. The concept of transfer hydrogenative carbonyl addition introduced by our laboratory (8-10) provides an important inroad to this problem. Using ruthenium(0) catalysts, vicinally oxygenated secondary alcohols serve dually as reductants and carbonyl precursors (11, 12) (Figure 2). Ethylene, propylene, 1-octene, styrene and a host of other terminal olefins were found to engage in highly regio- and diastereoselective C-C coupling with 3-hydroxy-2-oxindoles to form the corresponding tertiary alcohols (11). The scope of this process was extended through the use of related osmium(0) catalysts (12), which promote the C-C couplings of ethylene and 1-octene with diols, α-ketols or α-hydroxy esters by way of vicinal dicarbonyl intermediates. The collective data corroborate a catalytic mechanism involving oxametalacyclopentane formation via olefin-carbonyl oxidative coupling. Transfer hydrogenolysis of the metalacycle mediated by the reactant alcohol releases the product and regenerates the requisite carbonyl partner. As indicated in the general catalytic mechanism, carboxylic acid cocatalysts dramatically enhance rate and conversion in these processes (Figure 2); an effect that may be attributed to intervention of 6-centered transition structures for both protonolytic cleavage of the metalacycle and substitution of the carboxylate ligand by the reactant alcohol at the metal center. Related C-C couplings of vinyl carboxylates with activated secondary alcohols results in metalacycle fragmentation to form products of vinyl transfer (13) (not shown).
Figure 2.

Metal catalyzed reductive coupling of α-olefins with carbonyl partners.a
aAdCO2H refers to 1-adamantane carboxylic acid. X-Phos = 2-dicyclohexylphosphino-2’,4′,6′-triisopropylbiphenyl
Anhydrides represent an alternate class of carbonyl electrophile that have proven effective in catalytic reductive couplings with styrenes and certain α-olefins (Figure 3). Following initial observations by Miura (14), we reported a highly regioselective rhodium catalyzed olefin-anhydride reductive coupling mediated by elemental hydrogen (15). Subsequently, an enantioselective variant of this process involving copper catalysts was developed by Buchwald (16). Oxidative coupling-metalacycle fragmentation pathways are proposed for the hydrogen-mediated processes, whereas the copper catalyzed reactions are postulated to operate via olefin hydrometalation to form σ-benzyl copper intermediates. While hydroacylations employing aldehydes as acyl donors require chelating groups to suppress conversion of the transient acyl metal intermediates to catalytically inactive carbonyl complexes, the present anhydride reductive couplings overcome this limitation (17).
Figure 3.

Metal catalyzed reductive coupling of olefins with anhydrides.a
a(S,S)-Ph-BPE = 1,2-bis[(2S,5S)-2,5-diphenylphospholano]ethane. DMMS = dimethoxymethylsilane.
Recently, an iron catalyzed Prins-Meerwein-Ponndorf-Verley-type olefin-aldehyde reductive coupling mediated by 2-propanol was reported by Ye (18) (Figure 4). Deuterium labelling studies corroborate a catalytic mechanism wherein condensation of the aldehyde with 2-propanol triggers nucleophilic attack by the olefin. The nascent cation is reduced via internal hydride transfer. As anticipated on the basis of this non-concerted or asynchronous oxonia-ene mechanism, olefins that best stabilize the developing cation are the most efficient reactants. The use of unactivated α-olefins in these couplings would represent a major advance.
Figure 4.

Iron catalyzed Prins-Meerwein-Ponndorf-Verley-type olefin-aldehyde reductive coupling.
Dienes and Enynes
Butadiene (12 × 106 tons/year), isoprene (0.8 × 106 tons/year), and myrcene (30 × 103 tons/year) are important chemical feedstocks. The first intermolecular metal-catalyzed reductive coupling of dienes with carbonyl compounds, a process mediated by triethylborane, was reported by Kimura and Tamaru in 1998 (19, 20) (Figure 5). Diverse dienes may be converted to the respective homoallylation products in good yields and excellent levels of anti-1,3-diastereoselectivity. Regioselectivity in favor of coupling to the more substituted olefin moiety is observed. A mechanism involving diene-carbonyl oxidative coupling to form transient oxo-nickelacycles is postulated (19, 20). Corresponding ketone homoallylations were developed using diethylzinc as terminal reductant (21). Asymmetric variants of the nickel-catalyzed diene-aldehyde reductive couplings are limited to 1,4-diaryl-butadienes (22, 23). Whereas rhodium-catalyzed diene-carbonyl reductive coupling mediated by elemental hydrogen require use of α-ketoaldehydes (24), ruthenium catalysts promote the reductive coupling of diverse dienes to unactivated aldehydes via transfer hydrogenation (25). Here, 2-propanol or formic acid may serve as terminal reductant or, remarkably, the reactant itself may serve dually as reductant and carbonyl precursor. Enantioselective variants of the ruthenium catalyzed reductive couplings have been developed (26-29). Whereas initial studies relied on the use of 2-silyl-substituted dienes to direct syn-diastereo and enantioselectivity (26), chiral phosphate counterions (28) enable access to either the anti- or syn- diastereomers with good control of enantioselectivity (27, 28, 30). The collective data are consistent with a catalytic mechanism wherein alcohol dehydrogenation triggers diene hydrometalation (Figure 5).
Figure 5.

Metal catalyzed diene-carbonyl reductive coupling.
adppf = 1,1-bis-(diphenylphosphino)ferrocene. (R)-DM-SEGPHOS = (R)-(+)-5,5′−bis-[di(3,5-xylyl)phosphino]-4,4′-bi-1,3-benzodioxole. (S)-SEGPHOS = (S)-(−)-5,5′-bis-(diphenylphosphino)-4,4′-bi-1,3-benzodioxole. acac = acetylacetonate.
Ruthenium complexes that embody cationic character catalyze the reductive coupling of 2-substituted dienes to form all-carbon quaternary centers, as illustrated in 2-propanol mediated reductive couplings with paraformaldehyde (31, 32) (Figure 6). Here, a vacant or labile coordination site at the metal center facilitates reversible diene hydrometalation, enabling conversion of the kinetic π-allylruthenium isomer to the thermodynamically more stable terminally disubstituted π-allyl species. Related hydrohydroxymethylations that directly employ methanol (36 × 106 tons/year) as a coupling partner were reported for the first time using an iridium catalyst and 1,1-disubstituted allenes as pronucleophiles (33). Indeed, iridium complexes also catalyze the reaction of dienes with carbonyl compounds to form secondary homoallylic alcohols (33, 34). Cyclohexadiene (34) and butadiene (35) engage in either 2-propanol-mediated reductive coupling or, as shown, direct primary alcohol C-C coupling via hydrogen auto-transfer (Figure 6).
Figure 6.

Alternate regioselectivity and use of cyclic dienes in metal catalyzed carbonyl reductive coupling.a
adppb = bis-(diphenylphosphino)butane. biphep = 2,2′-bis-(diphenylphosphino)-1,1′-biphenyl.
Ruthenium(0) catalysis enables reductive coupling of dienes with activated ketones from the secondary alcohol oxidation level via hydrogen auto-transfer (36-38) (Figure 7). Mechanistic studies corroborate a catalytic mechanism involving diene-carbonyl oxidative coupling to form an oxaruthenacycle. Hydrogen transfer from the secondary alcohol reactant mediates metalacycle hydrogenolysis, releasing the products of C-C coupling and regenerating the activated ketone to close the catalytic cycle. The regioselectivity of C-C coupling at the diene C4-position is unique among diene-carbonyl reductive couplings. Beyond α-hydroxy esters (36), this process applies to 3-hydroxy-2-oxindoles (37) and heteroaryl substituted secondary alcohols (38). In the latter case, the oxaruthenacycle intermediate was isolated and characterized and reversible metalacycle formation was established through experiments involving diene exchange.
Figure 7.

Ruthenium(0) catalyzed diene-ketone reductive coupling via hydrogen auto-transfer.a
aPCy3 = tricyclohexylphosphine.
The reductive coupling of conjugated enynes with carbonyl compounds to form homopropargylic alcohols was first reported in 2008 (39) (Figure 8). Using the ruthenium catalyst derived in situ from HClRu(CO)(PPh3)3 and dppf, hydrogen is transferred from primary alcohols to 1,3-enynes to form aldehyde-allenylruthenium pairs that combine to deliver the products of C-C coupling as single regioisomers in the absence of stoichiometric byproducts. In corresponding 2-propanol mediated reductive couplings of aldehydes with 2-propoxy substituted enynes, good levels of anti-diastereoselectivity are achieved (40). The 2-propoxy group of the product readily eliminates acetone upon exposure to aqueous sodium hydroxide to reveal the terminal alkyne. More recently, using the chiral ruthenium complex derived in situ from (TFA)2Ru(CO)(PPh3)2 and (R)-BINAP, the enantioselective C-C couplings of diverse primary alcohols with the commercially available 1,3-enyne, TMSC≡CC(Me)=CH2, were reported (41). Metals other than ruthenium catalyze enyne-carbonyl reductive coupling. For example, an iridium catalyst with (R)-SEGPHOS or (R)-DM-SEGPHOS ligands catalyzes highly enantioselective enyne-carbonyl reductive coupling from the alcohol or aldehyde oxidation level (42). In the latter case, formic acid serves as terminal reductant. Finally, copper complexes recently were found to catalyze the silane mediated reductive coupling of 1,3-enynes with diverse ketones with excellent control of syn-diastereo- and enantioselectivity (43) (Figure 8).
Figure 8.

Metal catalyzed enyne-carbonyl reductive coupling.a
aDMMS = dimethoxymethylsilane. (R)-BINAP = (R)-(+)-2,2′-bis-(diphenylphosphino)-1,1′-binaphthyl
Acrylates, Enones and Vinyl Azines
The use of α,β-unsaturated carbonyl compounds as pronucleophiles in reductive couplings with carbonyl compounds is known as the “reductive aldol reaction” (44). Following seminal studies by Revis in 1987 on the rhodium-catalyzed reductive aldol reaction of acrylates with aldehydes and ketones mediated by silane (45), numerous processes of this type were developed using different metal catalysts. We focus here on enantioselective intermolecular reductive aldol reactions (Figure 9). Catalytic carbonyl reductive couplings of α,β-unsaturated carbonyl compounds that occur at the β-position are not covered (46).
Figure 9.

Enantioselective metal catalyzed reductive aldol reactions.a
aSee primary literature for the structures of indane-pybox, phebox, taniaphos and AbbasPhos-I.
The first enantioselective reductive aldol reaction was reported by Morken in 2000 (47). This reductive coupling of acrylic esters with aldehydes was catalyzed by a rhodium-BINAP catalyst using Et2MeSiH as the terminal reductant. High levels of enantioselectivity were accompanied by modest levels of syn-diastereoselectivity (Figure 9). Mechanistic studies implicate hydrometalative pathways en route to rhodium enolates. Using an Ir(pybox) catalyst, improved syn-diastereo- and enantioselectivities were observed; however, inductively activated aldehydes are required (48). A remarkably general Rh(phebox) catalyst for asymmetric reductive aldol addition was subsequently reported by Nishiyama (49). Uniformly high levels of anti-diastereoselectivity and enantioselectivity were observed across a diverse range of substrates, including additions to ketones (50). Ketone electrophiles are also accommodated by copper catalysts (51). The preceding examples of asymmetric reductive aldol coupling pair acrylate pronucleophiles with hydrosilane as terminal reductant. Vinyl ketones serve as pronucleophiles with rhodium catalysts and H2 as reductant (52). Substituting deuterium as the reductant leads to transfer of a single deuterium atom to the former enone β-position, consistent with a catalytic mechanism involving oxidative coupling followed by hydrogenolysis of the resulting metalacycle via σ-bond metathesis. Hydrometalative pathways cannot be excluded on the basis of these results; however, reversible hydrometalation-β-hydride elimination would be anticipated to diminish the extent of deuterium incorporation.
Vinyl azines are isostructural with respect to α,β-unsaturated carbonyl compounds and, as described in the review literature (53), display analogous reactivity. However, their use as pronucleophiles in catalytic reductive coupling has only recently begun to be explored. In 2008, the first example of vinyl azine reductive coupling to a π-electrophile was achieved via rhodium catalyzed hydrogenation of vinyl azines in the presence of imines (54) (Figure 10). Good levels of syn-diastereoselectivity were observed. The Lam laboratory subsequently reported a copper catalyzed coupling of vinyl azines to ketone electrophiles with good levels of syn-diastereoselectivity and excellent enantioselectivity (55). For both processes, optimal results are obtained using vinyl azines where both positions adjacent to nitrogen are substituted.
Figure 10.

Metal catalyzed reductive coupling of vinyl azines.a
Conclusion and Outlook
Intermolecular catalytic reductive coupling of simple linear α-olefins with unactivated carbonyl partners remains an important unmet challenge in chemical synthesis. For such abundant feedstocks, an additional consideration resides in identifying terminal reductants that are equally inexpensive and minimize or eliminate byproduct formation. Hence, byproduct-free processes mediated by elemental hydrogen or transfer hydrogenative C-C couplings of alcohol reactants are especially attractive. Conversely, processes mediated by reductants that are pyrophic (ZnEt2, BEt3) or those that are costly and mass-intensive (R3SiH) can only be viewed as interim solutions. Despite the many unrequited challenges, progress made in the area of metal catalyzed reductive coupling clearly show that classical methods for carbonyl addition that traditionally have exploited stoichiometric organometallic reagents may be replaced by catalytic processes that bypass premetalated reagents. It is our hope this monograph will inspire and guide future research aimed at unlocking carbonyl addition chemistry beyond stoichiometric metals.
Figure 11.

A departure from preformed organometallic reagents in carbonyl addition.
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for financial support. H.S gratefully acknowledges JASSO graduate student exchange fellowship.
References and Notes
- 1.Frohning CD, Kohlpaintner CW. In: Applied Homogeneous Catalysis with Organometallic Compounds. Cornils B, Herrmann WA, editors. Wiley-VCH; Weinheim: 1996. pp. 29–104. [Google Scholar]
- 2.van Leeuwen PWNM. Homogeneous Catalysis: Understanding the Art. Kluwer; Dordrecht: 2004. [Google Scholar]
- 3.Baier MC, Zuideveld MA, Mecking S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014;53:9722–9744. doi: 10.1002/anie.201400799. [DOI] [PubMed] [Google Scholar]
- 4.Cornell CN, Sigman MS. Recent Progress in Wacker Oxidations: Moving toward Molecular Oxygen as the Sole Oxidant. Inorg. Chem. 2007;46:1903–1909. doi: 10.1021/ic061858d. [DOI] [PubMed] [Google Scholar]
- 5.Kablaoui NM, Buchwald SL. Reductive Cyclization of Enones by a Titanium Catalyst. J. Am. Chem. Soc. 1995;117:6785–6786. [Google Scholar]
- 6.Kablaoui NM, Buchwald SL. Development of a Method for the Reductive Cyclization of Enones by a Titanium Catalyst. J. Am. Chem. Soc. 1996;118:3182–3191. [Google Scholar]
- 7.Crowe WE, Rachita MJ. Titanium-Catalyzed Reductive Cyclization of δ,ε-Unsaturated Ketones and Aldehydes. J. Am. Chem. Soc. 1995;117:6787–6788. [Google Scholar]
- 8.Bower JF, Krische MJ. Formation of C–C Bonds via Iridium-Catalyzed Hydrogenation and Transfer Hydrogenation. Top. Organomet. Chem. 2011;34:107–138. doi: 10.1007/978-3-642-15334-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hassan A, Krische MJ. Unlocking Hydrogenation for C–C Bond Formation: A Brief Overview of Enantioselective Methods. Org. Proc. Res. Devel. 2011;15:1236–1242. doi: 10.1021/op200195m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ketcham JM, Shin I, Montgomery TP, Krische MJ. Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem. Int. Ed. 2014;53:9142–9150. doi: 10.1002/anie.201403873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi E, Mowat J, Luong T, Krische MJ. Regio- and Diastereoselective C-C Coupling of α-Olefins and Styrenes to 3-Hydroxy-2-oxindoles by Ru-Catalyzed Hydrohydroxyalkylation. Angew. Chem. Int. Ed. 2013;52:8428–8231. doi: 10.1002/anie.201303552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park BY, Luong T, Sato H, Krische M. Osmium(0) Catalyzed C-C Coupling of Ethylene and α-Olefins with Diols, Ketols or Hydroxy Esters via Hydrogen Transfer. Submitted for Publication. 2016 doi: 10.1021/acs.joc.6b01923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park BY, Luong T, Sato H, Krische MJ. A Metallacycle Fragmentation Strategy for Vinyl Transfer from Enol Carboxylates to Secondary Alcohol C-H Bonds via Osmium- or Ruthenium-Catalyzed Transfer Hydrogenation. J. Am. Chem. Soc. 2015;137:7652–7655. doi: 10.1021/jacs.5b04688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokubo K, Miura M, Nomura M. Rhodium-Catalyzed Reaction of Benzoic Anhydride with Styrene under Molecular Hydrogen. Organometallics. 1995;14:4521–4524. [Google Scholar]
- 15.Hong Y-T, Barchuk A, Krische MJ. Branch-Selective Intermolecular Hydroacylation: Hydrogen-Mediated Coupling of Anhydrides to Styrenes and Activated Olefins. Angew. Chem. Int. Ed. 2006;45:6885–6888. doi: 10.1002/anie.200602377. [DOI] [PubMed] [Google Scholar]
- 16.Bandar JS, Ascic E, Buchwald SL. Enantioselective CuH-Catalyzed Reductive Coupling of Aryl Alkenes and Activated Carboxylic Acids. J. Am. Chem. Soc. 2016;138:5821–5824. doi: 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung JC, Krische MJ. Catalytic Intermolecular Hydroacylation of C-C π-Bonds in the Absence of Chelation Assistance. Chem. Sci. 2012;3:2202–2209. [Google Scholar]
- 18.Zheng Y-L, Liu Y-Y, Wu Y-M, Wang Y-X, Lin Y-T, Ye M. Iron-Catalyzed Regioselective Transfer Hydrogenative Couplings of Unactivated Aldehydes with Simple Alkenes. Angew. Chem. Int. Ed. 2016;55:6315–6318. doi: 10.1002/anie.201602130. [DOI] [PubMed] [Google Scholar]
- 19.Kimura M, Ezoe A, Shibata K, Tamaru Y. Novel and Highly Regio- and Stereoselective Nickel-Catalyzed Homoallylation of Benzaldehyde with 1,3-Dienes. J. Am. Chem. Soc. 1998;120:4033–4034. doi: 10.1021/ja0608904. [DOI] [PubMed] [Google Scholar]
- 20.Kimura M, Tamaru Y. Nickel Catalyzed Reductive Coupling of Dienes and Carbonyl Compounds. Top. Curr. Chem. 2007;279:173–207. [Google Scholar]
- 21.Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Nickel-Catalyzed Homoallylation of Aldehydes and Ketones with 1,3-Dienes and Complementary Promotion by Diethylzinc or Triethylborane. Angew. Chem. Int. Ed. 1999;38:397–400. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Zhu S-F, Duan H-F, Zhou C-Y, Wang L-X, Zhou Q-L. Asymmetric Reductive Coupling of Dienes and Aldehydes Catalyzed by Nickel Complexes of Spiro Phosphoramidites: Highly Enantioselective Synthesis of Chiral Bishomoallylic Alcohols. J. Am. Chem. Soc. 2007;129:2248–2249. doi: 10.1021/ja0693183. [DOI] [PubMed] [Google Scholar]
- 23.Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N. Nickel-Catalyzed Enantio- and Diastereoselective Three-Component Coupling of 1,3-Dienes, Aldehydes, and Silanes Using Chiral N-Heterocyclic Carbenes as Ligands. Org. Lett. 2007;9:5597–5599. doi: 10.1021/ol702543m. [DOI] [PubMed] [Google Scholar]
- 24.Jang H-Y, Huddleston RR, Krische MJ. A New Catalytic C-C Bond-Forming Hydrogenation: Reductive Coupling of Dienes and Glyoxals under Catalytic Hydrogenation Conditions. Angew. Chem. Int. Ed. 2003;42:4074–4077. doi: 10.1002/anie.200351986. [DOI] [PubMed] [Google Scholar]
- 25.Shibahara F, Bower JF, Krische M. Ruthenium Catalyzed C-C Bond Forming Transfer Hydrogenation: Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level Employing Acyclic 1,3-Dienes as Surrogates to Preformed Allyl Metal Reagents. J. Am. Chem. Soc. 2008;130:6338–6339. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zbieg JR, Moran J, Krische MJ. Diastereo- and Enantioselective Ruthenium Catalyzed Hydrohydroxyalkylation of 2-Silyl-Butadienes: Carbonyl syn-Crotylation from the Alcohol Oxidation Level. J. Am. Chem. Soc. 2011;133:10582–10586. doi: 10.1021/ja2046028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Enantioselective C-H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science. 2012;336:324–327. doi: 10.1126/science.1219274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McInturff EL, Yamaguchi E, Krische MJ. Chiral Anion Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium Catalyzed Butadiene Hydrohydroxyalkylation. J. Am. Chem. Soc. 2012;134:20628–20631. doi: 10.1021/ja311208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komanduri V, Krische MJ. Enantioselective Reductive Coupling of 1,3-Enynes to Heterocyclic Aromatic Aldehydes and Ketones via Rhodium Catalyzed Asymmetric Hydrogenation: Mechanistic Insight into the Role of Brønsted Acid Additives. J. Am. Chem. Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027. [DOI] [PubMed] [Google Scholar]
- 30.Grayson MN, Krische MJ, Houk KN. Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc. 2015;137:8838–8850. doi: 10.1021/jacs.5b04844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smejkal T, Han H, Breit B, Krische MJ. All-Carbon Quaternary Centers via Ruthenium-Catalyzed Hydroxymethylation of 2-Substituted Butadienes Mediated by Formaldehyde: Beyond Hydroformylation. J. Am. Chem. Soc. 2009;131:10366–10367. doi: 10.1021/ja904124b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sam B, Breit B, Krische MJ. Paraformaldehyde and Methanol as C1 Feedstocks in Metal-Catalyzed C-C Couplings of π-Unsaturated Reactants: Beyond Hydroformylation. Angew. Chem. Int. Ed. 2015;54:3267–3274. doi: 10.1002/anie.201407888. [DOI] [PubMed] [Google Scholar]
- 33.Moran J, Preetz A, Mesch RA, Krische MJ. Iridium Catalyzed Direct C-C Coupling of Methanol and Allenes. Nature Chem. 2011;3:287–290. doi: 10.1038/nchem.1001. [DOI] [PubMed] [Google Scholar]
- 34.Bower JF, Patman RL, Krische MJ. Iridium Catalyzed C-C Coupling via Transfer Hydrogenation: Carbonyl Addition from the Alcohol or Aldehyde Oxidation Level Employing 1,3-Cyclohexadiene. Org. Lett. 2008;10:1033–1035. doi: 10.1021/ol800159w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zbieg JR, Fukuzumi T, Krische MJ. Iridium Catalyzed Hydrohydroxyalkylation of Butadiene: Carbonyl Crotylation. Adv. Synth. Catal. 2010;352:2416–2420. doi: 10.1002/adsc.201000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung JC, Geary LM, Chen T-Y, Zbieg JR, Krische MJ. Direct, Redox-Neutral Prenylation and Geranylation of Secondary Carbinol C-H Bonds: C4-Regioselectivity in Ruthenium-Catalyzed C-C Couplings of Dienes to α-Hydroxy Esters. J. Am. Chem. Soc. 2012;134:15700–15703. doi: 10.1021/ja3075049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen T-Y, Krische MJ. Regioselective Ruthenium Catalyzed Hydrohydroxyalkylation of Dienes with 3-Hydroxy-2-oxindoles: Prenylation, Geranylation and Beyond. Org. Lett. 2013;15:2994–2997. doi: 10.1021/ol401184k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park BY, Montgomery TP, Garza VJ, Krische MJ. Ruthenium Catalyzed Hydrohydroxyalkylation of Isoprene Employing Heteroaromatic Secondary Alcohols: Isolation and Reversible Formation of the Putative Metallacycle Intermediate. J. Am. Chem. Soc. 2013;135:16320–16323. doi: 10.1021/ja4087193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patman RL, Williams VM, Bower JF, Krische MJ. Carbonyl Propargylation from the Alcohol or Aldehyde Oxidation Level Employing 1,3-Enynes as Surrogates to Preformed Allenylmetal Reagents: A Ruthenium-Catalyzed C-C Bond Forming Transfer Hydrogenation. Angew. Chem. Int. Ed. 2008;47:5220–5223. doi: 10.1002/anie.200801359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geary LM, Leung JC, Krische MJ. Ruthenium Catalyzed Reductive Coupling of 1,3-Enynes and Aldehydes by Transfer Hydrogenation: anti-Diastereoselective Carbonyl Propargylation. Chem. Eur. J. 2012;18:16823–16827. doi: 10.1002/chem.201202446. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen KD, Herkommer D, Krische MJ. Ruthenium-BINAP Catalyzed Alcohol C-H tert-Prenylation via 1,3-Enyne Transfer Hydrogenation: Beyond Stoichiometric Carbanions in Enantioselective Carbonyl Propargylation. J. Am. Chem. Soc. 2016;138:5238–5241. doi: 10.1021/jacs.6b02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geary LM, Woo SK, Leung JC, Krische MJ. Diastereo- and Enantioselective Iridium Catalyzed Carbonyl Propargylation from the Alcohol or Aldehyde Oxidation Level: 1,3-Enynes as Allenylmetal Equivalents. Angew. Chem. Int. Ed. 2012;51:2972–2976. doi: 10.1002/anie.201200239. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Perry IB, Lu G, Liu P, Buchwald SL. Copper-Catalyzed Asymmetric Addition of Olefin-Derived Nucleophiles to Ketones. Science. 2016 doi: 10.1126/science.aaf7720. 10.1126/science.aaf7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jang H-Y, Krische MJ. Metal-Catalyzed Reductive Aldol Coupling. In: Yamamoto H, Carreira EM, editors. Comprehensive Chirality. Vol. 4. Elsevier; Oxford: 2012. pp. 100–121. [Google Scholar]
- 45.Revis A, Hilty TK. Novel Synthesis of β-Siloxy Esters by Condensation of Carbonyls and Trimethylsilane with α,β-Unsaturated Esters Catalyzed by RhCl3. Tetrahedron Lett. 1987;28:4809–4812. [Google Scholar]
- 46.McInturff EL, Mowat J, Waldeck AR, Krische MJ. Ruthenium-Catalyzed Hydrohydroxyalkylation of Acrylates with Diols and α-Hydroxycarbonyl Compounds to Form Spiro- and α-Methylene-γ-Butyrolactones. J. Am. Chem. Soc. 2013;135:17230–17235. doi: 10.1021/ja410533y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor SJ, Duffey MO, Morken JP. Rhodium-Catalyzed Enantioselective Reductive Aldol Reaction. J. Am. Chem. Soc. 2000;122:4528–4529. [Google Scholar]
- 48.Zhao C-X, Duffey MO, Taylor SJ, Morken JP. Enantio- and Diastereoselective Reductive Aldol Reactions with Iridium-Pybox Catalysts. Org. Lett. 2001;3:1829–1831. doi: 10.1021/ol015859f. [DOI] [PubMed] [Google Scholar]
- 49.Nishiyama H, Shiomi T, Tsuchiya Y, Matsuda I. High Performance of Rh(Phebox) Catalysts in Asymmetric Reductive Aldol Reaction: High Anti-Selectivity. J. Am. Chem. Soc. 2005;127:6972–6973. doi: 10.1021/ja050698m. [DOI] [PubMed] [Google Scholar]
- 50.Shiomi T, Nishiyama H. Intermolecular Asymmetric Reductive Aldol Reaction of Ketones as Acceptors Promoted by Chiral Rh(Phebox) Catalyst. Org. Lett. 2007;9:1651–1654. doi: 10.1021/ol070251d. [DOI] [PubMed] [Google Scholar]
- 51.Deschamp J, Chuzel O, Hannedouche J, Riant O. Highly Diastereo- and Enantioselective Copper-Catalyzed Domino Reduction/Aldol Reaction of Ketones with Methyl Acrylate. Angew. Chem. Int. Ed. 2006;45:1292–1297. doi: 10.1002/anie.200503791. [DOI] [PubMed] [Google Scholar]
- 52.Bee C, Han SB, Hassan A, Iida H, Krische MJ. Diastereo- and Enantioselective Hydrogenative Aldol Coupling of Vinyl Ketones: Design of Effective Monodentate TADDOL-Like Phosphonite Ligands. J. Am. Chem. Soc. 2008;130:2746–2747. doi: 10.1021/ja710862u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Best D, Lam HW. C=N-Containing Azaarenes as Activating Groups in Enantioselective Catalysis. J. Org. Chem. 2014;79:831–845. doi: 10.1021/jo402414k. [DOI] [PubMed] [Google Scholar]
- 54.Komanduri V, Grant CD, Krische MJ. Branch-Selective Reductive Coupling of 2-Vinyl Pyridines and Imines via Rhodium Catalyzed C-C Bond Forming Hydrogenation. J. Am. Chem. Soc. 2008;130:12592–12593. doi: 10.1021/ja805056g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saxena A, Choi B, Lam HW. Enantioselective Copper-Catalyzed Reductive Coupling of Alkenylazaarenes with Ketones. J. Am. Chem. Soc. 2012;134:8428–8431. doi: 10.1021/ja3036916. [DOI] [PubMed] [Google Scholar]