

Abstract
Aquaporin-2 (AQP2) is essential to maintain body water homeostasis. AQP2 traffics from intracellular vesicles to the apical membrane of kidney collecting duct principal cells in response to vasopressin [arginine vasopressin (AVP)], a hormone released with low intravascular volume, which causes decreased kidney perfusion. Decreased kidney perfusion activates AMP-activated kinase (AMPK), a metabolic sensor that inhibits the activity of several transport proteins. We hypothesized that AMPK activation also inhibits AQP2 function. These putative AMPK effects could protect interstitial ionic gradients required for urinary concentration during metabolic stress when low intravascular volume induces AVP release. Here we found that short-term AMPK activation by treatment with 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR; 75 min) in kidney tissue prevented baseline AQP2 apical accumulation in principal cells, but did not prevent AQP2 apical accumulation in response to the AVP analog desmopressin (dDAVP). Prolonged AMPK activation prevented AQP2 cell membrane accumulation in response to forskolin in mouse collecting duct mpkCCDc14 cells. Moreover, AMPK inhibition accelerated hypotonic lysis of Xenopus oocytes expressing AQP2. We performed phosphorylation assays to elucidate the mechanism by which AMPK regulates AQP2. Although AMPK weakly phosphorylated immunoprecipitated AQP2 in vitro, no direct AMPK phosphorylation of the AQP2 COOH-terminus was detected by mass spectrometry. AMPK promoted Ser-261 phosphorylation and antagonized dDAVP-dependent phosphorylation of other AQP2 COOH-terminal sites in cells. Our findings suggest an increasing, time-dependent antagonism of AMPK on AQP2 regulation with AICAR-dependent inhibition of cAMP-dependent apical accumulation and AVP-dependent phosphorylation of AQP2. This inhibition likely occurs via a mechanism that does not involve direct AQP2 phosphorylation by AMPK.
Keywords: epithelial, PKA, water transport, metformin, mpkCCDc14
mammals conserve fluids largely due to the kidneys' ability to concentrate the urine. The cortico-medullary osmotic gradient is generated in the kidney parenchyma by the loop of Henle's countercurrent multiplication system, where water and solute transport play major roles (48). Aquaporins expressed in kidney epithelium are essential for the maintenance of total body volume homeostasis (31). In the kidney collecting duct, the water channel aquaporin-2 (AQP2) is expressed predominantly at the apical membrane. The hormone vasopressin [antidiuretic hormone or arginine vasopressin (AVP)] is synthesized in the hypothalamus and then stored and secreted from the posterior pituitary gland in response to increased serum osmolality or decreased blood volume (31). In kidney collecting duct principal cells, AVP binds the type II vasopressin receptor (V2R), inducing a cascade that involves increases in cyclic AMP (cAMP). This cascade results in insertion of AQP2-containing vesicles into the apical membrane of principal cells, water reabsorption from the urine, and reestablishment of a normal plasma osmolality and intravascular volume (31). Downstream of AVP, AQP2 subcellular localization is regulated by the phosphorylation status of several residues in its COOH-terminus (S256, S261, S264, and S269; reviewed in Ref. 37). The regulatory effects of differential phosphorylation at these sites are still being elucidated. In particular, the kinases downstream of AVP that are involved in the direct phosphorylation of AQP2 are still under active investigation (14, 27).
The transport of Na+, Cl−, and urea into the kidney's interstitium is essential for the establishment of the osmotic gradient that allows for passive reabsorption of water via AQP2 and urinary concentration. However, when the metabolic sensor AMP-activated protein kinase (AMPK) is activated under metabolic stress, epithelial cells may be unable to effectively maintain solute transport, and thus the maintenance of this gradient could be compromised (20, 40). Specifically, if there was unopposed and ongoing accumulation of AQP2 at the principal cell apical membrane under conditions when salt transport is inhibited (e.g., with AMPK activated secondary to metabolic stress from hypovolemia/hypoperfusion), AQP2-facilitated transepithelial water transport would tend to wash out the concentrated medullary interstitium via the vasa recta countercurrent exchange system, thus dissipating this metabolically expensive cortico-medullary gradient.
AMPK is a serine/threonine protein kinase that acts as a sensitive gauge of cellular energy status (23). Under conditions of metabolic stress like hypoxia and ischemia, slight drops in cellular [ATP] (where brackets denote concentration) result in significant rises in [ADP] and [AMP] and the activation of AMPK (20, 42). Once activated, AMPK phosphorylates many substrates, leading to a decrease of cellular ATP consumption and stimulation of ATP synthesis, thereby maintaining ATP levels in the face of energy depletion. Our group discovered that, in epithelial cells, AMPK regulates a number of transport proteins, such as the cystic fibrosis transmembrane conductance regulator, the epithelial sodium channel (ENaC), the creatine transporter, the vacuolar H+-ATPase (V-ATPase), and the K+ channel KCNQ1 (1, 4, 10, 18, 22, 34). In the kidney, AMPK is quickly and dramatically upregulated in ischemic injury (40). Moreover, we have shown that AMPK inhibits the PKA-mediated trafficking of the V-ATPase to the apical membrane in epididymal clear cells, proximal tubule S3 segment epithelial cells (1, 21), and in kidney intercalated cells (3, 18).
We hypothesized that AMPK could induce intracellular accumulation of AQP2 via changes in phosphorylation of the channel and inhibition of cAMP-mediated AQP2 trafficking to the membrane. Downregulation of AQP2 by AMPK would contribute to the AMPK-dependent protection of ionic gradients in the kidney, an effect that could be beneficial for kidney cells and for the entire organisms during the acute phases of kidney ischemia (20). In addition, we considered that AQP2 inhibition by AMPK could contribute to the well-documented polyuria and inability to concentrate the urine that occurs in patients recovering from ischemic acute kidney injury (AKI) (45). Indeed, the apical distribution and abundance of AQP2 decreases in rats recovering from AKI (15, 33).
In this study, we tested whether AMPK, which is activated during ischemia, prevents acute cAMP-mediated AQP2 apical accumulation in principal cells of kidney slices ex vivo and in the polarized mouse collecting duct mpkCCDc14 cell line. We also performed functional studies to determine the rates of AQP2-mediated swelling and lysis in Xenopus oocytes coexpressing AQP2 and AMPK mutants. Finally, we examined if AMPK can directly phosphorylate AQP2 or indirectly alter AQP2 phosphorylation status following modulation of AMPK activity. Taken together, the results from this study provide insight into a potential mechanism for the observed internalization and dysfunction of AQP2 that occurs during both acute kidney ischemia and recovery from this insult.
MATERIALS AND METHODS
Reagents and chemicals.
All chemical compounds used in the studies presented here were purchased from Sigma-Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Pittsburgh, PA), unless otherwise stated. The AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) was purchased from Toronto Research Chemicals (Toronto, Canada). PKA catalytic subunit (cPKA) was obtained from Promega (Madison, WI). Purified recombinant active human AMPK holoenzyme (α1-T172D,β1,γ1) was synthesized and purified as described previously (41, 50).
Plasmid constructs.
AQP2 plasmids were purchased from Open Biosystems (clone ID 4222942 for Mus musculus and clone ID 7109642 for Rattus norvegicus). Each plasmid was subcloned by PCR into the dual mammalian-oocyte expression vector pMO (21) using its EcoRI and HindIII restriction sites to generate the pMO-V5-AQP2 plasmids for each species. The forward primer was 5′TCAGAATTCACCATGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTAGTGGGAACTCAGATCCATA-3′. The reverse primer was 5′-CCCAAGCTTGGGTCA GGCCTTGCTG-3′.
All clones were confirmed by DNA sequencing. Complementary RNAs (cRNAs) for mouse AQP2 and either wild-type (WT) AMPK-α1, kinase-dead, dominant-negative AMPK (α1-K45R), or constitutively active AMPK (α1-R70Q) were generated as described (10).
Antibodies.
A goat anti-AQP2 antibody was obtained from Santa Cruz Biotechnology (Dallas, TX). The rabbit anti-AQP2 antibody was obtained from Alomone Labs (Jerusalem, Israel). The mouse anti-β-actin antibody was obtained from Sigma-Aldrich. The secondary antibodies against horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G and anti-mouse immunoglobulin G antibodies were obtained from GE Healthcare Biosciences (Pittsburgh, PA). The anti-AMPK-α-pThr-172 antibody (raised in rabbit) was obtained from Cell Signaling Technology, whereas the anti-V5 antibodies (unconjugated and conjugated to HRP) antibodies were obtained from Invitrogen (Carlsbad, CA). Secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA).
Kidney tissue preparation and confocal immunofluorescence microscopy.
All animal protocols were performed at the University of Pittsburgh and approved by the Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Kidney slices were prepared from kidneys harvested from male Sprague-Dawley rats that had been perfused via the left ventricle with Ringer pH 7.4, at 37°C (in 5% CO2/air mixture), as described previously (9, 43). Briefly, ex vivo kidney slices were prepared from freshly harvested rat kidneys using a Stadie-Riggs microtome. The slices (∼0.5 mm in thickness) were quickly transferred to fresh vials containing Ringer buffer with or without the AMPK activator AICAR (1 mM) for 75 min at 37°C in 5% CO2/air mixture, followed by fixation (18). Simultaneously, additional kidney slices were incubated in either Ringer buffer with AICAR or Ringer buffer without AICAR for 60 min, followed by the addition of AVP analog desmopressin (dDAVP; 10 nM) and fixation at the 75-min incubation time point. Immunofluorescence labeling was subsequently performed using antibodies against AQP2 raised in goat (1:400 dilution), followed by a secondary donkey anti-goat antibody coupled to CY3 (1:800 dilution). Tissues were also colabeled with an antibody raised in chicken against the E subunit of the V-ATPase, followed by a secondary antibody raised in goat coupled to Alexa 488 (1:800 dilution) to specifically mark intercalated cells (47). Immunolabeled tissues were mounted in VECTASHIELD mounting medium (Vector Laboratories), and imaged in a confocal laser scanning microscope using identical laser settings for all samples (Leica TCS SP5, model upright DM 6000S, Leica Microsystems, Buffalo Grove, IL) and a ×63 objective.
Quantification of AQP2 apical membrane accumulation in kidney slices.
AQP2 accumulation was quantified at the apical membrane of principal cells using confocal microscopy images and Metamorph software (Molecular Devices, Sunnyvale, CA) or NIH Image, using methods validated in previous studies by the authors and others (1, 5, 8, 9, 18). The mean pixel intensity (MPI) of AQP2-associated fluorescence was measured in the same shape and size of region of interest at the apical and the cytoplasmic area next to the nucleus within each selected cell. We evaluated 30–118 cells per treatment condition from 3–6 independent rat kidney slice experiments (n = 3–6). For each treatment, the apical-to-cytoplasmic ratio of the MPI of AQP2-associated fluorescence was used to measure AQP2 apical accumulation. This value was calculated for each cell, and then a mean was obtained for each kidney. The AQP2 apical membrane accumulation for each condition is expressed as the mean ± SE.
Cell culture.
Mouse cortical collecting duct (mpkCCDc14) cells were originally derived by microdissection of cortical collecting ducts of an SVPK/Tag transgenic mouse and have been used previously to study the regulation of the ENaC and more recently AQP2 (6, 10, 39). The cells in culture were maintained in a humidified 5% CO2/95% air incubator in defined “CCD” media containing equal volumes of Dulbecco's modified Eagle's medium and Ham's F-12, 60 nM sodium selenate, 5 μg/ml transferrin, 2 mM glutamine, 50 nM dexamethasone, 1 nM triiodothyronine, 10 ng/ml epidermal growth factor, 5 μg/ml insulin, 20 mM d-glucose, 2% (vol/vol) FBS, and 20 mM HEPES, pH 7.4 (all reagents from Invitrogen Life Technologies and Sigma-Aldrich). Cells were grown to ∼90% confluency in 75-cm2 plastic flasks and then seeded onto Transwell filters before experiments (24). Human embryonic kidney (HEK)-293 cells were maintained in Dulbecco's modified Eagle's medium (Lonza, Basel, Switzerland) with 4.5 g/l glucose and l-glutamine, 10% fetal bovine serum, and penicillin-streptomycin. Cells were grown to ∼90% confluency and then were seeded onto 60-mm Petri dishes. Both cell lines were used also in transfection experiments with a rat pMO V5 epitope-tagged AQP2 plasmid in the experiments described below where immunoprecipitation was required.
Cell surface biotinylation assays in polarized mpkCCDc14 cells.
Cells grown on Transwell filter supports (24) were treated with either 10 μM forskolin for 2 h to increase intracellular cAMP, 1 mM AICAR for 4 h to activate AMPK, or both. The monolayers were then subjected to apical surface biotinylation as previously described by our laboratory (1, 34), in which cells were first washed with ice-cold PBS containing Mg2+ and Ca2+ three times for 5 min. Then the apical membrane was then biotinylated using 1 mg/ml EZ link sulfo-NHS-SS-biotin (Thermo Fisher Scientific) in ice-cold PBS for two consecutive incubation periods of 10 min. Free NHS-biotin was then quenched by washing cells with ice-cold 10% FBS in PBS. Monolayers were then washed with ice-cold PBS containing Ca2+ and Mg2+ three times, and cells were harvested using PBS containing 1% Triton X-100, 50 mM EGTA, 10 mM Tris·Cl (pH 7.4), 0.4% deoxycholate, and one tablet of complete protease inhibitor cocktail (Roche, Basel, Switzerland), and complete phosphatase inhibitor cocktail (Roche). Protein concentration was determined using the Bradford technique, and 500 μg of protein were incubated with 30 μl of streptavidin-agarose beads (50%, Thermo Fisher Scientific) in a total volume of 1 ml overnight at 4°C, while kept in constant rotation. The beads were washed three times in lysis buffer, and the biotinylated proteins were eluted from the beads by boiling samples in 2 × Laemmli sample buffer containing 20% DTT. The protein samples were then separated by SDS-PAGE and subjected to immunoblot analysis using anti-AQP2 polyclonal anti-goat (Santa Cruz Biotechnology) and anti-β-actin monoclonal anti-mouse (Sigma) antibodies.
AQP2 in vitro phosphorylation assays.
HEK-293 cells were seeded onto 60-mm petri dishes at a density of 5 × 105 cells/dish (4). Cells were grown to ∼90% confluency and then transiently transfected according to the manufacturer's protocol using Lipofectamine 2000 (Invitrogen, Life Technologies, Carlsbad, CA) to express V5 epitope-tagged AQP2. Two days after transfection, the tagged AQP2 was immunoprecipitated from precleared lysates using an anti-V5 antibody (Invitrogen), as described previously (3, 5). The in vitro phosphorylation was performed using purified active AMPK holoenzyme or cPKA as a positive control with [γ-32P]ATP labeling (3, 5). The immunoprecipitation samples were subjected to SDS-PAGE and immunoblotting using anti-V5 antibody coupled to HRP (1:10,000). The probed protein and phosphorylated proteins were then visualized and quantified, as described above (5).
Mass spectrometry analysis of in vitro translated AQP2 after phosphorylation assays.
Peptides corresponding to the complete carboxyl-terminal tail of human AQP2 were subjected to in vitro phosphorylation assays using purified recombinant active human AMPK holoenzyme (α1-T172D,β1,γ1), as described previously (25). A purified SAMS peptide was used as a positive control. Subsequently, peptides were digested by either trypsin or Lys-C, and the resulting peptides (or nondigested full-length 44-amino acid peptides) subjected to nano-liquid chromatography (Ultimate 3000, Dionex)-tandem mass spectrometry (Q Exactive, Thermo Fisher Scientific) analysis, separately with a 30-min gradient, as previously described (11). Target sequences were appended to the total yeast proteome sequences as the database to prevent forced matching.
AQP2 phosphorylation assays in mpkCCDc14 cells.
Cells were seeded onto 60-mm Petri dishes (5 × 105 cells/dish), grown to 90% confluency, and then transiently transfected to express V5 epitope-tagged WT AQP2 (rat sequence). Two days after transfection, [32P]orthophosphate labeling in mpkCCDc14 cells was performed, as previously described (3, 5, 21), in the presence or absence of AMPK activator (AICAR, 2 mM, 20 h), with dDAVP (10 nM) being added during the last 30 min of the labeling incubation. After the labeling was completed, V5-tagged AQP2 was immunoprecipitated from precleared lysates using the anti-V5 antibody. Immunoblotting was performed with anti-V5 antibody directly coupled to HRP (1:10,000 dilution) and quantified using a Versa-Doc Imager with Quantity One software (Bio-Rad, Hercules, CA). Phosphorylated bands on the membrane were identified by exposure of the membrane to a phosphoscreen, and bands were quantitated using a Bio-Rad PhosphorImager with Quantity One software.
AQP2 expression in Xenopus oocytes and hypotonic shock.
Maintenance of Xenopus laevis frogs, surgical extraction of ovaries, and collagenase treatment of oocytes were performed as previously described, in accordance with a protocol approved by the University of Pittsburgh Institutional Animal Care and Use Committee (10, 28). cRNAs for AQP2 (mouse sequence) and different AMPK constructs were synthesized, as described previously (10, 22), using the T7 mMessage mMachine kit (Ambion, Austin, TX). The purity and quantity of cRNA were assessed by agarose gel electrophoresis. Stage V–VI oocytes were microinjected with cRNA for both AQP2 and either WT AMPK, kinase-dead, dominant-negative AMPK (α1-K45R), or constitutively active AMPK (γ1-R70Q) (10). After 3 days, the oocytes were subjected to hypotonic shock by placing them in deionized water. The time-to-lysis for each condition was measured by direct visualization under the microscope. Fifty-two to sixty oocytes from three separate batches were microinjected per condition.
AQP2 phosphorylation site evaluation in mpkCCDc14 cells.
Cells were grown in 60-mm dishes to ∼90% confluency and then transiently transfected with a pMO plasmid expressing rat V5-tagged AQP2 for 2 days. During the 2nd day, cells were treated with AICAR (2 mM) for 4 h, while dDAVP (15 nM) was added during the last 30 min of the incubation. After 24 h, the transfected cells were harvested and underwent immunoprecipitation using an anti-V5 antibody (raised in mouse) following methods previously described by our laboratory (7). Immunoblot analysis of immunoprecipitated V5-AQP2 was performed using the corresponding antibodies against phosphorylated AQP2 COOH-terminal sites: −S256, −S261, −S264, and −S269. Rabbit polyclonal phospho-specific antibodies were purchased from PhosphoSolutions (Aurora, CO). After each incubation of the membrane with a particular anti-phospho COOH-terminal antibody, the nitrocellulose membrane was stripped and reblotted using anti-AQP2 antibody (Alomone Labs). To confirm that immunoprecipitation had been successful, we also reblotted the membranes using an anti-V5 antibody coupled to HRP (Sigma). Differences in relative phosphorylation across conditions were assessed by comparing the phospho-specific band intensity normalized to the AQP2 expression level on reprobe of the membrane.
Statistical analysis.
All statistical analyses were performed using commercially available software (SIGMASTAT, Systat Software, CA; or Microsoft Excel Data Analysis Tools, Redmond, WA). Single comparisons were performed using two-tailed unpaired Student's t-test, assuming unequal variances. Group comparisons were performed using one-way ANOVA. When ANOVA showed significant differences among treatments, we performed post hoc analyses (Tukey) to determine whether the specific treatment was significantly different from the control. In all cases, P values <0.05 were considered significant.
RESULTS
AMPK inhibits AQP2 apical accumulation in principal cells in kidney tissue or in a collecting duct cell line.
To test whether AMPK activation had effects on AQP2 subcellular localization, we incubated kidney tissue slices ex vivo in the absence or presence of the AMPK activator AICAR. Our laboratory has routinely used the kidney slices method (9) to study the trafficking of transport proteins ex vivo, and we have previously shown that incubation with AICAR for 75 min induces acute AMPK activation in these slices (1, 4, 18). As shown in Fig. 1, A and B, AICAR-treated slices revealed cytosolic AQP2 immunolabeling in principal cells, while untreated slices displayed an apical distribution of AQP2. A significantly more diffuse cytosolic distribution of AQP2 following AMPK activation was quantified in several independent kidney slice preparations, as shown in Fig. 1E. Our laboratory has previously shown that incubation of kidney slices in bicarbonate-containing Ringer buffer gassed with 5% CO2 for 75 min induces PKA activation downstream of bicarbonate-induced activation of soluble adenylyl cyclase (18). We thus hypothesized that the observed baseline accumulation of AQP2 at the apical membrane occurred secondary to activation of PKA, which we tested by comparing the distribution of AQP2 in Ringer buffer for 75 min in the absence or presence of myristoylated protein kinase inhibitor, a specific inhibitor of PKA (Fig. 1, F and G). Of note, the subcellular distribution of AQP2 was more diffuse at earlier time points in Ringer buffer (not shown). We found that treatment with myristoylated protein kinase inhibitor prevented the apical accumulation of AQP2 in multiple replicate experiments (Fig. 1H). Together, these findings suggest that AMPK regulates the subcellular distribution of AQP2 in collecting duct principal cells, antagonizing the effects of cAMP/PKA, which acts to promote accumulation of AQP2 at the apical membrane.
Fig. 1.

AMPK activator AICAR induces intracellular redistribution of AQP2 in ex vivo kidney slices. A: confocal images of AQP2 immunofluorescence labeling (green) in kidney slices incubated in Ringer buffer alone for 75 min display an apical distribution of the water channel. Asterisks indicate the position of the lumen of the collecting ducts. B: addition of 1 mM AICAR for 75 min induced a cytosolic distribution of AQP2. C: incubation of kidney slices with dDAVP for the last 15 min of a 75-min incubation revealed apical accumulation of AQP2. D: however, preincubation of slices with AICAR did not prevent the dDAVP-induced apical accumulation of AQP2. E: regions of interest (ROIs; example shown in A) were outlined for each cell at apical and cytoplasmic regions for quantification using previously described methods (18). Quantification of the mean (±SE) AQP2-associated mean pixel intensity (MPI) of apical-to-cytoplasmic ratio [arbitrary units (AU)] relative to that of cells measured under the control condition was used as a measure of apical AQP2 accumulation. Additional incubations of kidney slices in Ringer buffer were performed in the absence (F) or presence (G) of the PKA inhibitor myristoylated protein kinase inhibitor (mPKI; 10 μM) for 75 min. H: the relative AQP2 mean pixel intensity apical-to-cytoplasmic ratio was reduced dramatically with mPKI treatment. Data were obtained from at least 3 separate kidney slice experiments, using kidneys from at least 3 animals, and measuring a total of at least 30 cells per condition. *P < 0.05 vs. control (Ringer pH 7.4 for 75 min; ANOVA followed by unpaired t-tests for E and unpaired t-test for H). Scale bars ∼10 μm.
We then studied whether AMPK has an inhibitory effect on the apical accumulation of AQP2 induced by the vasopressin analog dDAVP, which is reported to promote AQP2 trafficking to the apical membrane largely via increased cAMP (38). To test this, we incubated the slices ex vivo in Ringer buffer in the presence or absence of AICAR for 75 min, supplemented by addition of dDAVP for the last 15 min of the 75-min incubation to all slices. The confocal images of these tissues (Fig. 1, C and D) and the quantification of MPI of AQP2 associated fluorescence (Fig. 1E) revealed that either in the presence or absence of AICAR, dDAVP treatment was effective in inducing apical accumulation of AQP2 in principal cells. This result suggests that there are cAMP/PKA-independent effects of dDAVP on AQP2 that are insensitive to AMPK effects during this acute period.
To determine whether the AICAR-induced cytoplasmic redistribution of AQP2 observed in kidney slices represents changes in water channel expression at the apical plasma membrane, we performed surface biotinylation experiments in polarized mpkCCDc14 cells grown on permeable supports in the presence or absence of pharmacological PKA stimulation. These cells are a good model system to perform functional and biochemical studies on AQP2, as they exhibit several major functional properties of collecting duct principal cells, including AVP-stimulated alterations in AQP2 distribution and water permeability (35).
The polarized mpkCCDc14 cells grown on filter supports were treated with either forskolin to increase intracellular cAMP and as a surrogate for AVP activation, AICAR to activate AMPK, or both. Forskolin induces AQP2 trafficking to the apical membrane of several cell lines of principal cell origin (8). Moreover, the use of forskolin effectively eliminates any potential contributions of the treatment (i.e., AICAR) on membrane expression of the V2R. Surface biotinylation assays revealed that forskolin treatment (10 μM for 2 h) increased both the total cellular expression of AQP2 and the amount of AQP2 expressed at the apical membrane (Fig. 2). However, 1 mM AICAR treatment (4 h) in combination with forskolin for the last 2 h blocked the forskolin-induced increases in both total cellular expression and apical membrane expression of the core-glycosylated form of AQP2. AICAR also blunted the increase in the proportion of core-glycosylated AQP2 at the apical membrane relative to total cellular core-glycosylated AQP2 induced by forskolin (not shown). Treatment with AICAR and forskolin produced the expected increases in cellular AMPK and PKA activities, respectively, as evidenced by increased phosphorylation of AMPK-α at its Thr-172 activation site (Fig. 2C) and increased phosphorylation of multiple cellular PKA targets, as detected by the phospho-PKA substrate antibody (Fig. 2D) on immunoblots.
Fig. 2.

AMPK activator AICAR prevents apical membrane accumulation of AQP2. Polarized mpkCCDc14 cells were treated with vehicle (DMSO) or the AMPK activator AICAR (1 mM) for 4 h and then 10 μM forskolin (vs. vehicle) for the last 2 h before cell harvesting. 5% of the cell lysates were used for immunoblotting using antibodies against AQP2 or actin as a loading control. The rest of the lysates were affinity purified using streptavidin followed by immunoblotting. A: representative immunoblots of 3 separate surface biotinylation experiments are shown. B: summary densitometric data comparing the mean (±SE) amounts of apical AQP2 as a percentage of that in the control condition for nonglycosylated (solid bars), core glycosylated (open bars), and fully glycosylated (shaded bars) forms of AQP2 are shown. Nonglycosylated: *P < 0.05, control vs. forskolin; core-glycosylated: **P < 0.05, control vs. forskolin, #P < 0.05, forskolin vs. AICAR + forskolin; fully glycosylated: ***P < 0.05, control vs. forskolin (one-tailed unpaired t-tests). Representative immunoblots were performed using antibodies against total AMPK-α, pThr-172 AMPK-α, and β-actin (C) and phospho-PKA substrate (D) under the above treatment conditions.
AMPK inhibits AQP2 function in oocytes.
To assess whether AMPK activity has an effect on AQP2 functional activity in vitro, we expressed mouse AQP2 in Xenopus oocytes, along with either WT AMPK-α1, kinase-dead, dominant-negative AMPK (α1-K45R), or constitutively active AMPK (α1-R70Q) (10). Three days after injection of the AQP2 cRNA, the oocytes were subjected to hypotonic shock. This technique has long been used as a functional assay for water channels (13). The time to lysis for each condition was measured and normalized to the values obtained in oocytes expressing WT AMPK (Fig. 3). The dominant-negative AMPK-injected oocytes had a significant 15–20% shorter mean time to lysis, suggesting that blocking AMPK activity increased the rate of swelling due to water flow into the oocyte cytoplasm via AQP2. Taken together, the activation of water transport by AMPK inhibition in oocytes is consistent with the inhibitory effect of AMPK activation by AICAR on AQP2 apical membrane expression in both kidney slices and in polarized collecting duct cells.
Fig. 3.
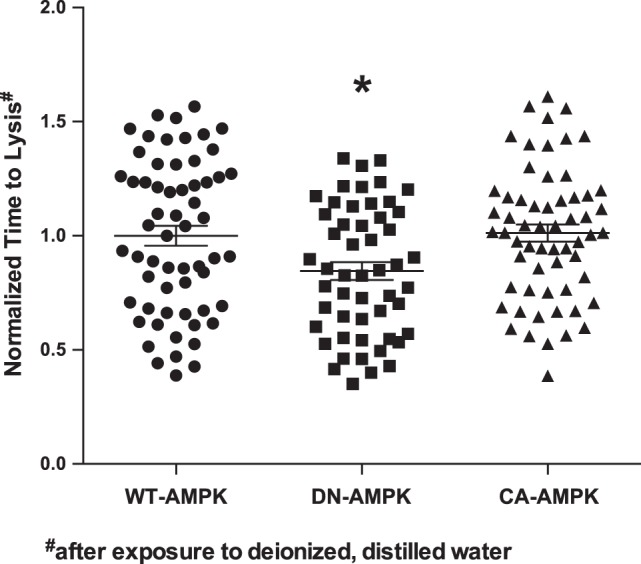
AMPK inhibition promotes AQP2-mediated oocyte swelling and shortens time to lysis. Oocytes were microinjected with cRNAs for AQP2 and the different indicated AMPK constructs [WT-AMPK-α (●), dominant-negative (DN) AMPK-α1-K45R (■), and constitutively active (CA) AMPK-γ1-R70Q (▲)]. After 3 days, the oocytes were subjected to hypotonic shock in deionized water. Oocyte times to lysis were assessed by microscopy, and the mean times to lysis (normalized to that of the WT-AMPK-expressing group for each batch) are shown for all of the data obtained for these 3 groups. Oocytes expressing DN-AMPK had a significantly reduced time to lysis relative to each of the other two groups. *P < 0.01; unpaired t-tests. The times to lysis ranged from 93 to 574 s for the whole population. 52–60 oocytes from 3 separate batches were measured per condition.
AQP2 phosphorylation was not detected in vitro.
We initially hypothesized that AMPK inhibited AQP2 plasma membrane expression via a mechanism that involved direct phosphorylation of the water channel by this kinase. To investigate this possibility, we first performed a traditional “in vitro” phosphorylation assay using immunoprecipitated rat AQP2 and purified kinases following protocols extensively used in our laboratory (3, 5). We expressed rat V5-tagged AQP2 in HEK-293 cells and incubated the immunoprecipitated water channel with [γ-32P]ATP and recombinant active purified AMPK holoenzyme (41). In a parallel reaction, we incubated AQP2 with the active cPKA. In our assay, PKA phosphorylated AQP2 as expected (4, 10, 37) (Fig. 4). However, purified AMPK only appeared to phosphorylate AQP2 very weakly. Moreover, in certain repeat in vitro phosphorylation experiments, we were unable to detect any phosphorylation of AQP2 by AMPK under the same conditions (not shown). We hypothesized that the weak, inconsistent phosphorylation by AMPK in these experiments might have represented background phosphorylation by other kinases that were inconsistently retained in the AQP2 immunoprecipitation step, before the addition of purified AMPK. To resolve this issue, we performed in vitro phosphorylation assays on a purified peptide corresponding to the COOH-cytoplasmic tail of human AQP2 (residues 225–271) in the presence or absence of purified AMPK holoenzyme (either WT AMPK-α1,β1,γ1, or a dominant-active AMPK-α1-T172D,β1,γ1) (Fig. 5). We confirmed the in vitro activity of the kinase by performing parallel experiments using the SAMS peptide, a positive-control target peptide for AMPK phosphorylation (12). The resulting phosphorylated proteins were subjected to mass spectrometry. Our results revealed that, while SAMS peptide was heavily phosphorylated by AMPK in these studies, AQP2 had only background phosphorylation at the COOH-terminus, in either the presence or absence of purified AMPK (Fig. 5).
Fig. 4.
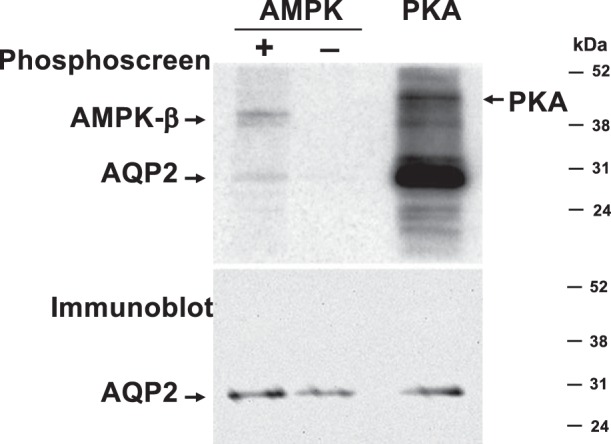
AMPK fails to significantly phosphorylate AQP2 in vitro. V5-tagged AQP2 was expressed in HEK-293 cells, immunoprecipitated, and incubated with [γ-32P]ATP in the presence of PKA catalytic subunit (positive control) or in the presence or absence of AMPK holoenzyme. A phosphoscreen image (top) and immunoblot (bottom) of the same membrane are shown (representative of 4 experiments). AQP2 gets robustly phosphorylated in the presence of PKA, but only weakly phosphorylated in the presence of AMPK (top). PKA catalytic subunit and the AMPK β-subunit get autophosphorylated, as indicated. The immunoblot (bottom) reveals similar AQP2 loading in all lanes.
Fig. 5.

Mass spectrometry analysis of synthetic peptides corresponding to the COOH-terminal tail of human AQP2 did not detect any significant phosphorylation of the water channel by AMPK. Protease-treated peptides (trypsin or LysC) or full-length peptides are indicated with bold underlined amino acids, indicating where phosphorylation was detected. The no. in parentheses indicates the adjusted spectral count of that particular phosphorylation site. The adjusted spectral count is obtained by multiplying the spectral count of each site with its phosphorylated probability reported by PhosphoRS. The last column indicates the percent sequence coverage. SAMS peptide was used as a positive control.
The AMPK activator AICAR inhibits dDAVP-mediated AQP2 phosphorylation in intact mpkCCDc14 cells.
Although there was no detectable direct phosphorylation of AQP2 by AMPK in vitro, we considered that AMPK could modulate AQP2 phosphorylation downstream of vasopressin signaling in intact cells. We, therefore, performed [32P]orthophosphate labeling in intact mpkCCDc14 cells transfected with rat V5-tagged AQP2. These labeling experiments were performed following treatment with dDAVP, AICAR, or both (Fig. 6). As expected, dDAVP treatment significantly increased the phosphorylation of cellular AQP2 to ∼2.5 times that of levels observed for vehicle control-treated cells (Fig. 6B). AMPK activation alone did not induce any significant changes in AQP2 phosphorylation status. However, treatment with the AMPK activator AICAR prevented the dDAVP-induced increase in AQP2 phosphorylation. These results can be explained by a regulatory model wherein AMPK activation, downstream of metabolic stress, indirectly modulates AVP-dependent phosphorylation by affecting other kinases and/or phosphatases. Moreover, our results confirm that there is a low, basal level of AQP2 phosphorylation in unstimulated cells.
Fig. 6.

AMPK activator blocks vasopressin-induced phosphorylation of AQP2 in mpkCCDc14 cells. mpkCCDc14 cells were transiently transfected with V5-tagged rat wild-type AQP2 plasmid. Twenty-four hours posttransfection, cells were incubated in the presence or absence of AICAR (2 mM) for 20 h, and then dDAVP (10 nM) or vehicle was added during the last 30 min of the incubation. [32P]orthophosphate labeling of cells before cell lysis, immunoprecipitation, SDS-PAGE, and immunoblotting and phosphoscreen exposure of the same membrane were then performed as described in materials and methods. A: typical phosphoscreen image (top) revealing the signal of AMPK in vivo phosphorylated AQP2. The immunoblots (bottom) confirm similar protein expression and loading of the gel for the different conditions. B: quantification of AQP2 phosphorylation signal normalized for protein loading, as assessed by densitometry of the immunoblot. While AMPK activation alone did not induce significant changes in AQP2 phosphorylation, treatment with the AMPK activator AICAR prevented the dDAVP-induced increase in AQP2 phosphorylation. Values are means ± SE of 4 independent experiments. *P < 0.05 relative to the vehicle control.
AMPK modulates the phosphorylation of AQP2 at various COOH-terminal residues.
To more carefully dissect the effects of AMPK on the phosphorylation status of AQP2, we performed immunoblotting using phospho-specific antibodies directed against various known COOH-terminal phosphorylation target residues (51). mpkCCDc14 cells were treated in the presence and absence of dDAVP to stimulate cAMP and the AMPK activator AICAR before cell lysis and immunoblotting (Fig. 7). As previously reported, AVP significantly enhanced AQP2 COOH-terminal tail phosphorylation at residues Ser-256 and Ser-269, and to a lesser degree at Ser-264 (Fig. 7) (25, 29). Of note, pretreatment with the AMPK activator AICAR prevented the dDAVP-dependent phosphorylation at Ser-269, but did not have an appreciable effect on Ser-256 phosphorylation. Rather, AICAR treatment alone enhanced Ser-261 phosphorylation, an effect that was prevented by the subsequent addition of dDAVP in AICAR-treated cells. In summary, the AMPK activator AICAR promoted phosphorylation of Ser-261 and antagonized some of the effects of dDAVP on AQP2 phosphorylation at other COOH-terminal sites.
Fig. 7.

Changes in AQP2 phosphorylation pattern following dDAVP treatment in the presence or absence of AMPK activator in mpkCCDc14 cells. Twenty-four hours posttransfection with wild-type AQP2 plasmid, cells were incubated in the presence or absence of AICAR (2 mM) for 4 h, and then dDAVP (10 nM) or vehicle was added for 30 min before cell harvesting. Immunoblot analysis of immunoprecipitated V5-AQP2 was performed using the corresponding rabbit polyclonal antibodies against phosphorylated AQP2 COOH-terminal sites: −S256, −S261, −S264, and −S269. To confirm successful immunoprecipitation, the membranes were reblotted using anti-AQP2 and anti-V5 antibodies coupled to HRP. A: representative set of immunoblots from these experiments using antibodies against AQP2 and its different COOH-terminal phosphorylation sites. B: quantification of AQP2 phosphorylation signal for different AQP2 COOH-terminal sites normalized for protein loading, as assessed by densitometry of the immunoblot. Values are means ± SE of 4 independent experiments. *P < 0.05 and #P = 0.07 relative to vehicle control.
DISCUSSION
Kidney ischemia and AKI are highly relevant public health concerns. However, the mechanisms that regulate kidney function under those conditions are not entirely understood, especially with respect to epithelial transport proteins. For example, rapid inhibition of AQP2 function occurs in kidney ischemia, both in animal models and in patients. Under conditions of kidney ischemia or hypoperfusion, plasma levels of the hormone AVP, which stimulates AQP2 insertion at the membrane, are often elevated in the circulation. However, this hormone may not be able to reach its receptors in principal cells under kidney ischemia or inflammation (33). Lack of AVP action could thus explain the decreased AQP2 expression at the principal cell membrane in kidney ischemia and the inability to concentrate the urine in patients with AKI.
As AMPK becomes rapidly activated in the ischemic kidney, we envisioned that AMPK could also play an important role in the downregulation of AQP2 in this setting. It is interesting that short-term treatment with the AMPK activator AICAR in kidney slices prevented apical accumulation of AQP2 in response to cAMP/PKA agonists, but not in response to dDAVP, suggesting that the AVP effect involves additional signaling pathways in this setting (Fig. 1). Of note, the AVP signaling pathway that induces AQP2 exocytosis has been reported to involve not only cAMP/PKA, but also other factors, including nitric oxide and atrial natriuretic peptide, likely via increased cGMP levels (8). Although different time points and conditions were required in the different experimental assays for technical reasons, our findings suggest that, in the short term, AMPK activation cannot overcome dDAVP-stimulated AQP2 apical accumulation. These data also suggest that the V2R can induce AQP2 trafficking independent of the cAMP/PKA pathway. In contrast to short-term effects, after longer time periods of AICAR treatment (>4 h), there are significant changes in AQP2 apical accumulation (Fig. 2) and in the AQP2 COOH-terminal phosphorylation pattern (Fig. 7). Overall, our results indicate that short-term effects of AMPK are to inhibit AQP2 function, by preventing apical membrane accumulation of the water channel and by decreasing AQP2 cellular expression levels. In cell culture models, this AMPK-mediated inhibition of AQP2 occurred even in the presence of agents that increase intracellular cAMP, such as forskolin. In vivo, we interpret these findings as suggesting that, if AMPK is sufficiently preactivated, the response of AQP2 to vasopressin is blunted. In general, the potential role of AMPK in dampening the effects of hormones released during hypoperfusion (AVP, aldosterone, angiotensin II) is an area worthy of further study. Of note, our preliminary studies suggest that AMPK also antagonizes the effects of these hormones on ENaC (not shown).
It has been shown that, after ischemia, there is inhibition of AQP2 function, resulting in diuresis (15). Given these findings, the regulation of AQP2 by AMPK may also make sense from a teleological standpoint. Based on our data, we propose that the regulation of AQP2 in the setting of renal hypoperfusion could have two main phases. In the first phase, when hypoperfusion begins, AVP is released and AMPK is acutely activated, yet AQP2 traffics to the membrane of principal cells. This scenario would result in conservation of free water reabsorption from the tubule lumen, as well as an attempt to defend overall intravascular volume and tissue perfusion. Of note, AMPK activation has been reported to increase Na+-K+-2Cl− cotransporter 2 activity in the thick ascending limb (16), which could concurrently promote salt reabsorption in that segment acutely. In a second phase (still relatively short term) as hypoperfusion persists, AVP actions in principal cells may become “exhausted” downstream of the V2R and/or due to reduced delivery of AVP. This lack of AVP signaling may inhibit AQP2 function. Concurrently, solute reabsorption is also inhibited by AMPK. Thus both hypoperfusion and inhibition of AQP2 by AMPK could be a contributing factor in the preservation of the interstitial gradient for future use (e.g., when the kidney starts to recover from ischemia) through a combined decrease in salt and water reabsorption. If AMPK were not to inhibit AQP2 during this second phase, then water reabsorption would become limited by dissipation of the medullary interstitial gradient via the vasa recta countercurrent exchange system with the sustained presence of AQP2 at the apical membrane. AMPK-mediated AQP2 inhibition may thus prevent dilution of this energetically expensive gradient and thereby conserve the metabolic energy required to maintain it. Furthermore, net water loss in the distal nephron during prolonged hypoperfusion may also be minimized by the proximal tubule, which may be maximally reabsorbing isotonic filtrate.
We tried to demonstrate consistent and significant AMPK-mediated phosphorylation of immunoprecipitated AQP2 in vitro assays. Our mass spectrometry findings confirmed that the carboxyl-terminal tail of AQP2 is not likely a direct target of the kinase activity of AMPK. Nonetheless, when AMPK was activated, dDAVP failed to induce the expected levels of AQP2 phosphorylation in intact cells. We observed an additional unexpected effect of AICAR on AQP2 posttranslational modifications (Fig. 2). AQP2 is an integral membrane protein that can be detected in different glycosylation states as it progresses through the biosynthetic pathway: unglycosylated (29 kDa), core glycosylated (32 kDa), and complex glycosylated (∼40–45 kDa). Increases in cAMP via forskolin induced increased levels of the core-glycosylated form, while AMPK activation antagonized this increase (32, 37).
The parallel reductions in both whole cell (lysate) and apical AQP2 expression (biotinylation) induced by the AMPK activator AICAR could reflect either decreased synthesis, or enhanced AQP2 degradation, or a combination of both. The decreases in core-glycosylated forms of AQP2 in response to AMPK activation suggest a slower rate of progression of the water channel through the biosynthetic pathway. Also, as AICAR treatment decreased overall levels of AQP2, we speculated that AMPK could be increasing ubiquitination of AQP2. However, we were unable to show significant changes when detecting endogenous AQP2 ubiquitination in the presence of AICAR (not shown).
While this paper was in preparation, another study was published examining the effects of the AMPK activator metformin on the activities of both AQP2 and the urea transporter UT-A1 in inner medullary collecting ducts in vivo and in ex vivo tubule preparations (30). In contrast to our findings, Klein et al. (30) observed an apparent increase in functional AQP2 expression and activity following metformin treatment. A number of factors may account for the apparent differences in their study. First, the study used metformin, a different pharmacological agent that is reported to activate AMPK through a mechanism distinct from AICAR and also has other pleiotropic, AMPK-independent effects [e.g., inhibiting complex I in the mitochondrial respiratory chain and inhibiting gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase (36, 44, 53)]. Moreover, the time course of treatments differed from our studies reported here. In the cited paper by Klein et al., it is not clear what the effects were on AMPK activity under the conditions of their experimental maneuvers, as phospho-AMPK activity was not measured. Of note, although they reported an increase in AQP2 phosphorylation following metformin treatment, any AMPK effect on phosphorylation status of the water channel is likely indirect and mediated through other kinases (or phosphatases), as we have found that AMPK does not significantly phosphorylate AQP2 directly (Figs. 4 and 5).
Regarding the changes in phosphorylation status of AQP2 following AMPK activation, it appears that the AMPK activator AICAR has inhibitory effects on the previously described vasopressin/cAMP-stimulated phosphorylation sites at Ser-256 and Ser-269, while conversely promoting phosphorylation at Ser-261, a site previously shown to be inhibited by AVP/cAMP signaling over 30 min in ex vivo rat inner medullary collecting duct preparations (26). Of note, Ser-256 and Ser-269 are reported to be important in the exocytosis of AQP2 and its retention at the apical membrane, respectively (17, 25). It has been reported that Ser-261 is phosphorylated by the MAP kinases ERK1/2 and JNK (46). Although it is conceivable that AMPK promotes signaling via the MAPK/ERK/JNK pathway in collecting duct cells, previous studies suggest that AMPK is actually an upstream inhibitor of this pathway via phosphorylation of BRAF in cutaneous squamous carcinoma cells, promoting its association with 14-3-3 proteins (49). However, another study suggests that AICAR treatment in a hepatic cell line causes ERK1/2 activation and stabilization of the LDL receptor, so the effects may be cell line or context specific (52). An additional likely possibility is that acute AMPK stimulation antagonizes the cAMP/PKA pathway, as our laboratory has previously reported in its antagonistic effects on the PKA stimulation of cystic fibrosis transmembrane conductance regulator and V-ATPase in relevant epithelial systems (1, 18). Indeed, our results (Figs. 1 and 2) suggest that AMPK antagonizes the effects of the cAMP/PKA pathway on AQP2 apical membrane accumulation. Further work will be required to elucidate the precise signaling pathways involved in the AMPK-dependent regulation of AQP2 phosphorylation and function.
Of potential clinical significance, it would be relevant to test whether treatment with AICAR in rodents and humans with increased AVP levels has the expected diluting effect on urine. If so, treatment with AICAR or similar compounds could be beneficial in the treatment of various diseases of AVP excess, including severe heart failure, cirrhosis, and the syndrome of inappropriate antidiuresis.
GRANTS
This work was supported by the Danish Medical Research Council and the Lundbeck Foundation (to R. A. Fenton), a VIDI-Innovational Research Grant from the Netherlands Organization of Scientific Research (NWO-ALW Grant no. 864.10.007; to D. Neumann), the Chinese Scholarship Council (to X. Zhu), and a Sanofi Fellowship Grant (to M. M. Al-bataineh). This research was also supported by the following National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grants: F32 DK097889 (to M. M. Al-bataineh), R01 DK084184 (to N. M. Pastor-Soler), R01 DK075048 (to K. R. Hallows), P30 DK079307 Pittsburgh Center for Kidney Research (to N. M. Pastor-Soler and K. R. Hallows), and the University of Southern California/University Kidney Research Organization Kidney Research Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.M.A.-b., H.L., R.A.F., N.M.P.-S., and K.R.H. conception and design of research; M.M.A.-b., H.L., K.O., F.G., A.M., S.N., X.Z., D.N., Q.W., L.C., R.A.F., N.M.P.-S., and K.R.H. performed experiments; M.M.A.-b., H.L., K.O., F.G., Q.W., L.C., R.A.F., N.M.P.-S., and K.R.H. analyzed data; M.M.A.-b., H.L., K.O., F.G., D.N., R.A.F., N.M.P.-S., and K.R.H. interpreted results of experiments; M.M.A.-b., H.L., K.O., A.M., R.A.F., N.M.P.-S., and K.R.H. prepared figures; M.M.A.-b., H.L., K.O., R.A.F., N.M.P.-S., and K.R.H. drafted manuscript; M.M.A.-b., H.L., K.O., D.N., R.A.F., N.M.P.-S., and K.R.H. edited and revised manuscript; M.M.A.-b., H.L., K.O., D.N., R.A.F., N.M.P.-S., and K.R.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Christy Smolak, Heather Bladek, and Ahmed Basim Abduljabar for expert technical assistance.
REFERENCES
- 1.Al-Bataineh MM, Gong F, Marciszyn A, Myerburg MM, Pastor-Soler NM. Regulation of the proximal tubule vacuolar H+-ATPase by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 306: F981–F995, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alzamora R, Al-Bataineh MM, Liu W, Gong F, Li H, Thali RF, Joho-Auchli Y, Brunisholz RA, Satlin LM, Neumann D, Hallows KR, Pastor-Soler NM. AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am J Physiol Renal Physiol 305: F943–F956, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alzamora R, Gong F, Rondanino C, Lee JK, Smolak C, Pastor-Soler NM, Hallows KR. AMP-activated protein kinase inhibits KCNQ1 channels through regulation of the ubiquitin ligase Nedd4-2 in renal epithelial cells. Am J Physiol Renal Physiol 299: F1308–F1319, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alzamora R, Thali RF, Gong F, Smolak C, Li H, Baty CJ, Bertrand CA, Auchli Y, Brunisholz RA, Neumann D, Hallows KR, Pastor-Soler NM. PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the a subunit in kidney cells. J Biol Chem 285: 24676–24685, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol 10: 923–934, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Bhalla V, Oyster NM, Fitch AC, Wijngaarden MA, Neumann D, Schlattner U, Pearce D, Hallows KR. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem 281: 26159–26169, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Bouley R, Breton S, Sun T, McLaughlin M, Nsumu NN, Lin HY, Ausiello DA, Brown D. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J Clin Invest 106: 1115–1126, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouley R, Pastor-Soler N, Cohen O, McLaughlin M, Breton S, Brown D. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra). Am J Physiol Renal Physiol 288: F1103–F1112, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Carattino MD, Edinger RS, Grieser HJ, Wise R, Neumann D, Schlattner U, Johnson JP, Kleyman TR, Hallows KR. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J Biol Chem 280: 17608–17616, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Cheng L, Wu Q, Kortenoeven ML, Pisitkun T, Fenton RA. A systems level analysis of vasopressin-mediated signaling networks in kidney distal convoluted tubule cells. Sci Rep 5: 12829, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies SP, Carling D, Hardie DG. Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem 186: 123–128, 1989. [DOI] [PubMed] [Google Scholar]
- 13.Deen PM, Verdijk MA, Knoers NV, Wieringa B, Monnens LA, van Os CH, van Oost BA. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science 264: 92–95, 1994. [DOI] [PubMed] [Google Scholar]
- 14.Fenton RA, Pedersen CN, Moeller HB. New insights into regulated aquaporin-2 function. Curr Opin Nephrol Hypertens 22: 551–558, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Llama P, Andrews P, Turner R, Saggi S, Dimari J, Kwon TH, Nielsen S, Safirstein R, Knepper MA. Decreased abundance of collecting duct aquaporins in post-ischemic renal failure in rats. J Am Soc Nephrol 10: 1658–1668, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405: 85–93, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 272: 14800–14804, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Gong F, Alzamora R, Smolak C, Li H, Naveed S, Neumann D, Hallows KR, Pastor-Soler NM. Vacuolar H+-ATPase apical accumulation in kidney intercalated cells is regulated by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 298: F1162–F1169, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallows KR. Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Curr Opin Nephrol Hypertens 14: 464–471, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Hallows KR, Alzamora R, Li H, Gong F, Smolak C, Neumann D, Pastor-Soler NM. AMP-activated protein kinase inhibits alkaline pH- and PKA-induced apical vacuolar H+-ATPase accumulation in epididymal clear cells. Am J Physiol Cell Physiol 296: C672–C681, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest 105: 1711–1721, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 67: 821–855, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Hasler U, Mordasini D, Bens M, Bianchi M, Cluzeaud F, Rousselot M, Vandewalle A, Feraille E, Martin PY. Long term regulation of aquaporin-2 expression in vasopressin-responsive renal collecting duct principal cells. J Biol Chem 277: 10379–10386, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Hoffert JD, Fenton RA, Moeller HB, Simons B, Tchapyjnikov D, McDill BW, Yu MJ, Pisitkun T, Chen F, Knepper MA. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem 283: 24617–24627, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffert JD, Nielsen J, Yu MJ, Pisitkun T, Schleicher SM, Nielsen S, Knepper MA. Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am J Physiol Renal Physiol 292: F691–F700, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Hoffert JD, Pisitkun T, Saeed F, Song JH, Chou CL, Knepper MA. Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol Cell Proteomics 11: M111.014613, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Q, Mak D, Devidas S, Schwiebert EM, Bragin A, Zhang Y, Skach WR, Guggino WB, Foskett JK, Engelhardt JF. Cystic fibrosis transmembrane conductance regulator-associated ATP release is controlled by a chloride sensor. J Cell Biol 143: 645–657, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katsura T, Gustafson CE, Ausiello DA, Brown D. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am J Physiol Renal Physiol 272: F817–F822, 1997. [PubMed] [Google Scholar]
- 30.Klein JD, Wang Y, Blount MA, Molina PA, LaRocque LM, Ruiz JA, Sands JM. Metformin, an AMPK activator, stimulates the phosphorylation of aquaporin 2 and urea transporter A1 in inner medullary collecting ducts. Am J Physiol Renal Physiol 310: F1008–F1012, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kortenoeven ML, Fenton RA. Renal aquaporins and water balance disorders. Biochim Biophys Acta 1840: 1533–1549, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Kortenoeven ML, Li Y, Shaw S, Gaeggeler HP, Rossier BC, Wetzels JF, Deen PM. Amiloride blocks lithium entry through the sodium channel thereby attenuating the resultant nephrogenic diabetes insipidus. Kidney Int 76: 44–53, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Kwon TH, Frokiaer J, Fernandez-Llama P, Knepper MA, Nielsen S. Reduced abundance of aquaporins in rats with bilateral ischemia-induced acute renal failure: prevention by alpha-MSH. Am J Physiol Renal Physiol 277: F413–F427, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Thali RF, Smolak C, Gong F, Alzamora R, Wallimann T, Scholz R, Pastor-Soler NM, Neumann D, Hallows KR. Regulation of the creatine transporter by AMP-activated protein kinase in kidney epithelial cells. Am J Physiol Renal Physiol 299: F167–F177, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loo CS, Chen CW, Wang PJ, Chen PY, Lin SY, Khoo KH, Fenton RA, Knepper MA, Yu MJ. Quantitative apical membrane proteomics reveals vasopressin-induced actin dynamics in collecting duct cells. Proc Natl Acad Sci U S A 110: 17119–17124, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez JP, Lee HY, Cline GW, Samuel VT, Kibbey RG, Shulman GI. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510: 542–546, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moeller HB, Olesen ET, Fenton RA. Regulation of the water channel aquaporin-2 by posttranslational modification. Am J Physiol Renal Physiol 300: F1062–F1073, 2011. [DOI] [PubMed] [Google Scholar]
- 38.Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev 34: 278–301, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moeller HB, Slengerik-Hansen J, Aroankins T, Assentoft M, MacAulay N, Moestrup SK, Bhalla V, Fenton RA. Regulation of the water channel aquaporin-2 via 14-3-3theta and -zeta. J Biol Chem 291: 2469–2484, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mount PF, Hill RE, Fraser SA, Levidiotis V, Katsis F, Kemp BE, Power DA. Acute renal ischemia rapidly activates the energy sensor AMPK but does not increase phosphorylation of eNOS-Ser1177. Am J Physiol Renal Physiol 289: F1103–F1115, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Neumann D, Woods A, Carling D, Wallimann T, Schlattner U. Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr Purif 30: 230–237, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Pastor-Soler NM, Hallows KR. AMP-activated protein kinase regulation of kidney tubular transport. Curr Opin Nephrol Hypertens 21: 523–533, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Pastor-Soler NM, Hallows KR, Smolak C, Gong F, Brown D, Breton S. Alkaline pH- and cAMP-induced V-ATPase membrane accumulation is mediated by protein kinase A in epididymal clear cells. Am J Physiol Cell Physiol 294: C488–C494, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia 56: 1898–1906, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richards WO, Shin B. Massive diuresis after acute renal failure. Crit Care Med 12: 202–203, 1984. [DOI] [PubMed] [Google Scholar]
- 46.Rinschen MM, Yu MJ, Wang G, Boja ES, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc Natl Acad Sci U S A 107: 3882–3887, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sands JM, Layton HE. The physiology of urinary concentration: an update. Semin Nephrol 29: 178–195, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, Zheng B. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell 52: 161–172, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem 281: 32207–32216, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Xie L, Hoffert JD, Chou CL, Yu MJ, Pisitkun T, Knepper MA, Fenton RA. Quantitative analysis of aquaporin-2 phosphorylation. Am J Physiol Renal Physiol 298: F1018–F1023, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yashiro T, Nanmoku M, Shimizu M, Inoue J, Sato R. 5-Aminoimidazole-4-carboxamide ribonucleoside stabilizes low density lipoprotein receptor mRNA in hepatocytes via ERK-dependent HuR binding to an AU-rich element. Atherosclerosis 226: 95–101, 2013. [DOI] [PubMed] [Google Scholar]
- 53.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]