

Abstract
The structural analysis of a peptide-based catalyst for the Baeyer-Villiger oxidation (BVO) is reported. This unique structure is then analyzed in the context of its previously documented facility to control selectivity (both enantioselectivity and migratory aptitude) in catalytic reactions. The effects of additives on the solution conformation of the peptide are found to be dramatic, revealing substrate-specific interactions and a possible “induced fit” model. The experimental observation of dynamic behavior supports the notion that flexibility in stereoselective catalysts can be an advantageous feature.
Graphical Abstract
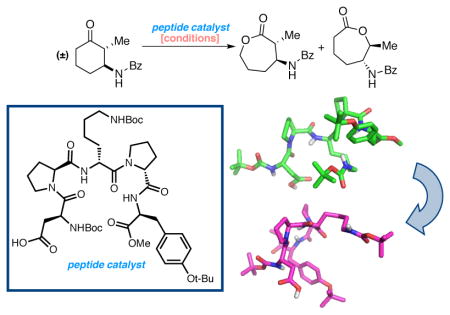
Since its discovery, the Baeyer-Villiger oxidation (BVO)1 has taken its place as a highly utilized and interesting reaction in organic synthesis. The use of a peroxyacid or peroxide as a stoichiometric reagent to transform a ketone or aldehyde into an ester or lactone (Figure 1a) is the best-known implementation of this reaction. Catalysts for this transformation, on the other hand, are not numerous. Early contributions to catalytic BVOs were dominated by enzymes, and in fact many enantioselective BVOs have been disclosed employing so-called Baeyer-Villiger monoxygenases.2 Small molecule catalysts have also been published. Transition metal complexes,3 Lewis acids,4 and chiral phosphoric acids5 have been reported, and variants of each that achieve enantioselctivity for various BVOs are known.6 Our lab pursued a complementary approach based on the use of aspartyl peptides as catalysts.7 These studies recently led to the discovery of catalyst 1, which is enantioselective and regioselective for the BVO of substrates such as 2 (Figure 1b).8 Interestingly, catalyst 1 allows for the kinetic resolution of 2 such that lactones 4 and 5 can be obtained with significant enantioenrichment. Even more striking is the fact that catalyst 1 produces regioisomer 4 preferentially, reversing the intrinsic migratory aptitude preferences exhibited by ketone 2 when simple peracids such as mCPBA are used as stoichiometric oxidants. Catalyst 1 was identified through analysis of combinatorial peptide libraries.7, 9
Figure 1.

(a) Generic Baeyer-Villiger oxidation reaction. (b) Baeyer-Villiger oxidation of substrate 2. The observed migratory aptitude is reversed when mCPBA is used in comparison to catalyst 1.
As a result, the basis of its stereochemical influence was unknown at the point of its discovery. Moreover, peptide 1 possesses a primary sequence for which prediction of the solution conformation is not straightforward. Thus, we set out to learn about the conformation of peptide 1 in solution. Additionally, we were able to use NMR solution structures to glean insights about the importance of the D-Lys to the catalyst and its structure.
Ground state solution structure of 1
Our NMR investigations of the solution structure of 1 were based on full assignment of the proton NMR spectrum, followed by 2D ROESY experiments. Thus, a solution of 1 at a reaction-relevant concentration (0.01 M in CDCl3) was fully characterized by COSY and 1H NMR experiments. Three-dimensional structural features of 1 were further investigated through measurement the ROESY spectrum. Distance restraints for through-space interactions between protons on 1 were extracted and factored into a computationally simulated annealing protocol.10 The average of the ten lowest-energy-scored conformations was calculated, and then subjected to DFT optimization (see SI for details) to produce a proposed snap-shot of 1 in solution (Figure 2a). 8a–b, 11
Figure 2.

(a) Calculated structure of 1 showing parallel pyrollidine ring and proximal aspartic acid and lysine side chain structural features. (b) Zoomed-in section of 1 exhibiting γ-turn, a OLys to NHTyr(t-Bu) contact. (c) Zoomed-in section of 1 showing OLys to εNHLys hydrogen bond.
Two particularly notable features of the experimentally restrained, calculated structure are the likely hydrogen bonds within 1. These include an NHTyr(t-Bu) to OD-Lys interaction (2.8 Å) and second, an εNHLys to OD-Lys putative contact (3.1 Å) (Figure 2). The former interaction meets the definition of a γ-turn in terms of its seven-membered H-bond defined ring (Figure 2b).12 The second interaction involves a side chain to backbone interaction of the D-Lys (Figure 2c) in a manner that may portend its functional role (vide infra). Noting structural rigidity that is separate from the peptide’s active residue, we observed that the two pyrrolidine rings, Pro at i+1 and D-Pro at i+3, may be considered as two separate sectors of the peptide, segregated by the intervening D-Lys residue. In addition, the Pro-D-Lys-D-Pro array does not mimic the backbone dihedral angles of tripeptide segments associated with known motifs for poly-proline helices.13 Finally, one provocative feature in the ground state structure is the proximal nature of the aspartic acid side chain at the i position and the i+2 D-Lys side chain. Taken together, the experimentally restrained solution structure of 1 provides a catalytic scaffold for consideration that is not reminiscent of known catalytic peptide sequences, nor other types of organocatalysts for any reaction studied to date.
Studies of catalyst in the presence of substrates and products
We then turned our attention to the identification of possible catalyst-substrate complexes. These experiments were also undertaken with the idea in mind that catalyst substrate interactions might produce conformational changes,14 as catalyst dynamics for peptide-based catalysts have been implicated previously.8a, 15 For these studies, we elected to examine the interactions of substrate 6 (eq 1), which participates in the BVO reaction with near total regioselectivity. We felt this substrate was a good choice given its high intrinsic preference for one lactone product, and its enantioselective response to the catalyst. The results of these studies are presented below.
NMR titration experiments with mismatched substrate (and favored product)
Shown in Figure 3a is an NMR titration experiment that measured chemical shift changes in the presence of the stereochemically mismatched starting material 6 (i.e., the slow reacting enantiomer in the 1-catalyzed BVO reaction). At reaction-relevant concentrations, minimal perturbation was observed in the overlay of the spectrum of 1 and the spectrum of 1 in the presence of the less reactive enantiomer of 6 (Figure 3a). Similarly, the analogous titration experiment in the presence of favored product 8 (present in a 1:1 ratio) resulted in no significant changes to the proton resonances of either 8 or 1 alone (Figure 3b). The spectra of both mixed samples were nearly identical to the proton spectra of each individual component.
Figure 3.

(a) Stacked spectra of mismatched substrate 6, peptide 1, and a 1:1 mixture of both. (b) Stacked spectra of lactone BVO-product 8, peptide 1 and a 1:1 mixture of both.
Solution structure of 1 in the presence of weakly interacting substrates
ROESY spectra of all three samples (1, 1+6, and 1+8) were also quite similar, although global differences in cross-peak volumes are noted (see SI for details). Two particularly notable differences when comparing 1 to the samples of 1+6/8 are the loss of a HαAsp to HαD-Lys contact, and the absence of a HαPro to HβD-Pro contact detected for 1 alone. Despite these similarities, the calculated structures of the two mixed samples vary from that of peptide 1 alone (Figure 4). These similarities and differences, when subjected to computationally simulated annealing and DFT optimization, culminate in solution structures that show 1 to be different from 1+6 (or 8). Yet, quite notably, 1+6 and 1+8 are themselves quite similar when the peptide components are overlayed, with a root mean squared deviation (RMSD) of 0.02 Å. On the other hand, when 1 itself is introduced into the overlay, an RMSD of 0.709 Å is calculated, reflecting the differences between the parent conformation and the other two structures. Our interpretation of these observations is that both the mismatched substrate and the product interact with 1 in a manner that is quite similar, and perhaps not overly specific. It may be that these compounds present their H-bond donor/acceptor functionality to the peptide, and disrupt the intramolecular D-Lys side chain-to-main chain interaction in a manner that is reversible, leaving the catalyst available for preferential interactions with the matched substrate as the ensemble proceeds through the reaction coordinate.
Figure 4.

Overlay of calculated structures for 1. Green structure was calculated from NMR restraints from 1 alone, blue structure was calculated from restraints extracted from spectra of 1 and 6, and orange structure was calculated from spectra of 1 and product 8.
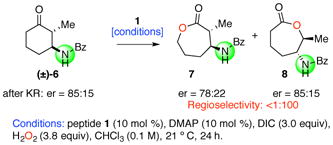 |
(Eq 1) |
NMR titration experiments with matched substrate
We then investigated the structure of the peptide in the presence of the stereochemically-matched substrate employing the analogous spectroscopic experiments.16 Importantly, the impact of the matched substrate on the conformation of ent-1 proved to be more profound in this stereochemically matched case. Several remarkable changes to the proton chemical shifts in its proton spectrum are evident (Figure 5a). The most noteworthy changes in the structure of ent-1 occur in the vicinity of the aspartic acid, which houses the catalytically active residue in the BVO. Moreover, the proximal and adjacent Lys side chain appears to have lost its side chain to back-bone interaction, exhibited in the ground state solution structure of 1 alone. This is consistent with the Δδ of εNHD-Lys, which was measured at 0.66 ppm in the upfield direction. This suggests the shielding of a proton and disengagement in a hydrogen bond. These observations further highlight the importance of the Lys to catalysis, previously reported to be significant based on catalyst analog synthesis.7
Figure 5.

(a) Stacked 1D proton spectra of peptide 1 alone and a 1:1 mixture of ent-1 and 6. (b) Structural change in 1 upon exposure to matched substrate 6.
Solution structure of 1 in the presence of the stereochemically matched subsrate
The changes in the NMR spectra of ent-1 are paralleled by significant changes in the overall computed structure of the peptide (Figure 5b). NMR and computational analysis of the co-mixed sample led to a structure that maintains the proximity of the pyrrolidine that is observed in the parent structure; however, perhaps most notably, the structure of ent-1 in the presence of 6 shows a remarkable displacement of the Lys side chain away from the backbone. It is possible that this conformational change occurs to accommodate a docked substrate.
Comparison of chemical shift perturbations of catalyst 1 in the presence of matched and mismatched substrate
A comparison of the proton chemical shift analyses for both matched and mismatched samples supports these conclusions (Figure 6). The most significant changes in the chemical shifts of our BVO-competent peptide occur in the presence of the matched enantiomer of 6 in the protons of the D-Lys residue. For example, the NHD-Lys (Δδ=1.33 ppm), both Hβ1D-Lys and Hβ2D-Lys (Δδ = 1.27 and 1.31 ppm, respectively) each exhibit major chemical shift perturbations in the presence of the matched substrate. The most pronounced change in the mismatched sample is comparatively meager (Hγ1Pro; (Δδ = −0.10 ppm). Taken together, these observations support a role for the D-Lys residue engaging in a productive association with 6 that is associated with a selective BVO.
Figure 6.

Proton Chemical Shift Analysis of Titrated Baeyer-Villiger Peptide. Histogram comparing the proton shifts of 1 and ent-1 in the presence of 6.
Our studies have culminated in a look at the three-dimensional solution structure of a peptide-based catalyst with a unique primary sequence and unique catalytic behavior. Discovered through combinatorial chemistry, its sequence was unobvious and lacked precedent in terms of experimental structure determination. Interestingly, the ground state structure of 1, and its conformation in the presence of additives are different. Interactions with the preferred substrate stereoisomer appear to be strong and specific. Interactions with the disfavored substrate appear to be less dramatic. Among the implications of these studies is the role of dynamics. In general, it is well known from studies of enzymology that conformationally flexible catalysts can interact with substrates at various points in a complicated reaction coordinate. The conformational requirements at each position may indeed be different, and our results suggest that catalyst 1 exhibits dynamics of this type. The binding of the catalyst to stereochemically matched and mismatched substrates is different. In this case, the specific interactions seem to parallel reactivity in a manner that is consistent, although of course the scenarios associated with Curtin-Hammett analysis dictate that this need not be so.17 Nonetheless, while these studies do not yet reveal all of the aspects of stereochemistry determining steps at the transition state, they do provide an experimental look at allowable conformations for catalyst 1 on its own, and in the presence of relevant substrates. The documentation of these dynamic, and substrate-specific interactions improves our understanding of this unusual catalyst. Combined experimental and computational approaches could refine hypotheses about the basis of stereocontrol with 1 further and efforts along these lines are underway.
Supplementary Material
Acknowledgments
We are grateful to the National Institute of General Medical Sciences of the NIH (GM-096403) for support. We also thank Dr. Eric K. Paulson, Anthony J. Metrano, Sean M. Colvin and Dr. David K. Romney (each of Yale University) for advice and materials.
Footnotes
The authors declare no competing financial interest.
Experimental procedures, characterization data for 1, NMR data for all mixed samples, and four .mol files for the NMR structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Baeyer A, Villiger V. Berichte de deutschen chemischen Gesellschaft. 1899;32:3625–3633. [Google Scholar]
- 2.de Gonzalo G, van Berkel WJH, Fraaije MW. Science of Synthesis, Biocatalysis in Organic Synthesis. 2015;3:187–233. [Google Scholar]
- 3.Evan PA, Lawler MJ. J Am Chem Soc. 2004;126:8642–8643. doi: 10.1021/ja049080n. [DOI] [PubMed] [Google Scholar]
- 4.Michelin RA, Sgarbosa P, Scarso A, Strukul G. Coord Chem Rev. 2010;254:646–660. [Google Scholar]
- 5.Xu S, Wang Z, Zhang X, Ding K. Eur J Org Chem. 2011;1:110–116. [Google Scholar]
- 6.Zhou L, Lui X, Ji J, Zhang Y, hu X, Lin L, Feng X. J Am Chem Soc. 2012;134:17023–17026. doi: 10.1021/ja309262f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Peris G, Miller SJ. Org Lett. 2008;10:3049–3052. doi: 10.1021/ol8010248. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romney DK, Colvin SM, Miller SJ. J Am Chem Soc. 2014;136:14019–14022. doi: 10.1021/ja508757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Abascal NC, Lichtor PA, Giuliano MW, Miller SJ. Chem Sci. 2014;5:4504–4511. doi: 10.1039/C4SC01440E. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lichtor PA, Miller SJ. J Am Chem Soc. 2014;136:5301–5308. doi: 10.1021/ja410567a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lichtor PA, Miller SJ. Nature Chem. 2012;4:990–995. doi: 10.1038/nchem.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Peris G, Jakobsche CE, Miller SJ. J Am Chem Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Copeland GT, Miller SJ. J Am Chem Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]; b) Lichtor PA, Miller SJ. ACS Comb Sci. 2011;13:321–326. doi: 10.1021/co200010v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Brunger AT, Adams PD, Clore GM, Gros P, Kunstleve-Grosse RW, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]; b) Brunger AT. Nature Protocols. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 11.Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Miller SJ. Chem Commun. 2016;52:4816–4819. doi: 10.1039/c6cc01428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nementhy G, Printz MP. Macromolecules. 1972;5:755–758. [Google Scholar]
- 13.Adzhubei AA, Stemberg MJ, Makarov AA. J Mol Biol. 2013;12:2100–2132. doi: 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Zhou H-X. Chem Rev. 2016;116:6503–6515. doi: 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grunenfelder CE, Kisunzu JK, Trapp N, Kastl R, Wennemers H. Biopolymers (Pept Sci) 2016 doi: 10.1002/bip.22912. [DOI] [PubMed] [Google Scholar]
- 16.For these experiments, the enantiomer of peptide 1 (ent-1) was mixed in a 1:1 ratio with 6. This modus operandi was adopted as it was experimentally more convenient to isolate 6 as the illustrated enantiomer following kinetic resolution of 6 with 1, which consumes the “matched enantiomer” of 6 to deliver lactone 8. The enantiomer of 1 was prepared in a trivial manner with the corresponding enantiomeric amino acid residues. For ease of comparison to the other, previously discussed experiments (vide supra), the ROESY contacts were mapped onto the structure of 1 in our computational protocol. Figures are shown in the original enantiomeric series to facilitate visualization.
- 17.Bures J, Armstrong A, Blackmond DG. J Am Chem Soc. 2012;134:6741–6750. doi: 10.1021/ja300415t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.