

Abstract
Ischemia-reperfusion (IR)-induced kidney injury is a major clinical problem, but its underlying mechanisms remain unclear. The transcription factor known as nuclear factor, erythroid 2-like 2 (NFE2L2 or Nrf2) is crucial for protection against oxidative stress generated by pro-oxidant insults. We have previously shown that Nrf2 deficiency enhances susceptibility to IR-induced kidney injury in mice and that its upregulation is protective. Here, we examined Nrf2 target antioxidant gene expression and the mechanisms of its activation in both human and murine kidney epithelia following acute (2 h) and chronic (12 h) hypoxia and reoxygenation conditions. We found that acute hypoxia modestly stimulates and chronic hypoxia strongly stimulates Nrf2 putative target HMOX1 expression, but not that of other antioxidant genes. Inhibition of AKT1/2 or ERK1/2 signaling blocked this induction; AKT1/2 but not ERK1/2 inhibition affected Nrf2 levels in basal and acute hypoxia-reoxygenation states. Unexpectedly, chromatin immunoprecipitation assays revealed reduced levels of Nrf2 binding at the distal AB1 and SX2 enhancers and proximal promoter of HMOX1 in acute hypoxia, accompanied by diminished levels of nuclear Nrf2. In contrast, Nrf2 binding at the AB1 and SX2 enhancers significantly but differentially increased during chronic hypoxia and reoxygenation, with reaccumulation of nuclear Nrf2 levels. Small interfering-RNA-mediated Nrf2 depletion attenuated acute and chronic hypoxia-inducible HMOX1 expression, and primary Nrf2-null kidney epithelia showed reduced levels of HMOX1 induction in response to both acute and chronic hypoxia. Collectively, our data demonstrate that Nrf2 upregulates HMOX1 expression in kidney epithelia through a distinct mechanism during acute and chronic hypoxia reoxygenation, and that both AKT1/2 and ERK1/2 signaling are required for this process.
Keywords: acute kidney injury, ischemia-reperfusion, NRF2, antioxidant genes, ERK1/2, AKT1/2
in native kidneys, acute kidney injury (AKI) is associated with a mortality of up to 50% in patients in intensive care units and it worsens outcome in transplanted kidneys (22, 34). Although mechanisms involved in the pathogenesis of AKI are complex and multifactorial, oxidative stress generated during ischemia reperfusion (IR) is a major mechanism that has been shown to contribute to AKI (7, 25, 27). Thus, understanding the mechanisms regulating oxidative stress during IR is of clinical importance. Changes in the intracellular redox state actively regulate several signaling pathways that control essential biological processes. The endogenous cellular defense system consists of a number of antioxidant enzymes and detoxifying enzymes that maintain the “reducing” environment of the body. The transcriptional induction of these genes is largely mediated by the nuclear factor, erythroid 2-like 2 (NFE2L2 or Nrf2) via the antioxidant response element (ARE), with a core binding sequence of 5′-TGAG/CnnnGC-3′ (13, 16, 26). Nrf2 transcription factor plays a crucial role in preventing cellular injury in response to oxidant and toxicant insults (14). Previously, we have demonstrated that mice with genetic disruption of Nrf2 (Nrf2−/−) are more susceptible to IR-induced kidney injury than the wild-type (Nrf2+/+) mice, and upregulation of Nrf2 activity improves outcome of AKI (20). Nrf2 deficiency caused deregulated antioxidant and cytokine gene expression, whereas antioxidant supplementation significantly improved renal structure and function in Nrf2−/− mice, demonstrating an important role for Nrf2-driven transcriptional response in conferring protection against IR-induced kidney injury and cisplatin-induced nephrotoxicity (20, 21). We therefore hypothesized that perturbation of Nrf2 activation during hypoxia and hypoxia-reoxygenation can result in lower levels of antioxidant enzyme expression, thereby contributing to and/or enhancing susceptibility to development of kidney injury and pathogenesis. In the current study, we determined the nature of Nrf2 activation and its target antioxidant gene expression during acute and chronic hypoxia as well as reoxygenation conditions, using a cell system consisting of established human kidney proximal tubular epithelial cells (HK-2) and primary cultured mouse kidney epithelial cells with and without Nrf2 deficiency. Here, we report that acute and chronic hypoxia largely activates Nrf2 target HMOX1 expression, but not other putative target genes in kidney epithelial cells. We demonstrate that Nrf2 regulates HMOX1 induction in kidney epithelia indirectly and directly during acute and chronic hypoxia and hypoxia-reoxygenation, respectively, and that both AKT1/2 and ERK1/2 signaling pathways regulate this process.
METHODS
Cell culture.
Human normal renal proximal tubular epithelial cells, HK-2 (CRL-2190; American Type Culture Collection), were cultured in DMEM with Ham's F-12 nutrient mixture (DMEM/F-12; Life Technologies) in the presence of 10% fetal bovine serum (FBS) and antibiotics. Primary murine kidney epithelial (pMKE) cells were isolated from adult naïve male mice (aged 6–9 wk) as described previously (3) and the animal protocol was approved by the animal care and use committee at the University of Illinois at Chicago. Briefly, kidney cortical tissue was minced in serum-free media containing collagenase (1 mg/ml; Sigma-Aldrich, St. Louis, MO). The tubules were incubated at 37°C for 45 min with frequent mixing. Tubule fragments were sieved through a 100-μm filter and washed twice with complete media, followed by centrifugation for 5 min at 4°C, and the cells were resuspended in DMEM/F-12 medium with 10% FBS and cultured to 80% confluence before being used for experiments.
Hypoxia exposure.
Cells at subconfluence were replaced with DMEM/F-12 medium with 1% serum and placed in a hypoxia incubator for 2 h (referred as acute hypoxia) or 12 h (referred as chronic hypoxia). The chamber was filled with a gas mixture of 5% CO2 and nitrogen to obtain 1% oxygen concentration, and monitored continuously. After exposure, cells were maintained in complete medium and incubated at 37°C in an atmosphere of 21% O2 and 5% CO2 for reoxygenation for 0–360 min.
Transfections and reporter gene analyses.
Cells were transfected with the luciferase reporter construct (100 ng) bearing the AREs of the AB1 (located at −9.0 kb) or E1 (or formerly known as SX2, located at −4.0 kb) enhancer linked to a minimal promoter (−33/+73 bp) of murine heme oxygenase 1 (Hmox1) (30) (kindly provided by Jawed Alam). Renilla luciferase plasmid, pRL-TK (5 ng; Promega, Madison, WI) was used as a reference. After 24 h of transfection, cells were exposed to hypoxia or hypoxia-reoxygenation, lysed, and the extracts were assayed for firefly and Renilla luciferase activities using a dual luciferase kit (Promega). Luciferase activity of individual samples was normalized to that of Renilla luciferase activity.
Gene expression analysis.
Quantitative RT-PCR (qRT-PCR) was performed by SYBR-green based assays (Applied Biosystems), and immunoblot analysis was performed using indicated antibodies by standard methods.
Small interfering RNA transfection.
Cells were transfected with Silencer Select small interfering RNA (siRNA) specific for Nrf2 or NFE2L2 (20 nM, 4392420; ThermoFisher Scientific, Waltham, MA) and nontargeting scrambled siRNA (20 nM) using DharmaFECT1 reagent (Dharmacon, Lafayette, CO), and after 24 h of posttransfection the medium was replaced and exposed to room air or hypoxia.
Nuclear extracts preparation.
Nuclear extracts were isolated using an NI-PER kit (ThermoFisher Scientific). Nuclear protein was separated on a 10% SDS-PAGE membrane, and the membrane was probed with anti-Nrf2 or Nuclear Matrix Protein p84 (Nmp-p84) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
Chromatin immunoprecipitation assays.
Chromatin immunoprecipitation (ChIP) assays were carried out using a commercially available kit (Upstate Biotechnology, Lake Placid, NY) as detailed elsewhere (30). Briefly, cells (∼1 × 107) were exposed to hypoxia and hypoxia-reoxygenation conditions, and chromatin was cross-linked with formaldehyde (1%) for 10 min at 37°C. A fraction of the soluble chromatin (1%) was saved for measurement of total chromatin input. Precleared chromatin was incubated with Nrf2 antibodies for 18 h at 4°C. DNA recovered from the immunoprecipitated products was used as a template for quantitative PCR with HMOX1 enhancer and proximal promoter-specific primers as follows: AB1 enhancer, forward, CCCTGCTGAGTAATCCTTTCCCGA and reverse, ATGTCCCGACTCCAGACTCCA; SX2 enhancer, forward, CCTCATCCCCTCGTGCAGC and reverse, GAGAAGCTGAGGAGGCACTG; and proximal promoter, forward, GCCAGAAAGTGGGCATCAGC and reverse, CTGAGGACGCTCGAGAGGAG.
Statistical analyses.
Data are presented as means ± SE. A value of P ≤ 0.05 was considered significant and was determined using the t-test.
RESULTS
Acute and chronic hypoxia stimulate HMOX1 expression, but not other Nrf2-regulated antioxidant genes.
To determine Nrf2-regulated antioxidant gene expression during hypoxia and hypoxia-reoxygenation, HK-2 cells were exposed to hypoxia (1% O2) for 2 h (hereafter notated as acute hypoxia) and then allowed to recover under normoxic conditions up to 6 h. Nrf2 putative target genes NQO1, HMOX1, GCLC, and GCLM were analyzed by qRT-PCR. We chose those genes because they are upregulated by cellular stress in an Nrf2-dependent manner and mitigate oxidative stress (5). Acute hypoxia modestly but significantly (approximately 1.8-fold) stimulated HMOX1 mRNA expression, whereas its expression returned to basal levels during reoxygenation (Fig. 1A). In contrast, acute hypoxia did not result in increased expression levels of GCLC, NQO1, or GCLM. In fact, we noted modestly diminished levels of GCLC and NQO1 mRNA expression during hypoxia reoxygenation. To further verify these results obtained in HK-2 cells, we performed analogous experiments in freshly isolated pMKE cells. As shown in Figure 1B, acute hypoxia modestly but significantly induced (∼twofold) Hmox1 mRNA expression in pMKE cells (Fig. 1B), but its expression level was returned to the basal state during reoxygenation in a manner similar to that observed in HK-2 cells. Consistent with data for HK-2 cells, hypoxia did not increase Nqo1, Gclc, or Gclm mRNA expression in pMKE cells. A similar increase in HMOX1 protein was observed by Western blot analysis in HK-2 and pMKE cells exposed to hypoxia (Fig. 1C). As expected, hypoxia-inducible factor 1α (HIF1α) was induced by acute hypoxia in both HK2 and pMKE cells, whereas Nrf2 levels were decreased in hypoxia-exposed HK-2 and pMKE cells (Fig. 1D). Reoxygenation resulted in the restoration of HIF1α and Nrf2 levels to the basal state.
Fig. 1.

Effects of acute hypoxia on Nrf2-regulated antioxidant gene expression in kidney epithelia. HK-2 (A) and primary murine kidney epithelial (pMKE) (B) cells were exposed to room air (RA), 1% hypoxia for 2 h (0hR), or hypoxia-reoxygenation for 6 h (6hR). RNA was isolated and Nrf2 target gene expression was analyzed by quantitative RT-PCR (qRT-PCR). Data are presented as means ± SE (n = 3–4). *RA vs. hypoxia or hypoxia-reoxygenation. C: immunoblot analysis of HMOX1 levels in HK-2 (left) and pMKE (right) cells exposed to hypoxia and hypoxia-reoxygenation. The band intensity was quantitated using β-actin as a reference, and the value of RA controls was considered as one unit. Relative fold change (RFC) shown is from a representative blot of three independent samples (n = 3). D: immunoblot analysis of hypoxia-inducible factor 1α (HIF1α) and Nrf2 expression in HK-2 and pMKE cells exposed to hypoxia and hypoxia-reoxygenation. A representative blot of two independent experiments is shown.
We next investigated whether severe hypoxia is required for optimal induction of antioxidant genes regulated by Nrf2. To evaluate this aspect, HK-2 cells were exposed to 1% O2 for 12 h (i.e., chronic hypoxia) and the magnitude of Nrf2 target gene induction was analyzed as detailed above. As anticipated, HMOX1 mRNA expression levels were markedly (approximately 10-fold) higher in cells exposed to chronic hypoxia (Fig. 2A) than acute hypoxia (approximately 1.8-fold, see Fig. 1A). Moreover, HMOX1 mRNA expression levels remained elevated above basal levels during reoxygenation. A similar result was obtained in freshly cultured pMKE cells that were exposed to acute hypoxia and hypoxia-reoxygenation (Fig. 2B). Increased expression of HMOX1 protein in HK-2 and pMKE cells exposed to hypoxia was verified by Western blot analysis (Fig. 2C). Chronic hypoxia also did not stimulate other Nrf2-target gene expression in HK-2 or pMKE cells. In fact, expressions were modestly but significantly decreased in HK-2 (GCLC and GCLM) and pMKE (Nqo1, Gclc, and Gclm) cells exposed to chronic hypoxia. However, we noted that decreased expression was modestly elevated above basal levels during hypoxia-reoxygenation. As shown in Figure 2D, increased HIF1α activation and decreased Nrf2 expression were observed in both HK2 and pMKE cells exposed to chronic hypoxia, whereas reoxygenation restored expression levels to basal states in a manner similar to that observed in acute hypoxia (Fig. 1D).
Fig. 2.

Effects of chronic hypoxia on Nrf2-regulated antioxidant gene expression in kidney epithelia. HK-2 (A) and pMKE (B) cells were exposed to RA, 1% hypoxia for 12 h (0hR), or hypoxia-reoxygenation for 6 h (6hR), RNA was then isolated and subjected to qRT-PCR. Data are presented as means ± SE (n = 3–4). *RA vs. hypoxia or hypoxia-reoxygenation. C: immunoblot analysis of HMOX1 expression in HK-2 (left) and pMKE (right) cells exposed to hypoxia and reoxygenation. The band intensity was quantitated using β-actin and shown from a representative blot (n = 3). D: HIF1α and Nrf2 expression was analyzed by Western blot analysis in HK-2 and pMKE cells exposed to chronic hypoxia and hypoxia-reoxygenation. A representative blot of two independent experiments is shown.
Inhibition of AKT1/2 signaling blocks hypoxia-inducible Nrf2 nuclear accumulation and HMOX1 expression.
Because phosphoinositide 3-kinase (PI3K)-AKT1/2 signaling regulates Nrf2 activity in response to stressful stimuli (28), we examined whether this pathway modulates Nrf2-mediated HMOX1 induction by hypoxia. HK-2 cells were exposed to hypoxia for 2 h and then reoxygenated at room air for 0 to 6 h, and activation of AKT1/2 signaling was assessed by immunoblot analysis (Fig. 3A). Hypoxia resulted in modestly increased AKT1/2 phosphorylation, which remained elevated above the basal level during reoxygenation up to 360 min. To examine whether AKT1/2 regulates Nrf2 nuclear accumulation in response to hypoxia, cells were treated with PI3K-AKT1/2 inhibitor (LY294002) or an AKT1/2-specific inhibitor (AKTi-II) and exposed to hypoxia. Nuclear extracts were prepared and immunoblotted with Nrf2 antibody (Fig. 3B). We found decreased levels of nuclear Nrf2 in hypoxia-exposed cells, but these levels were restored to basal states during reoxygenation. However, AKT1/2 inhibition attenuated nuclear Nrf2 levels under the basal state and during hypoxia-reoxygenation (Fig. 3B). Consistent with this result, inhibition of AKT1/2 blocked hypoxia-stimulated HMOX1 mRNA expression (Fig. 3C). These results suggest that AKT1/2 signaling controls hypoxia-stimulated HMOX1 induction by affecting nuclear Nrf2 levels.
Fig. 3.
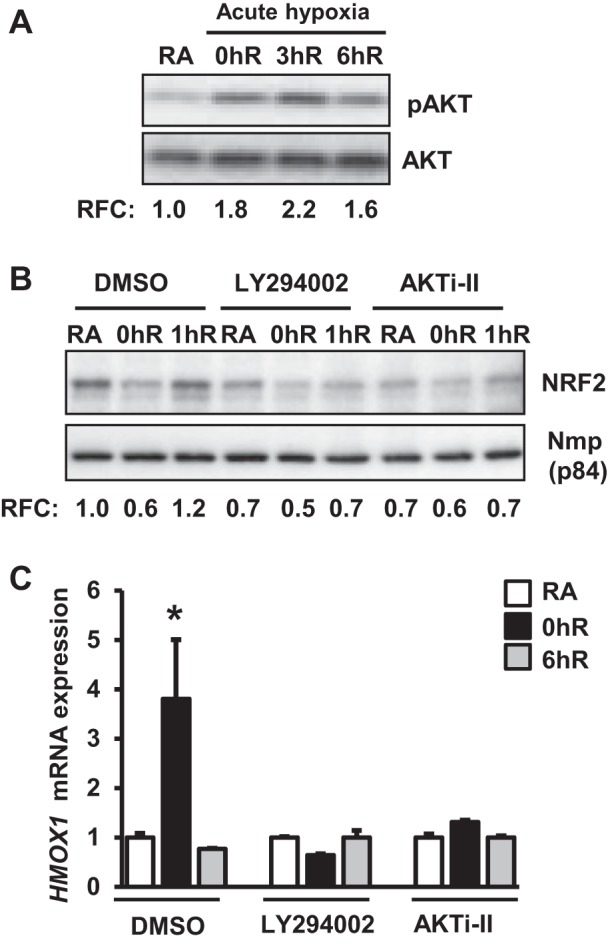
AKT1/2 signaling regulates Nrf2 levels and acute hypoxia-inducible HMOX1 expression. A: HK-2 cells were exposed to hypoxia (0hR) and then reoxygenated for 3 h (3hR) or 6 h (6hR), lysed, and immunoblotted with the phospho-specific AKT (pAKT) or total AKT antibodies. pAKT levels were quantified and expressed relative to RA controls. pAKT levels were quantitated using total AKT as reference and values from a representative blot are shown (n = 3). B: cells pretreated with DMSO, LY294002 (10 μM), or AKTi-II (an AKT1/2-specific inhibitor, 10 μM) for 60 min were exposed to hypoxia and hypoxia-reoxygenation for 1 h (1hR). Nuclear extracts (∼15 g) were separated on an SDS-PAGE membrane, and probed with Nrf2 or Nmp-p84 antibodies. Nrf2 band intensity was quantified using Nmp-p84 as a reference and expressed to relative to RA controls. Values shown are from a representative blot (n = 3). C: cells were pretreated with DMSO or AKTi-II and then exposed to RA, 1% hypoxia for 2 h (0hR), or hypoxia-reoxygenation for 6 h (6hR). HMOX1 mRNA expression was analyzed by qRT-PCR. Data are presented as means ± SE (n = 3). *RA vs. hypoxia or hypoxia-reoxygenation.
We next investigated whether AKT1/2 signaling regulates Nrf2 nuclear accumulation and HMOX1 induction in chronic hypoxia and reoxygenation conditions (Fig. 4A). AKT1/2 phosphorylation in cells exposed to chronic hypoxia (Fig. 4A, lane 2) was nearly identical to that of room air-exposed samples (Fig. 4A, lane 1), but it was markedly elevated during reoxygenation (Fig. 4A, lanes 3 and 4). Inhibition of AKT1/2 signaling had no effect on Nrf2 nuclear accumulation during reoxygenation (Fig. 4B), although it significantly attenuated hypoxia-induced HMOX1 mRNA expression (Fig. 4C). These results demonstrate that AKT1/2 regulates hypoxia-induced HMOX1 induction in chronic hypoxia and reoxygenation conditions without affecting nuclear Nrf2 levels.
Fig. 4.
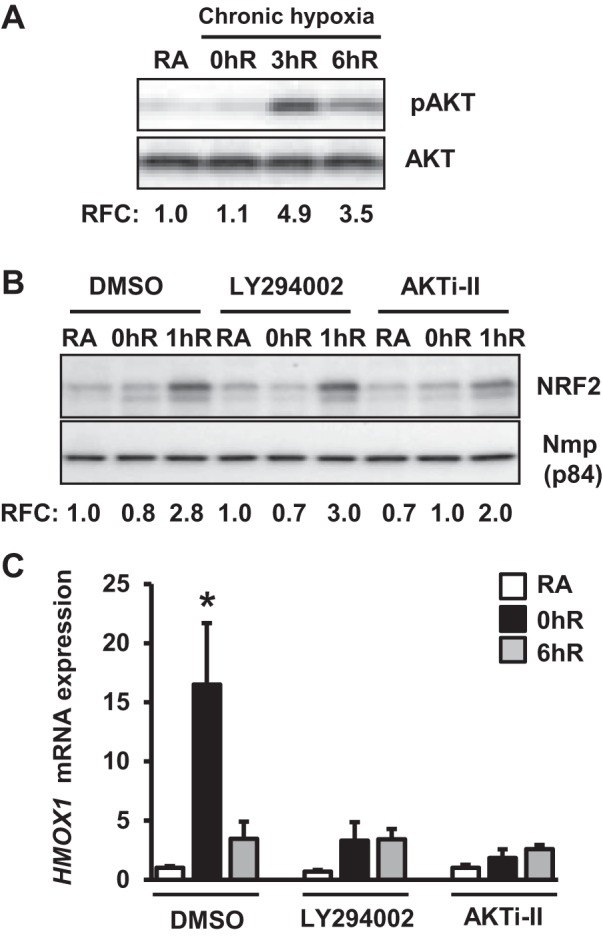
AKT1/2 signaling regulates chronic hypoxia-inducible HMOX1 expression but does not affect Nrf2 levels. A: HK-2 cells were exposed to chronic hypoxia and then reoxygenated for 3 h (3hR) or 6 h (6hR), and pAKT/AKT levels were analyzed as in Fig. 3A. Band intensities shown are from a representative blot (n = 3). B: cells were treated with DMSO, 10 μM LY 294002, or 10 μM AKTi-II for 60 min and then exposed to RA, 1% hypoxia for 12 h (0hR), or hypoxia-reoxygenation for 1 h (1hR). Nuclear extracts were isolated and immunoblotted with anti-Nrf2 or Nmp-p84 antibodies, and Nrf2 levels were quantified as descried above. Band intensity values shown are from a representative blot (n = 3). C: HMOX1 mRNA expression in cells exposed to RA, chronic hypoxia, or hypoxia-reoxygenation in the presence and absence of AKTi-II. Data are presented as means ± SE (n = 2–3). *RA vs. hypoxia or hypoxia-reoxygenation.
ERK1/2 stimulates HMOX1 induction without altering Nrf2 nuclear accumulation.
Because the MEK1/2-ERK1/2 pathway regulates AKI (12), we examined whether it modulates Nrf2 activation and HMOX1 induction in response to hypoxia. Cells were exposed to hypoxia for 2 h and then reoxygenated for 3 or 6 h, and ERK1/2 activation was analyzed by immunoblot analysis. Hypoxia stimulated ERK1/2 phosphorylation (Fig. 5A, lane 2), which remain elevated above the basal level (Fig. 5A, lane 1) during reoxygenation up to 6 h (Fig. 5A, lanes 3 and 4). We found that in contrast to AKT1/2 inhibition, treatment of cells with U0126—an ERK1/2 inhibitor—did not attenuate nuclear Nrf2 levels during hypoxia or hypoxia-reoxygenation (Fig. 5B). However, U0126 blocked hypoxia-stimulated HMOX1 mRNA expression (Fig. 5C).
Fig. 5.
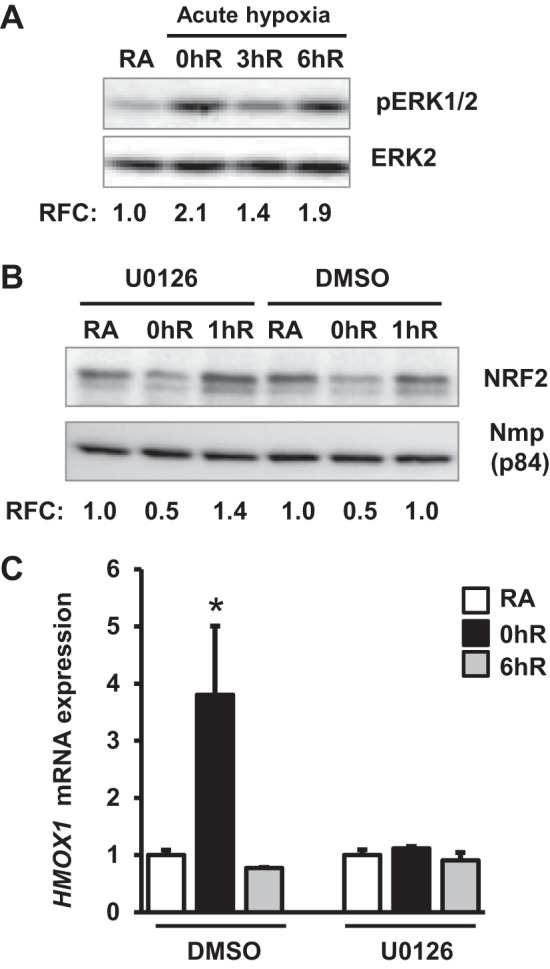
ERK1/2 signaling regulates acute hypoxia-inducible HMOX1 expression without affecting Nrf2 levels. HK-2 cells were exposed to hypoxia (0hR) and then reoxygenated for 3 h (3hR) or 6 h (6hR), lysed, and immunoblotted with pERK1/2 and ERK2 antibodies. pERK1/2 levels were quantitated using total ERK2 as reference, and values from a representative blot are shown (n = 3). ERK1/2 activation was analyzed as described in Fig. 3A. B: cells were treated with DMSO or 10 μM U0126 (an ERK/1/2 inhibitor) for 60 min and exposed to RA, hypoxia for 2 h (0hR), and hypoxia-reoxygenation for 1 h (1hR). Nuclear extracts were isolated and immunoblotted with Nrf2 or Nmp-p84 antibodies. Values from a representative blot are shown (n = 3). C: HMOX1 mRNA expression in cells exposed to RA, hypoxia (0hR), or allowed to recover at room air for 1 h (6hR) in the presence of DMSO or U0126. Data are presented as means ± SE (n = 2–3). *RA vs. exposure.
In contrast to AKT1/2, levels of ERK1/2 phosphorylation in cells exposed to chronic hypoxia (Fig. 6A, lane 2) were markedly higher than those in room-air control cells (Fig. 6A, lane 1). ERK1/2 phosphorylation was elevated during reoxygenation (Fig. 6A, lanes 3 and 4) following chronic hypoxia in a manner to similar to that of AKT1/2 signaling (Fig. 6A). ERK1/2 inhibition had no effect on Nrf2 nuclear accumulation during reoxygenation (Fig. 6B), but it significantly attenuated hypoxia-induced HMOX1 mRNA expression (Fig. 6C). These results demonstrate that ERK1/2 signaling regulates hypoxia-induced HMOX1 induction not by altering Nrf2 levels.
Fig. 6.
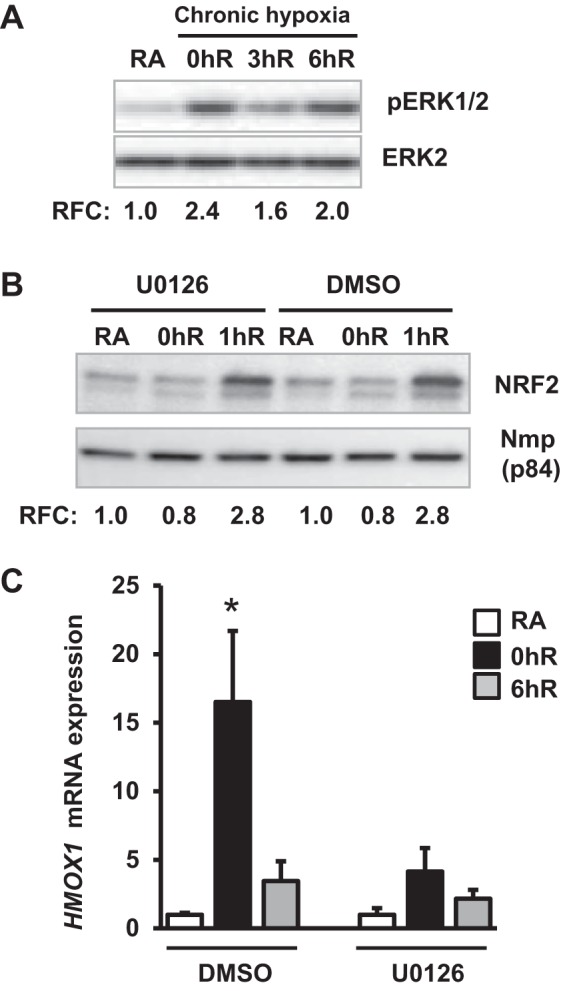
Effects of ERK1/2 inhibition on chronic hypoxia-inducible HMOX1 expression. HK-2 cells were exposed to chronic hypoxia and reoxygenation conditions in the presence and absence of U0126 (10 μM). Levels of ERK1/2 phosphorylation (A, n = 3), nuclear Nrf2 (B, n = 3), and HMOX1 gene expression (C, n = 3–4) were analyzed as described in Fig. 5.
Diminished Nrf2 binding at the HMOX1 promoter in acute and chronic hypoxia.
In transient transfection assays, we found that luciferase activity driven by the Hmox1 promoter bearing the perfect AREs of the AB1 (−9.0 kb) enhancer (see schema, Fig. 7A, top) was significantly lower in cells exposed to acute hypoxia than in room-air controls (Fig. 7A, left). Hmox1 promoter activity in reoxygenation was restored back to that of room-air exposed samples. We next assessed Hmox1 promoter activity in cells exposed to chronic hypoxia and reoxygenation conditions. Hmox1 promoter was not altered by chronic hypoxia, but it was significantly elevated in chronic hypoxia-exposed reoxygenated cells compared with room-air counterparts (Fig. 7A, right).
Fig. 7.

HMOX1 promoter activity and Nrf2 binding to the antioxidant response elements (AREs) in HK-2 cells exposed to hypoxia and hypoxia-reoxygenation. A: HMOX1 promoter reporter construct (100 ng) with pRL-TK plasmid (5 ng) were transfected into cells, exposed to hypoxia (0hR) and hypoxia-reoxygenation for 6 h (6hR). Luciferase activity was analyzed and expressed related to RA-exposed cells. *RA vs. exposure (n = 3). B: Chromatin immunoprecipitation (ChIP) analysis of Nrf2 binding to the functional AREs of the HMOX1 promoter. HK-2 cells were exposed to RA, hypoxia, or hypoxia-reoxygenation, and chromatin was cross-linked and immunoprecipitated with IgG or anti-Nrf2 antibodies. Immunoprecipitated DNA was subjected to qRT-PCR using SYBR primers. Top: positions of the ARE sites and forward (F) and reverse (R) primers of the HMOX1 enhancer used in ChIP assays. Nrf2 binding to the HMOX1 promoter in cells exposed to acute (left) or chronic (right) hypoxia and hypoxia-reoxygenation. Data are presented as means ± SE (n = 3). *RA vs. exposure.
We next performed ChIP assays to analyze the nature of Nrf2 recruitment to the endogenous AB1 enhancer AREs (see schema, Fig. 7B, top) in cells exposed to acute or chronic hypoxia as well as reoxygenation conditions (Fig. 7B). Nrf2 binding at the AB1 enhancer was significantly lower in cells exposed to acute hypoxia and reoxygenation conditions (Fig. 7B, left). Likewise, the Nrf2 binding to the HMOX1 promoter in cells exposed to chronic hypoxia was modestly lower than that in room air controls (Fig. 7B, right). However, recruitment of Nrf2 to the HMOX1 promoter during chronic hypoxia-reoxygenation was increased at the 3-h time point and returned to the basal state by 6 h (Fig. 7B, right).
The E1 (formerly known as SX2) enhancer located at −4.0 kb and the proximal promoter (−44 bp) regions consisting of AREs and E-box have been shown to regulate HMOX1 promoter transactivation in renal epithelial cells (8, 9) and are conserved in humans and mice (2, 33). We therefore examined the relevance of these sites in the regulation of hypoxia-inducible HMOX1 expression using promoter reporter constructs, or ChIP assays, or both. In transient transfection assays, we found that luciferase activity driven by the E1 enhancer was significantly lower in cells exposed to acute hypoxia than in room-air controls (Fig. 8A, left), and activity remained low during hypoxia-reoxygenation. In contrast, Hmox1 promoter activity was significantly elevated in chronic hypoxia-exposed cells and remained high in reoxygenated cells compared with room-air counterpart cells (Fig. 8A, right).
Fig. 8.

Promoter activity and Nrf2 binding of the E1 (or SX2) enhancer following acute and chronic hypoxia and reoxygenation. A: cells were transfected with Hmox1 promoter reporter bearing E1 (formerly known as SX2) enhancer, but lacking the −9.0 kb AB1 enhancer elements (100 ng). pRL-TK plasmid (5 ng) was used as an internal reference to monitor transfection efficiency. Cells were exposed to acute (2 h) or chronic (12 h) hypoxia and hypoxia-reoxygenation for 6 h, and luciferase activity was determined as described in Fig. 7. B: ChIP analysis of Nrf2 binding to the E1 enhancer. Chromatin immunoprecipitated with IgG or Nrf2 antibodies was subjected to qRT-PCR using forward (F) and reverse (R) primers encompassing AREs located at −4.0 kb. C: ChIP analysis of Nrf2 binding to the proximal promoter (−44 bp) region. Data are presented as means ± SE (n = 3). *RA vs. exposure. Arrows indicate position of the primers.
We next analyzed Nrf2 recruitment at the E1 enhancer encompassing the functional AREs following acute or chronic hypoxia and reoxygenation conditions (Fig. 8B). Nrf2 binding to the E1 region was significantly lower in cells exposed to acute hypoxia and reoxygenation conditions (Fig. 8B, left). However, Nrf2 binding in cells exposed to chronic hypoxia was higher than in room-air counterpart cells (Fig. 8B, right). Nrf2 recruitment during reoxygenation was returned to the basal state as early as 1 h. Note that increased Nrf2 binding to the E1 enhancer at 0 h of reoxygenation correlates with Hmox1 promoter activation in chronic hypoxia settings. Likewise, ChIP analysis of the proximal promoter −44 bp region revealed lower levels of Nrf2 binding in acute hypoxia and reoxygenation conditions, but the binding at this region was not altered in chronic hypoxia and reoxygenation conditions (Fig. 8C).
Nrf2 is required for acute and chronic hypoxia-inducible HMOX1 expression.
Expression of several Nrf2 putative targets such as NQO1, GCLC, and GCLM, with the exception of HMOX1, was not significantly induced by acute or chronic hypoxic in either established or primary cultured kidney epithelial cells. Thus, we evaluated whether HMOX1 induction by hypoxia is mediated in an Nrf2-dependent or -independent manner. HK-2 cells were transfected with scrambled siRNA or Nrf2-siRNA and then exposed to acute (Fig. 9A, left) or chronic (Fig. 9A, right) hypoxia. Acute hypoxia-stimulated HMOX1 mRNA expression was significantly lower in Nrf2-siRNA transfected cells than in scrambled siRNA-transfected cells. Likewise, Nrf2 knockdown markedly attenuated chronic hypoxia-induced HMOX1 expression, Nrf2 knockdown was verified by assessing its expression levels in scrambled siRNA and Nrf2-siRNA transfected cells (Fig. 9B).
Fig. 9.

Effects of Nrf2 knockdown on hypoxia stimulated HMOX1 expression. qRT-PCR analysis of HMOX1 (A) and Nrf2 (B) mRNA expression in HK-2 cells transfected with either scrambled (Scr)-silent interfering (si) RNA or Nrf2-siRNA and subsequently exposed to acute or chronic hypoxia. Values from the Scr-siRNA transfected cells are considered as 1. *RA vs. hypoxia; **Scr-siRNA vs. Nrf2-SiRNA (n = 3).
To further validate siRNA knockdown results, kidney epithelial cells isolated from wild-type (Nrf2+/+) mice and Nrf2-null (Nrf2−/−) mice were exposed to hypoxia and hypoxia-reoxygenation conditions, and Hmox1 expression was analyzed by qRT-PCR (Fig. 10). The magnitude of Hmox1 expression induced by acute and chronic (Fig. 10A) hypoxia was significantly lower in Nrf2−/− cells compared with Nrf2+/+ counterparts. These results were verified by immunoblot analysis (Fig. 10B). Together, the results obtained from Nrf2-knockdown and Nrf2-null kidney epithelial cells clearly demonstrate that HMOX1 induction by hypoxia is mediated through Nrf2.
Fig. 10.

Hypoxia-inducible Hmox1 expression is blunted in Nrf2-deficient kidney epithelia. Primary kidney epithelial cells from Nrf2+/+ or Nrf2−/− mice were isolated and then exposed to RA, acute or chronic hypoxia, and hypoxia-reoxygenation conditions. Cells were lysed for RNA and protein extractions. A: Hmox1 expression analyzed by qRT-PCR analysis. *RA vs. hypoxia; **Nrf2+/+ vs. Nrf2−/−. Data are presented as means ± SE (n = 4). B: Hmox1 levels were analyzed by immunoblot analysis and quantified as detailed above. Values from a representative blot (n = 2) are shown.
DISCUSSION
We previously reported that genetic disruption of Nrf2 worsens IR-induced and nephrotoxin-induced AKI, accompanied by decreased levels of antioxidant gene expression and increased inflammatory response (20). Furthermore, our studies and others have shown that augmentation of Nrf2 activity protects IR-induced AKI in mice (21, 36). Nrf2 plays a crucial role in the transcriptional activation of HMOX1 induction in response to pro-oxidant and toxicant stimuli. HMOX1 has been shown to confer protection against various experimental models of AKI (18, 23, 24). Genetic disruption of Hmox1 led to impaired renal function and systemic inflammation in response to IR (29), whereas its overexpression conferred protection against renal tissue injury (1, 4, 32). In the current study, using Nrf2-depleted HK-2 cells (Fig. 9) and Nrf2-null kidney epithelia (Fig. 10), we now demonstrate that Nrf2, a major effector of oxidative stress signaling, is required for acute or chronic hypoxia-inducible HMOX1 induction hypoxia in kidney epithelia. Unexpectedly, however, we found that several putative transcriptional targets of Nrf2, such as NQO1, GCLC, and GCLM, are not induced by either acute or chronic hypoxia in both human and murine kidney epithelia (Figs. 1 and 2). Our results are consistent with a previous report that showed lack of strong activation of Nrf2 target gene expression in lungs of mice exposed to hypoxia (35). In fact, we noted their expression was modestly reduced in response to chronic hypoxia and hypoxia-reoxygenation. Induction of HMOX1 but no other putative transcriptional targets of Nrf2 implies that selective upregulation of Nrf2-regulated antioxidant gene transcription occurs in hypoxia, but the relevance of this differential induction in acute hypoxic response remains unclear and warrants a detailed investigation.
Previously, our studies and others have shown induction of Nrf2-mediated antioxidant gene (such as Hmox1, Nqo1, and Gclc) expression in lung epithelia following acute oxidant or toxicant exposure (28). But these genes, with the exception of Hmox1, are not induced in kidney epithelia in response to acute or chronic hypoxia, suggesting the requirement of other transcription factor(s), in addition to Nrf2, for antioxidant gene expression in hypoxia. The lack of other Nrf2 putative target gene (e.g., Nqo1 and Gclc) induction by hypoxia could be attributable to the lack of hypoxia response elements such as HIF binding sites in their promoter regions. Indeed, Hmox1 bears a functional HIF binding site and HIF signaling has been shown to regulate its expression (17), whereas the presence of functional HIF sites that could contribute to Nqo1 or Gclc regulation have not been reported thus far.
Hypoxia stimulates Nrf2-dependent HMOX1 expression via the AKT1/2 pathway (38), and induction of HMOX1 by a brown algae compound, ekcol, is mediated by AKT1/2 and ERK1/2 signaling in an Nrf2-dependent manner (15). Our current studies revealed that both AKT1/2 and ERK1/2 signaling are required for acute or chronic hypoxia-inducible HMOX1 transcription (Figs. 5 and 6). AKT1/2 inhibition decreases Nrf2 levels in both the basal and hypoxia-reoxygenation states following acute hypoxia, but it had no effect on Nrf2 levels in chronic hypoxia-reoxygenation. In contrast, ERK1/2 inhibition, although blocked HMOX1 induction, had no effect on Nrf2 levels in basal, acute, or chronic hypoxia, and hypoxia-reoxygenation conditions. These results suggest that AKT1/2 and ERK1/2 signaling regulate HMOX1 induction through a distinct mechanism. Although the effects of AKT1/2 inhibition on hypoxia-stimulated HMOX1 expression could be attributable in part to the reduced levels of Nrf2, how ERK1/2 controls HMOX1 induction remains unclear. The AP-1 (JUN and FOS) family of proteins, the downstream targets of ERK1/2 signaling, are induced by hypoxia and regulate the transcriptional activation of HMOX1 by hypoxia (8, 37). Thus, we speculate that ERK1/2 inhibition blocks the activation of other factors such as the AP-1 family of proteins and/or the recruitment of one or more coactivators (11, 19), which are required for optimal HMOX1 induction in our experimental settings.
Our studies revealed reduced levels of Nrf2 in cells exposed to hypoxia, which correlates with reduced activation or a lack of activation of several of its target gene expressions. Consistent with these observations, ChIP analysis showed decreased levels of Nrf2 binding both at distal (AB1) and proximal (E1) enhancer as well as at the proximal promoter (−44 bp) regions in acute hypoxia and reoxygenation conditions. Enhanced recruitment of Nrf2 to the ARE is crucial for the transcriptional induction of its target genes by toxicant and pro-oxidant stimuli (31). Thus, it is counterintuitive that despite decreased levels of nuclear Nrf2 and its reduced binding at the endogenous HMOX1 promoter, siRNA-mediated knockdown of endogenous Nrf2 expression profoundly inhibited hypoxia-inducible HMOX1 expression. Consistent with this result, Hmox1 induction by hypoxia in primary kidney epithelial cells lacking Nrf2 was markedly lower than in their wild-type counterparts (Fig. 9), demonstrating the requirement of Nrf2 for hypoxia-inducible HMOX1 transcription. It was demonstrated that Nrf2-regulated ferritin gene induction by hypoxia is modulated through a posttranscriptional mechanism, and not through the ARE (10). However, mRNA stability experiments have ruled out involvement of posttranscriptional regulation (data not shown), suggesting that Nrf2, through modulation of other transcription factor(s), regulates HMOX1 induction in response to acute hypoxia in kidney epithelia. The Hmox1 promoter reporter constructs show lack of inducible activity in acute (2 h) hypoxia, although Hmox1 mRNA expression was significantly increased by hypoxia. Thus, it is likely that the discrepancy between promoter activity and HMOX1 mRNA expression results could be attributable to inherent differences between luciferase (mRNA and protein) expression and/or activity and HMOX1 expression. It is also possible that the promoter reporter constructs used in our studies (which optimally respond to oxidant stimuli) lack other upstream and downstream genomic elements (6, 17) required for an optimal HMOX1 promoter activation in acute hypoxia.
Notably, our studies revealed increased Nrf2 recruitment at the AB1 and E1 enhancers during chronic hypoxia and reoxygenation, which corresponds with enhanced reporter gene expression driven by the HMOX1 promoter bearing the functional AREs of AB1 (Fig. 7) or E1 (Fig. 8) enhancer. Nrf2 binding at E1 was increased in cells exposed to chronic hypoxia, but it was returned to the basal state as early as 1 h during reoxygenation (Fig. 8B). In contrast, increased Nrf2 binding can be detectable at the distal AB1 enhancer during reoxygenation (Fig. 7B), suggesting a differential binding of Nrf2 to the proximal and distal enhancer regions during chronic hypoxia and reoxygenation in kidney epithelia. Thus, we propose that Nrf2 indirectly upregulates HMOX1 expression during acute hypoxia, whereas it binds directly to the AREs and drives its (latter) transcription during chronic hypoxia and reoxygenation.
In summary, our studies demonstrate that Nrf2 is a critical positive regulator of HMOX1 induction both in acute and chronic hypoxia and hypoxia-reoxygenation conditions, and that it mediates this regulation indirectly and directly, respectively. Induction of HMOX1 expression but no other putative transcriptional targets of Nrf2 imply selective upregulation of antioxidant gene transcription during hypoxia and hypoxia-reoxygenation in kidney epithelia.
GRANTS
Support for this study was provided by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-084445 to H. Rabb and S. P. Reddy.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.R.P., H.R., and S.P.R. conceived and designed research; H.R.P., C.R.T., and R.M. performed experiments; H.R.P., N.M.R., and S.P.R. analyzed data; H.R.P., S.N., and S.P.R. interpreted results of experiments; H.R.P. prepared figures; H.R.P. and S.P.R. drafted manuscript; H.R.P., S.N., H.R., and S.P.R. edited and revised manuscript; H.R.P., C.R.T., R.M., N.M.R., S.N., H.R., and S.P.R. approved final version of manuscript.
REFERENCES
- 1.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol 11: 965–973, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol 36: 166–174, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bell CL, Tenenhouse HS, Scriver CR. Initiation and characterization of primary mouse kidney epithelial cultures. In Vitro Cell Dev Biol 24: 683–695, 1988. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Wei SY, Li JS, Zhang QF, Wang YX, Zhao SL, Yu J, Wang C, Qin Y, Wei QJ, Lv GX, Li B. Overexpression of heme oxygenase-1 prevents renal interstitial inflammation and fibrosis induced by unilateral ureter obstruction. PLoS One 11: e0147084, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornblatt BS, Ye L, Dinkova-Kostova AT, Erb M, Fahey JW, Singh NK, Chen MS, Stierer T, Garrett-Mayer E, Argani P, Davidson NE, Talalay P, Kensler TW, Visvanathan K. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 28: 1485–1490, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Deshane J, Kim J, Bolisetty S, Hock TD, Hill-Kapturczak N, Agarwal A. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J Biol Chem 285: 16476–16486, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Himmelfarb J, McMonagle E, Freedman S, Klenzak J, McMenamin E, Le P, Pupim LB, Ikizler TA; The PICARD Group. Oxidative stress is increased in critically ill patients with acute renal failure. J Am Soc Nephrol 15: 2449–2456, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Hock TD, Liby K, Wright MM, McConnell S, Schorpp-Kistner M, Ryan TM, Agarwal A. JunB and JunD regulate human heme oxygenase-1 gene expression in renal epithelial cells. J Biol Chem 282: 6875–6886, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Hock TD, Nick HS, Agarwal A. Upstream stimulatory factors, USF1 and USF2, bind to the human haem oxygenase-1 proximal promoter in vivo and regulate its transcription. Biochem J 383: 209–218, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang BW, Miyazawa M, Tsuji Y. Distinct regulatory mechanisms of the human ferritin gene by hypoxia and hypoxia mimetic cobalt chloride at the transcriptional and post-transcriptional levels. Cell Signal 26: 2702–2709, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janknecht R, Nordheim A. MAP kinase-dependent transcriptional coactivation by Elk-1 and its cofactor CBP. Biochem Biophys Res Commun 228: 831–837, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Jo SK, Cho WY, Sung SA, Kim HK, Won NH. MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis. Kidney Int 67: 458–466, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Kang KW, Lee SJ, Kim SG. Molecular mechanism of Nrf2 activation by oxidative stress. Antioxid Redox Signal 7: 1664–1673, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Kim KC, Kang KA, Zhang R, Piao MJ, Kim GY, Kang MY, Lee SJ, Lee NH, Surh YJ, Hyun JW. Up-regulation of Nrf2-mediated heme oxygenase-1 expression by eckol, a phlorotannin compound, through activation of Erk and PI3K/Akt. Int J Biochem Cell Biol 42: 297–305, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal 7: 385–394, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272: 5375–5381, 1997. [PubMed] [Google Scholar]
- 18.Lever JM, Boddu R, George JF, Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxid Redox Signal 25: 165–183, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li QJ, Yang SH, Maeda Y, Sladek FM, Sharrocks AD, Martins-Green M. MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300. EMBO J 22: 281–291, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, Reddy SP, Rabb H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int 76: 277–285, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Liu M, Reddy NM, Higbee EM, Potteti HR, Noel S, Racusen L, Kensler TW, Sporn MB, Reddy SP, Rabb H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int 85: 134–141, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molitoris BA. Ischemic acute renal failure: exciting times at our fingertips. Curr Opin Nephrol Hypertens 7: 405–406, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Nath KA. Heme oxygenase-1 and acute kidney injury. Curr Opin Nephrol Hypertens 23: 17–24, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nath KA. Human AKI and heme oxygenase-1. J Am Soc Nephrol 23: 971–974, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nath KA, Norby SM. Reactive oxygen species and acute renal failure. Am J Med 109: 665–678, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 43: 233–260, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Noiri E, Nakao A, Uchida K, Tsukahara H, Ohno M, Fujita T, Brodsky S, Goligorsky MS. Oxidative and nitrosative stress in acute renal ischemia. Am J Physiol Renal Physiol 281: F948–F957, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Papaiahgari S, Zhang Q, Kleeberger SR, Cho HY, Reddy SP. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid Redox Signal 8: 43–52, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: pathophysiologic correlates. Kidney Int 68: 611–622, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Potteti HR, Reddy NM, Hei TK, Kalvakolanu DV, Reddy SP. The NRF2 activation and antioxidative response are not impaired overall during hyperoxia-induced lung epithelial cell death. Oxid Med Cell Longev 2013: 798401, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy SP. The antioxidant response element and oxidative stress modifiers in airway diseases. Curr Mol Med 8: 376–383, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278: F726–F736, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: molecular regulation of the heme oxygenase-1 gene in renal injury. Am J Physiol Renal Physiol 286: F425–F441, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Terasaki PI, Cecka JM, Gjertson DW, Takemoto S. High survival rates of kidney transplants from spousal and living unrelated donors. N Engl J Med 333: 333–336, 1995. [DOI] [PubMed] [Google Scholar]
- 35.Ueda K, Xu J, Morimoto H, Kawabe A, Imaoka S. MafG controls the hypoxic response of cells by accumulating HIF-1alpha in the nuclei. FEBS Lett 582: 2357–2364, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA, Grossman E, Chen J, Zhou XJ, Hartono J, Winterberg P, Chen B, Agarwal A, Lu CY. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am J Physiol Renal Physiol 300: F1180–F1192, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Bedard EL, Potter R, Zhong R, Alam J, Choi AM, Lee PJ. Mitogen-activated protein kinases regulate HO-1 gene transcription after ischemia-reperfusion lung injury. Am J Physiol Lung Cell Mol Physiol 283: L815–L829, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Zhao R, Feng J, He G. Hypoxia increases Nrf2-induced HO-1 expression via the PI3K/Akt pathway. Front Biosci (Landmark Ed) 21: 385–396, 2016. [DOI] [PubMed] [Google Scholar]