

Abstract
Pulmonary hypertension (PH) is a condition marked by a combination of constriction and remodeling within the pulmonary vasculature. It remains a disease without a cure, as current treatments were developed with a focus on vasodilatory properties but do not reverse the remodeling component. Numerous recent advances have been made in the understanding of cellular processes that drive pathologic remodeling in each layer of the vessel wall as well as the accompanying maladaptive changes in the right ventricle. In particular, the past few years have yielded much improved insight into the pathways that contribute to altered metabolism, mitochondrial function, and reactive oxygen species signaling and how these pathways promote the proproliferative, promigratory, and antiapoptotic phenotype of the vasculature during PH. Additionally, there have been significant advances in numerous other pathways linked to PH pathogenesis, such as sex hormones and perivascular inflammation. Novel insights into cellular pathology have suggested new avenues for the development of both biomarkers and therapies that will hopefully bring us closer to the elusive goal: a therapy leading to reversal of disease.
Keywords: pulmonary arterial hypertension, remodeling, vascular
pulmonary hypertension (PH), defined hemodynamically via right heart catheterization as an increase in pulmonary arterial pressure (PPA) to ≥ 25 mmHg at rest, is a complex and progressive condition arising from a variety of etiologies. Often fatal, PH can be divided into five major categories based on underlying cause and hemodynamic parameters: 1) pulmonary arterial hypertension (PAH), including idiopathic, heritable, and associated PH; 2) PH due to left heart disease; 3) PH due to interstitial lung diseases and/or hypoxia, including high-altitude and chronic obstructive pulmonary disease (COPD); 4) chronic thromboembolic PH (CTEPH); and 5) PH with unclear and/or multifactorial origin, including hematologic and systemic disorders (14, 83). While most forms of PH are adult onset, PH is also prevalent in the pediatric population. PH in newborns and infants remains a disease with high morbidity and mortality with multiple etiologies (122). Abnormal vascular architecture and function occur, whether due to persistent fetal circulation, secondary to lung hypoplasia, or consequent to premature birth or neonatal infections leading to chronic lung disease.
Sustained vasoconstriction and vascular remodeling contribute to all forms of PH to varying degrees, increasing right ventricular (RV) afterload and inducing RV hypertrophy (RVH) and eventually leading to mortality from RV failure. The structural changes that occur in the vasculature include reduced compliance, or stiffening of the proximal, elastic arteries (85, 210); thickening of the intimal and/or medial layer of muscular, resistance arteries; and muscularization of the precapillary arterioles due to proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) and in some cases endothelial-mesenchymal transformation (219). In severe cases, vaso-occlusive lesions arise from exuberant proliferation and reduced apoptosis of PASMCs and endothelial cells (ECs) within the vessels. It should be noted that the source of highly migratory/proliferative ECs found in occlusive lesions is still under investigation, as recent work suggests that cells from the vaso vasorum, the network of vessels that supply blood to the wall of the pulmonary arteries (PAs), undergo significant proliferation in the setting of PH (169). ECs isolated from the vaso vasorum of chronically hypoxic calves contained a high density of colony-forming ECs, which possessed high proliferative capacity and may contribute to vascular proliferation and remodeling elsewhere in the “pan-vasculopathic” PA in PH.
The development of PH can be associated with various pathogenic or genetic causes, and perhaps because of this complexity the cellular mechanisms causing disease are still incompletely understood. Over the past few decades, tremendous progress has been made in identifying contributing factors, including alterations in the production of vasodilator and vasoconstrictor mediators, various ion channels, bone morphogenetic protein (BMP) signaling, and mitochondrial dysfunction. In this review, we highlight advances made within the last 2 yr that have informed our understanding of the basis of this deadly disease, further delineating the molecular pathways in pulmonary ECs, PASMCs, and fibroblasts contributing to alterations in pulmonary vascular structure and identifying new therapeutic uses for medications as well as biomarkers for disease prognostication.
Models of PH
Given the limited availability of patient tissues and inability to perform mechanistic studies in humans, several animal models of PH have been developed to allow investigation into the functional and structural changes that occur during the development of PH and the underlying causes. Unfortunately, to date no single preclinical model perfectly replicates human PAH. Nonetheless, these models provide the opportunity to characterize the development and progression of PH, perform mechanistic studies, and evaluate potential therapeutic treatments.
To model pediatric PH, a variety of approaches have been developed. Exposure based models include short-term neonatal hyperoxia, fetal and/or post-natal hypoxia and a two-hit model of prenatal hypoxia followed by postnatal hyperoxia (reviewed in Ref. 20). All of these approaches recapitulate changes in lung structure, pulmonary vascular dysfunction and PH often observed in humans. For example, antenatal and/or postnatal hypoxic exposure leads to elevated PPA, impaired relaxation and increased contractility of PAs, PASMC hyperplasia and increased actin polymerization, and adventitial fibroblast proliferation (23, 74, 78, 269, 274, 279). Short-term hyperoxia in mice, which serves as a model of bronchopulmonary dysplasia (BPD), produces rarefaction of the microvasculature and increased smooth muscle actin expression in distal PAs, changes that are associated with downregulation of the BMP signaling pathway in affected lungs (278).
Interestingly, recent findings suggest that short-term neonatal exposure to hyperoxia, even if not directly inducing PH, may predispose adults to PH (95), due to persistent alterations in lung structure. In contrast, neonatal hyperoxia was beneficial for RVH, suggesting that while this exposure can cause vascular abnormalities that predispose the lung to PH later in life, an adaptive mechanism is invoked in the RV that improves tolerance.
While the former approaches are easy to implement and are typically used in small animals, surgical models often used in large animals include congenital diaphragmatic hernia, where a hernia is surgically created and the abdominal bowel is positioned into the chest (reviewed in Ref. 245) and partial ligation of the ductus arteriosus in utero (3). Both of these models result in elevated PPA after birth, mimicking failure to transition from fetal circulation after birth or persistent PH of the newborn (PPHN) observed in human infants. A large animal model using chronically ventilated lambs also exhibits increased pulmonary vascular resistance (PVR) and pulmonary vascular remodeling (7).
The most widely used model of adult PH remains the chronic hypoxia (CH) model. The phenotype observed in CH models is consistent with PH that develops in humans as a result of high altitude (reviewed in Refs. 219, 228). Both rats and mice are common choices for exposure to CH. Housing rodents at an FiO2 of 10% for 2–4 wk produces reliable PH. Rats exhibit substantial increases in PPA and RV mass, accompanied by increased vascular wall thickness, due to PASMC hypertrophy and hyperplasia (224, 225). Mice also develop PH upon exposure to CH, although elevations in RV systolic pressure (RVSP) and vascular remodeling are milder (18, 225). The most consistent finding in both rats and mice is muscularization of distal vessels and thickening and functional stiffening of proximal, conduit arteries (219, 225). Early studies using contrast media or microspheres to visualize blood flow suggested that rarefaction, or vascular pruning, occurs in this model (192). However, these results may have been contributed to by excessive hypoxic pulmonary vasoconstriction, as acute exposure to Rho kinase (ROCK) inhibitors almost completely normalizes PPA (166) and vascular remodeling appears to occur in an outward manner and is associated with angiogenesis (116).
The model that perhaps best reflects severe human PAH combines a single dose of the vascular endothelial growth factor (VEGF) receptor inhibitor SU5416, followed by exposure to hypoxia (SuHx), resulting in initial EC apoptosis with subsequent proliferation of a subset of apoptosis-resistant ECs (168, 234). In rats, this model develops robust EC and PASMC proliferation and occlusive lesions reminiscent of PAH in humans (2, 234). PH induced in the SuHx model is irreversible even after return to normoxia. The severity of PH that develops in response to the SuHx exposure is strain-dependent in rats, making strain selection an important consideration (125). A murine model has also been described, with increased arterial muscularization, vaso-occlusive lesions, collagen deposition in the media and adventitia, and RV dysfunction (48); however, mice required multiple injections of SU5416 and PH resolved upon removal of the mice from hypoxia (48, 251).
Debate continues as to the exact cause of endothelial dysfunction, vascular remodeling, and enhanced contractile mechanisms in all models of PH. Recent work using cells exposed to cyclic stretch to mimic hemodynamic forces to which pulmonary cells might be exposed in vivo suggests that mechanical forces alone may be sufficient to trigger cellular dysfunction (259). Similarly, PH induced in the SuHx model is irreversible even after return to normoxia, and ROCK inhibitors only partially reversed PPA (240), suggesting a significant portion of the increased PVR may be due to remodeling and occlusion of flow in small vessels. Temporal examination of the SuHx model demonstrated the presence of vaso-occlusive lesions early in the course of exposure; however, concentric neointimal, or plexiform, lesions did not form until later time points (2, 240). More recently, PA banding was used to alter blood flow in SuHx rats, resulting in diminished occlusive lesion formation in lobes with reduced blood flow (1). These results would suggest that the plexiform lesions observed in PAH patients, while perhaps contributing to progression/severity of the rise in PPA, may in fact be a consequence of EC damage and/or increased hemodynamic stresses during PH rather than the inciting cause. Intriguingly, both studies from sequential human lung biopsies as well as evaluation of the temporal sequence of remodeling in the SuHx rat model suggest that medial remodeling may precede intimal remodeling in PH (242), suggesting abnormal PASMC signaling could be a potential primary locus of disease pathology under certain conditions.
It should be noted that in assessing the effect of various pathways on the pathobiology of PH, careful histologic and hemodynamic assessment in animal models of PH is critical. Such in vivo data provide crucial correlates to in vitro study of endothelial function, but methodologic limitations exist. Specifically, assessment of alveolar capillary rarefaction (or angiogenesis) can be difficult using traditional histologic specimens. However, newer stereological assessments of the alveolar capillary network provide more accurate assessment of alveolar capillary volume, including a recent methodology for estimating capillary number (266). Additional areas of concern with respect to morphology include tissue distortion, as the fixative and embedding material used can alter tissue properties, a critical factor when determining structural remodeling. Most studies assessing morphometric analyses have used inflation at a fixed pressure and/or volume with formalin or low-melting point agarose. Recent studies suggest that preservation of lung tissue dimensions can be improved compared with formalin fixation and paraffin embedding by initial fixation with a mixture of glutaraldehyde and formaldehyde followed by embedding the samples in glycol methacrylate and treating them with osmium tetroxide and uranyl acetate (212). Thus, for quantitative histological studies, fixative and embedding protocols should be chosen carefully and adherence to published guidelines for evaluating lung structure (117) will help improve reproducibility and interpretations of results.
Pathways and Targets
The exact mechanisms governing abnormal migration and proliferation of vascular cells in PH are not fully known, but imbalances in normal antimigratory and/or antiproliferative mechanisms, activation of deleterious signaling pathways due to paracrine release of inflammatory substances, as well as increased endogenous and exogenous production of reactive oxygen species (ROS) are all thought to play a role (191). In this section, we will highlight some of the pathways that have recently been shown to contribute to abnormal cell function during PH.
Cell metabolism.
Altered mitochondrial function and metabolism (including fatty acid oxidation, glucose oxidation, and glutaminolysis) in the pulmonary vasculature has been an area of intense recent focus in PH research (reviewed in Ref. 203). A cancer-like metabolic shift known as the Warburg effect, in which glycolysis is uncoupled from glucose oxidation (referred to as aerobic glycolysis), has been noted in both the pulmonary vasculature and the RV in PH. This shift is related to impaired mitochondrial function resulting in inhibited glucose oxidation. There have been numerous recent advances in understanding the mechanisms of mitochondrial dysfunction in PH. Genetic loss of sirtuin 3 (SIRT3), a mitochondrial deacetylase, in mice results in impaired mitochondrial function and spontaneous development of PH, and further a loss-of-function SIRT3 polymorphism is associated with human PAH (179). PASMC-specific knockout of uncoupling protein 2 (UCP2), which conducts Ca2+ from the endoplasmic reticulum to the mitochondria, leads to low mitochondrial Ca2+ levels and mitochondrial hyperpolarization in addition to the development of pulmonary vascular remodeling and PH (63). Furthermore, the BMP receptor 2 (BMPR2) axis has been linked to mitochondrial function, such that mice with EC-specific deletion of BMPR2 have sustained PH following hypoxia then reoxygenation that is associated with lowered mitochondrial membrane potential and ATP production as well as accumulation of ΔmtDNA4977, a mitochondrial DNA deletion known to be associated with cancer (59).
Glycolysis represents the initial step in glucose metabolism, in which glucose is converted to pyruvate in a cytosolic pathway involving glucose-6-phosphate (G6P). In the glycolytic pathway, G6P usage results in the generation of pyruvate, which can either be transported into the mitochondria or converted to lactate in the cytosol (Fig. 1). Alternatively, G6P can be utilized in the pentose phosphate pathway, in which G6P dehydrogenase (G6PD) catalyzes the conversion of NADP+ to NADPH, which protects against oxidative damage, and results in the subsequent generation of ribose sugars that are used in the synthesis of nucleotides. Recent papers have shed further light on the contribution of G6P and G6PD to pulmonary vascular remodeling.
Fig. 1.
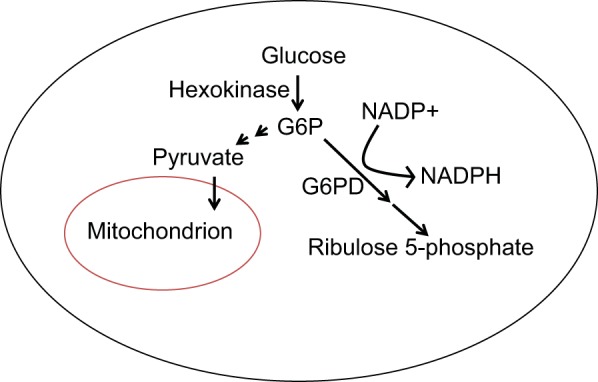
Glucose-6-phosphate (G6P) serves as a substrate that can be used in the glycolytic pathway resulting in generation of pyruvate. Alternatively, it can be used in the pentose phosphate pathway, in which glucose-6-phosphate dehydrogenase (G6PD) catalyzes a reaction resulting in formation of NADPH as well as precursors for nucleotide synthesis (such as ribulose 5-phosphate).
Prior work showed that hypoxic exposure increased G6PD activity within PASMCs, which modulated expression of smooth muscle phenotype proteins such as SM22α (44). Recent work has further unveiled the pathways via which G6PD modulates PASMC function in response to moderate hypoxia. The hypoxia-induced increase in G6PD activity was found to be necessary for the rise in rat PASMC proliferation (42). This G6PD-induced increase in proliferation was associated with increased Sp1 and hypoxia-inducible factor-1α (HIF-1α) expression and a decrease in contractile protein (SM22α and myocardin) expression combined with increased expression of proproliferative (cyclin A and phospho-histone H3) proteins. G6PD inhibition in vivo prevented the development of PH in the SuHx rat, with reduced remodeling associated with increased SM22α and decreased HIF-1α expression. Thus these data suggest G6PD serves as an upstream regulator that switches PASMCs from a contractile to a proliferative phenotype in response to hypoxia. Interestingly, although this paper demonstrated a correlation between increased HIF-1α and reduced SM22α levels, it was not directly tested whether SM22α was regulated by HIF-1. Perhaps surprising in the face of these findings, SM22α was initially identified from proteomics analysis of hypoxic PASMCS as a protein that was markedly upregulated (283). In human PASMCs, likely from conduit vessels, hypoxia upregulated SM22α via HIF-dependent induction of trasnforming growth factor-β (TGF-β) (282). In this case, SM22α induction was mediated by HIF-2 rather than HIF-1 (282). Similarly, PASMCs isolated from CH rats showed elevated SM22α levels that were attenuated, along with PH, in animals receiving lentivirus to reduce SM22α levels (282). However, others also observed reduced SM22α expression in hypoxic PASMCs (126), in PAs exposed to hypoxia (44), and in whole lung tissue from hypoxic rats (36). In contrast, SM22α downregulation was not observed in vivo in rats exposed to either CH or SuHx (42). While these results would seem at odds, variations in culture conditions, species (rat vs. human), location in the lung from which cells were derived (proximal vs. conduit), and level of hypoxia (moderate vs. severe) could all contribute to the differences observed.
G6PD also appears to be important in the hypoxia-induced proliferation and differentiation of CD133+ cells (43), bone-marrow derived progenitor cells that are recruited to the pulmonary vasculature in response to hypoxia and differentiate into mesenchymal or smooth muscle-like cells. CD133+ proliferation in response to hypoxia required G6PD, which modulated HIF-1α, cyclin A, and phospo-histone H3 expression (43). Furthermore, in coculture with PASMCs, differentiation of CD133+ cells into smooth muscle-like cells required G6PD-dependent production of H2O2 by the PASMCs. In vivo inhibition of G6PD in the SuHx rat decreased the number of CD133+ cells accumulating around occlusive lesions, as well as decreased the number of lesions (43). Thus G6PD is important to the function of both PASMCs and CD133+ cells, potentially via the stimulation of NADPH oxidase (Nox)-induced ROS production.
Altered cell metabolism is not restricted to the pulmonary vasculature in PH, as the RV has also become an area of recent interest. Under normal conditions, 95% of ATP is produced from oxidative phosphorylation, with 60–90% of the substrate from β-oxidation of fatty acids and the rest from glucose metabolism (222). Several studies have now shown RV mitochondrial and metabolic dysregulation in PH models (reviewed in Refs. 203, 229). In the SuHx model of PH, severe RV dysfunction was associated with a shift in metabolic substrate utilization, measured via imaging for 2-deoxy-2-[18F]fluoroglucose (glucose analog) and 14-(R,S)-[18F]fluoro-6-thia-heptadecanoic acid (fatty acid analog), demonstrating increased glucose, and reduced fatty acid, uptake (96). These results were consistent with data in patients indicating increased RV glucose uptake that correlated with RV dysfunction (33, 150, 275).
While it would seem clear that advanced and/or severe disease is associated with a metabolic shift in the RV, exactly when the glycolytic switch is “flipped” is unknown. In the neonatal calf model, where PH can be induced by short-term exposure to hypoxia, elevated PPA is accompanied by exuberant structural remodeling of the pulmonary vasculature and RV; however, overall hemodynamic function appears compensated with preserved cardiac output (29, 143, 257). Consistent with previous work demonstrating metabolic dysregulation in the left ventricle (LV) during heart failure (19, 204, 215), it was speculated that similar mitochondrial and metabolic abnormalities might be present in the RV. Surprisingly, following 2 wk of hypoxic exposure, mitochondrial function appeared to be largely maintained, with no significant changes in mitochondrial structure or dynamics and only small reductions in mitochondrial number and complex I activity (30). Interestingly, all differences noted in the RV were also observed in the LV, suggesting an overall response to hypoxia rather than chamber-specific adaptation. A small shift in metabolism consistent with an early switch to glycolytic metabolism was also noted, although again this was observed in both ventricles (30), suggesting that at this early stage of PH where RV function is largely compensated, mitochondrial and metabolic pathways do not contribute to the response to pressure overload.
Oxidative stress.
Closely related to dysfunctional mitochondrial metabolism, oxidative stress has also received significant attention with regards to the pathogenesis of pulmonary vascular remodeling (76). Perturbations in ROS levels have a myriad of effects on the pulmonary vasculature, ranging on a spectrum of oxidant-mediated signaling to oxidant-induced injury, depending on the type and concentration of ROS involved. In the case of models of PH in neonatal animals, increases in pulmonary blood flow and impaired mitochondrial respiration trigger ROS production, which in turn causes a vasoconstriction response (118). Recent work has begun to delineate mechanisms whereby altered ROS induces changes in vascular function. In ECs from PH fetal lambs, superoxide dismutase-2 (SOD2) was chaperoned into the mitochondrial matrix in an ATP-dependent manner by inducible heat shock protein 70 (iHSP70) (4). In this model, impaired SOD2 function with resultant increased mitochondrial oxidative stress led to impaired relaxation of PA rings in the presence of the vasodilator, nitric oxide (NO). Restoration of normal mitochondrial function with an oxygen radical scavenger returned relaxation of PA rings to baseline. In normal fetal lamb PASMCs, cyclic stretch, thought to mimic mechanical forces that contribute to PPHN, was found to induce increased Nox4 expression (259), which was mediated by mitochondrial complex III and NF-κB, and resulted in increased ROS levels in the PASMC cytosol as well as increased cyclin D1 expression. Thus ROS generation appears to be a key component of pathways through which fetal vascular function is altered in response to changes in flow.
In addition to pathologic ROS generation intracellularly, elevations in ROS can also be due to circulating inflammatory cells. Smooth muscle-specific deletion of extracellular superoxide dismutase (EC-SOD, or SOD3), the primary defender against elevations of ROS originating in the extracellular space, augmented CH-induced PH (CHPH) in mice, arguing for a protective role for this enzyme (174). While overexpression of EC-SOD attenuated (172), and total body loss of EC-SOD accentuated (271), hypoxic PH, the results from conditional EC-SOD deletion suggest that EC-SOD bound to SMC may be a key regulator of oxidant-induced injury in the CH model of PH. These findings are consistent with recent work demonstrating reduction in EC-SOD expression in lung tissue and PASMCs from PAH patients mediated by histone deacetylation (173).
One downstream effector of changes in ROS homeostasis is a nonselective cation channel, acid-sensing ion channel 1a (ASIC1a), which is a redox-sensitive channel that contributes to hypoxia-induced PH via augmentation of store-operated calcium entry (SOCE) in PASMCs (171). Recent work has shown that CH decreased H2O2 levels in rat PAs, likely due to a combination of decreased Cu/Zn superoxide dismutase (SOD1) activity and increased glutathione peroxidase activity (186). H202 decreases ASIC1a-dependent SOCE, suggesting that inhibition of H2O2 in CH could be a mechanism whereby SOCE is augmented, resulting in hypoxic elevations in intracellular calcium.
Peroxisome proliferator-activated receptor-γ.
Peroxisome proliferator-activated receptor-γ (PPARγ), a ligand-activated transcription factor, has been found to regulate adipogenesis and both glucose and lipid metabolism by modulating gene transcription through binding to PPAR response elements. Thiazolidinediones, such as rosiglitazone, are PPARγ agonists that have been widely used in the clinical treatment of diabetes. PPARγ expression is decreased in the pulmonary vasculature of patients with PH (10). The exact mechanisms resulting in PPARγ downregulation in PH are still under investigation, although proinflammatory mediators, such as IL-6 and TNF-α, may play a role (112). Conditional deletion of PPARγ in either SMCs (105) or ECs (100) results in the development of PH, whereas PPARγ agonism attenuates PH and pulmonary vascular remodeling in animal models (50, 170). PPARγ signaling results in antiproliferative effects (22) and is mediated by a number of pathways important to PH, such as miR-21 (98) and NF-κB (149), to list only a couple.
With the use of the model of PH where fetal sheep undergo partial ligation of the ductus arteriosus during late gestation, PPARγ signaling was shown to be crucial to normal endothelial function and angiogenesis in a NO-dependent pathway and was inhibited by endothelin-1 (ET-1), a potent vasoconstrictor known to be elevated in clinical PH (270). ET-1 receptor blockade or PPARγ agonism increased endothelial nitric oxide synthase (eNOS) protein and activity. In PASMCs from the same model, decreased PPARγ levels were associated with increased ROCK activity and cell numbers (91). In these cells, ROCK inhibition or PPARγ agonism was able to normalize cell growth.
p38 MAPK.
p38 kinases are members of the stress-activated kinase family that are induced by an array of stressors including inflammatory cytokines, osmotic changes, and mechanical deformation, controlling adaptive responses as well as driving pathologic signaling in varied disease states. In the case of PH, p38 has been implicated in maladaptive signaling in both the pulmonary vasculature and RV. In ECs, p38 has been found to function as a downstream effector of BMPR2 signaling (13). In pulmonary ECs, short-term BMPR2 silencing with siRNA was sufficient to increase migration and proliferation, providing evidence of the important role of ligand-independent BMPR2 in regulating EC behavior. Furthermore, BMPR2 silencing increased basal levels of ERK1/2 and p38 MAPK and BMPR2 silencing-induced increases in migration and proliferation could be attenuated through inhibition of the p38 MAPK pathway. These results argue a role for BMPR2 as a tonic suppressor of promigratory, proproliferative pathways in a noncanonical, ligand-independent fashion and for p38 MAPK as a downstream component of this signaling pathway.
The significance of p38 signaling to vascular cell function appears to not be limited to the endothelium. In lung sections from PAH patients, increased p38 was observed in all layers of the vessel walls (47), and in PASMCs from PAH patients, abnormal growth is reduced when p38 is inhibited (268). Moreover, migration of PASMCs from PAH patients is associated with increased p38 activation (267). Similar to ECs, downregulation of BMPR2 in PASMCs is associated with upregulation of p38 (157), further solidifying the link between BMPR2 loss of function and p38 activation in PH vascular remodeling.
Altered fibroblast function, both in the pulmonary vasculature and the RV, is an important contributor to PH pathogenesi (226) and also appears to be related to p38 signaling. In pulmonary fibroblasts, activation of p38 promotes fibroblast proliferation under hypoxic conditions (165). p38 can also contribute to release of mitogens from the fibroblast that stimulate adjacent PASMC proliferation and recruit proinflammatory cells (200). Inhibiting p38, in particular the α-isoform, in vitro prevented fibroblast production of IL-6 in response to hypoxia, while in vivo inhibition of p38 attenuated PH, as well as reduced lung and circulating levels of IL-6 (47). A number of studies have demonstrated a role for IL-6 in mediating PH (92, 108, 209, 223), with circulating levels of IL-6 correlating with outcome in PAH patients (124). IL-6 is also known to promote PASMC proliferation, migration, and contractility (112, 209). Together, these data suggest an important role for p38 not only in mediating fibroblast proliferation but also in promoting a proinflammatory milieu that can also contribute to PASMC dysfunction.
In the remodeling RV, p38 signaling appears to be important for hypertrophy. While compensatory hypertrophy is necessary for RV adaptation to increased afterload in PH, persistently elevated afterload eventually leads to RV fibrosis and failure (24). Several reports have indicated that increased RV afterload and hypertrophy are associated with alterations in RV, but not LV, PKC isoform expression, with increased PKCα (26, 244), PKCβ (46), and PKCδ (46, 244). The increase in PKC expression is likely downstream of increased angiotensin II (ANG II) signaling (46, 247), which can exert effects through either AT1 or AT2 receptors. In cardiac fibroblasts, ANG II leads to collagen production via inactivation of p38 (46), similar to the role of reduced p38 described in LV hypertrophy (27). AT2 receptors are abundantly expressed in the neonatal lung and, in a model of BPD, AT2 activation reduced RVH (254). Interestingly, low levels of the AT2 antagonist PD123319 provided a similar benefit for RVH, and reduced influx of inflammatory cells into the lung (255). At higher doses, the protective effect of the antagonist was lost, and it blocked the beneficial effects of the agonists as well. Whether the effects of the low dose of PD123319 were due to a partial-agonist effect remains to be determined.
Arginase.
The arginases, of which there are two isoforms (I, localized to the cytosol, and II, localized to the mitochondria), are emerging proteins of interest with regards to PH. These enzymes metabolize l-arginine to l-ornithine and urea, a step in the polyamine and proline synthesis pathways relevant to cell proliferation. Prior work has shown that arginase II protein expression and arginase activity were increased in cultured human PASMCs in response to hypoxia and that arginase II inhibition prevented hypoxia-induced PASMC proliferation (38). Furthermore, arginase activity was shown to be increased in lung tissue from chronically hypoxic mice (128) and in serum from PAH patients relative to controls (272), the latter being linked to decreased NO synthesis in pulmonary arterial endothelial cells (PAECs) from PAH patients. Both NO synthase (NOS) and arginase II share l-arginine as a substrate such that increased activity of one enzyme may result in decreased availability of l-arginine for the other. Arginase II expression, as well as that of arginase I, was increased in lung tissue from neonatal rats exposed to the chemotherapeutic agent bleomycin (97), which induces a robust inflammatory response, lung morphology resembling emphysema, and severe PH (87, 97, 123). In this model, the arginase inhibitor amino-2-borono-6-hexanoic acid enhanced NO production (despite decreased NOS2 expression) and prevented development of PH and pulmonary collagen deposition (97).
More recently, HIF-2α has been shown to control hypoxic upregulation of arginase I and II in PAECs (49). Pulmonary endothelial deletion of either HIF-2α or arginase I in mice resulted in blunted hypoxic increase in RVSP and vascular remodeling. Furthermore, plasma NO levels were increased in the arginase I-deficient mice. This suggests that HIF-2α and arginase I are necessary for hypoxia-induced PH, via regulation of NO production.
Resveratrol, a natural polyphenol found in red grapes and berries that inhibits arginase activity, has been found to prevent the development of pulmonary hypertension in the monocrotaline rat (51). Resveratrol inhibited hypoxia-induced arginase activity as well as proliferation in human PASMCs (39) and in vivo resveratrol treatment attenuated the development of hypoxia-induced RVH in rats. The effect of resveratrol upon arginase II was mediated via inhibition of the PI3K-Akt signaling pathway, findings consistent with other recent reports demonstrating a role for Akt signaling in PASMC proliferation in response to hypoxia (233). The Akt family contains three isoforms, and the use of mice with deficiency for Akt1 or Akt2 revealed that loss of Akt1 prevented hypoxia-induced proliferation in vitro and development of hypoxia-induced PH. Although the exact isoform inhibited by resveratrol is not clear, it is likely that Akt1 was the target.
Further insight into the regulation of arginase revealed the contribution of microRNA (miR) to the upstream and downstream signaling mechanisms of arginase II within human PASMCs (129). Specifically, hypoxic induction of miR-17-5p, which is known to induce PASMC proliferation, was necessary for increased arginase II expression. Conversely, inhibition of arginase also prevented hypoxia-induced increases in miR-17-5p expression, suggesting that hypoxia results in an arginase II/miR-17-5p feedback loop.
Sex hormones.
Female sex is perhaps one of the most well-established risk factors for development of PH. Registries of PAH patients report a female:male ratio of between 1.4–4.1:1 (reviewed in Ref. 139). Paradoxically, those men that develop PAH have worse outcomes and increased risk of mortality than female patients (139), perhaps due to preserved RV ejection fraction and function and better RV response to therapy in females (121, 134). The discrepancy in both risk and severity of disease between males and females has prompted intense research into the sex-based differences in PAH, with recent studies shedding new light into the mechanisms that may underlie both risk and protection.
While both male and female rats develop severe PH in response to SuHx, RV function is better preserved in females (82), consistent with the data obtained in patients. Treatment with E2 in ovarectomized rats normalized RVSP, RVH, and RV function, likely a consequence of reduced oxidative stress, proapoptotic signaling, metabolic dysfunction, and proinflammatory cytokine expression (82). Interestingly, E2 therapy attenuated RVH and RV function in male rats but did not impact RVSP, suggesting a cardiac-specific effect.
Estrogen signaling also appears to play a protective role in hypoxia-induced PH, where females are protected (159, 193, 197) and administration of estradiol improves PH (197). However, in other models, female sex clearly confers susceptibility. For example, PAH that developed in association with use of diet medications that increased serotonin availability can be modeled in mice with administration of anorexigens (57) or forced overexpression of the serotonin transporter (SERT), which facilitates both cellular uptake and efflux of serotonin (151). Increased uptake of serotonin into PASMCs is associated with proliferation, and SERT expression is upregulated in PASMCs from PAH patients (151). Female mice exposed to dexfenfluramine or with increased SERT expression (SERT+) exhibited vascular remodeling and enhanced PH at baseline or in response to hypoxia (57, 101, 152, 263) that was not observed in male mice (57, 263). Interestingly, in the SuHx rat model, loss of SERT had no effect (55). It should be noted, however, that since SERT overexpression appears to preferentially play a role in females, and the study using SERT deletion in the SuHx model was performed only in male animals, the impact of loss of SERT in female SuHx may be different and needs to be determined.
Recent studies have attempted to elucidate some of the mechanisms by which estrogen may mediate its effects in the patient population, with focus shifting to the enzymes that mediate estrogen metabolism. In particular, expression of the gene encoding cytochrome P450 1B1 (CYP1B1) was markedly downregulated in female PAH patients (12) while genetic polymorphisms in CYP1B1, which catalyzes estrogen to 2-hydroxy-estrogen (2-OHE) (Fig. 2), were associated with reduced disease penetrance in females with BMPR2 mutations (12). BMPR2 mutation carriers with wild-type alleles not only had fourfold higher disease penetrance but had diminished CYP1B1 activity and produced lower levels of 2-OHE, resulting in an imbalance between 2-OHE, a metabolite with protective actions, and 16α-hydroxyoestrone (16α-OHE1) (12), a highly mitogenic and genotoxic metabolite (130). Further evidence that altering the balance between 2-OHE/16α-OHE1, with increased 16α-OHE1, is detrimental can be found in studies where administration of 16α-OHE1 caused PH in mice (264). Although the exact actions of 16α-OHE1 are still being uncovered, one of the mechanisms by which 16α-OHE1 may facilitate PAH is via upregulation of miR-29, which in turn downregulates PPARγ and alters molecular and functional indexes of cellular energy metabolism (40). Perhaps surprisingly, increased expression of CYP1B1 was found in the PAs from animal models and PAH patients in a different registry (264). Consistent with these findings, CYP1B1 was increased in female SERT+ mice, and inhibiting CYP1B1 reduced development of PH (130). In these studies, elevated 16α-OHE1 levels were also observed (130, 264), suggesting that while more work will need to be done to understand the complex regulation of estrogen metabolism, any perturbations that result in increased 16α-OHE1 are likely to be detrimental.
Fig. 2.
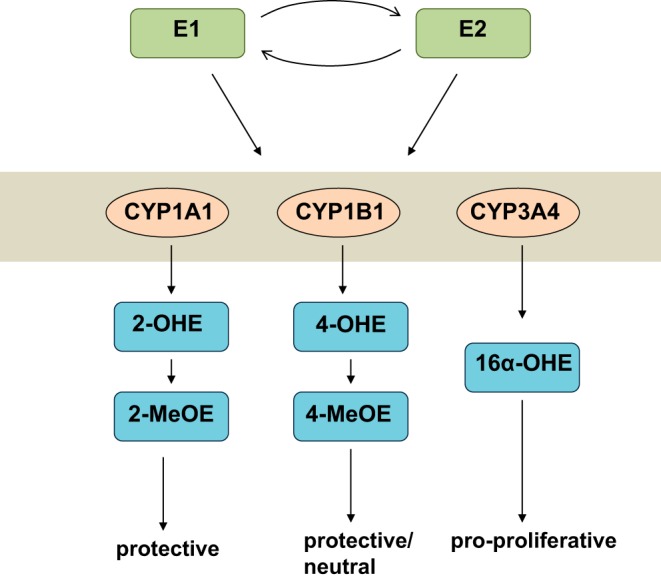
Schematic of simplified estrogen metabolism. Estrone (E1) and estradiol (E2) can be metabolized through oxidation pathways by cytochrome P450 (CYP) 1A, 1B, and 3A producing 2-, 4- and 16α-hydroxyestrogen (2-OHE, 4-OHE and 16α-OHE, respectively). 2-OHE and 4-OHE derivatives can be further converted to 2- and 4-methoxyestrogen metabolites (2-MeOE and 4-MeOE, respectively), which can have beneficial, antiproliferative properties. 16α-OHE, on the other hand, exerts proproliferative effects.
Inflammation.
It is now recognized that perivascular inflammation is a common contributing factor in almost all forms of PH (69, 188, 189, 194, 243). Macrophages (81, 237, 239, 248) and mast cells (17, 52, 114) have been shown to play a critical role in promoting vascular dysfunction in preclinical PH models. In samples from PAH patients, accumulation of immune cells, including T cells, B cells, macrophages, dendritic cells, and mast cells has been observed in and around sites of vascular lesions (52, 208, 243) and high levels of procontractile and proproliferative cytokines are observed in both patients and animal models of PH (reviewed in Refs. 69, 188, 194). In addition, anti-endothelial (58, 230), anti-fibroblast (231), and anti-nuclear (199) autoantibodies have been identified in PAH patients, although the extent to which these autoantibodies initiate or facilitate progression of disease is unknown. Interestingly, severe PH developed in rats given SU5416 following sensitization with ovalbumin (162), even in the absence of a hypoxic stimulus, suggesting that an allergic inflammatory phenotype can facilitate development of PH. Taken together these data providing compelling evidence for a link between inflammation and the vascular hallmarks of PH.
While the exact mechanisms by which inflammation might facilitate progression of PH are still under investigation, recent evidence has pointed to a role of inflammasomes. Inflammasome activation and assembly lead to caspase-1-mediated cleavage of pro-IL-18 and pro-IL-1β, leading to the release of activated IL-1β and IL-18. Increased circulating levels of IL-1β and IL-18 in PAH patients (120, 201) and animal models (250) suggest inflammasome activation. Interestingly, conflicting results have been reported with IL-18 loss, with some reporting protection (164) and others finding no effect (31), although it should be noted that in the latter study, control mice were housed at Denver altitude, and both male and female mice were used for the study, which could influence outcomes. Thus the downstream mediator of inflammasome activation, at least with respect to the development of PH, remains to be determined. Nonetheless, recent work demonstrated that loss of the common inflammasome component, apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), blunted RVSP and RVH and lowered IL-18 levels in mice exposed to CH (34). Interestingly, although the NLRP3 inflammasome has been shown to be activated in CHPH and antioxidant treatment reduced both NLRP3 activation and PH (250), loss of NLR pyrin domain-containing 3 (NLRP3), which is involved in the NLRP3 inflammasome only, did not recapitulate the effects of loss of ASC, suggesting multiple inflammasomes may participate in the pathogenesis of PH.
Many recent investigations have focused on the role played by exogenous signaling molecules produced by inflammatory cells in driving abnormal vascular cell function in PAH (68, 228, 252). Hypoxia-induced mitogenic factor (HIMF, FIZZ1), a protein secreted by M2 macrophages, promotes a proinflammatory milieu in the lung vasculature. Exogenous administration of HIMF was sufficient to induce lung endothelial apoptosis in vivo and this action of HIMF was dependent on IL-4 (273), suggesting that vascular inflammation in the context of PH may require both endogenous activators of EC apoptosis as well as immune cell cytokine responses to effect initial EC apoptosis and subsequent migration and proliferation of both ECs and SMCs. In addition, a metabolite of the 15-lipoxygenase pathway, 15-HETE, upregulates several cytokines such as IL-6 and TNF-α (37, 262), both of which have been implicated in PH. Macrophages, ECs, and platelets are all sources of 15-HETE, and inhibition of 15-HETE reduced platelet aggregation and thrombosis in SuHx and CH rats (216). In PBMCs from PAH patients, 15-HETE levels correlated with IL-6 and PVR (216). Incubating PAs with IL-6 or TNF-α induces a procontractile phenotype and enhances PASMC migration (112). Treatment with the omega-3 fatty acid-derived E-series resolvin, resolvin E1, reduced markers of inflammation (i.e., NF-κB activation), contractility and migratory capacity (112), suggesting a potential role for resolvins in reducing PASMC dysfunction caused by inflammation.
Whereas inflammation seems to clearly play a role in promoting PH, there is also evidence that immune pathways may be protective as well. For example, it was hypothesized that athymic rats exposed to SU5416 would develop less inflammation and thus diminished PH. Instead, these animals exhibited severe PH, even in the absence of hypoxia, and augmented inflammation (235), which was determined to be due to loss of regulatory T cells, which limit perivascular inflammation and vascular injury (232).
Having gained traction in the cancer field, immunotherapy may represent a promising avenue for treatment of PAH. However, with such a complex balance between detrimental and beneficial immune functions, any future immunomodulatory approaches aimed at blocking immune cell recruitment or function will likely require highly specific targeting of individual cell populations within the lung. The need for precise timing of immune targeting and/or the ability to target individual subpopulations of cells will also likely come into play. For example, lung-specific delivery of clondronate, to deplete macrophages, has been achieved using liposomal formulations (107, 158). However, since macrophages can be injurious or protective/anti-inflammatory, depending on subtype (11, 72, 110, 158), the differential contributions from macrophage subsets may represent a dynamic response to disease or injury, a factor that will need to be considered in any immunomodulatory regimen.
Extracellular matrix.
The extracellular matrix (ECM) is clearly important in the development of the pulmonary vasculature, as neonatal mice haploinsufficient for elastin are known to develop PH as adults (73, 217, 256). When challenged with a course of mechanical ventilation, these mice developed increased collagen and fibrillin deposition, which was associated with decreased respiratory system elastance (111). While the number of alveoli remained stable, the number of pulmonary microvessels was significantly decreased, providing important insight into how changes in neonatal anatomic development, lung growth arrest, and impaired respiration may impact long-term outcomes (Fig. 3). In rat pups with PH induced by chronic bleomycin infusion, increased perivascular collagen content was noted that was abrogated by arginase inhibition (97), suggesting links between the NO pathway and matrix remodeling.
Fig. 3.

Schematic illustrating impaired formation of alveoli and lung capillaries and extracellular matrix (ECM) remodeling in wild-type (Eln+/+) and elastin haploinsufficient (Eln+/−) neonatal mice subjected to mechanical ventilation. Eln+/− mice exhibit attenuated lung elastic fibers, increased collagen-1 and fewer capillaries at baseline. In wild-type mice, mechanical ventilation increases lung elastase activity and elastin degradation, leading to fragmented elastic fibers; however, no significant increase of lung elastase activity occurred in Eln+/− mice. Instead, Eln+/− mice exhibit reduced elastin synthesis and increased fibrillin production. In both genotypes, mechanical ventilation reduces abundance of the growth factors, vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) and causes apoptosis and reduces the numbers of capillaries and alveoli, resulting in diminished lung growth and abnormal lung function. [Reproduced with permission from Ref. 97.]
Accumulation of extracellular matrix components, including collagen, is a key feature of the vascular remodeling that contributes to RV afterload as increased collagen deposition and fibrosis causes stiffening of the proximal arteries (176, 195, 238, 241). Indeed, inhibiting new collagen synthesis using a proline analog prevented the effects of CH on RV hypertrophy and RVSP and attenuated collagen deposition in both the main PA and the distal vasculature (213). Interestingly, while most studies have found perivascular collagen deposition, examination of PAs from idiopathic pulmonary AH patients revealed collagen accumulation mainly in the intima, which correlated with increased expression of collagens (COL4A5, COL14A1, and COL18A1), matrix metalloproteinase (MMP) 19, and a disintegrin and metalloprotease (ADAM) and reduced expression of MMP10, ADAM17, tissue inhibitor of metalloproteinase 1 (TIMP1), and TIMP3 (113). Interestingly, collagen 4A1 and 4A2 expression was reduced in ECs from PAH patients, secondary to BMPR2 downregulation (198). Reductions in collagen 4A1 and 4A2 were associated with EC dysfunction (198), suggesting the potential for increased susceptibility to vascular injury with loss of BMPR2 signaling.
The mechanisms regulating collagen turnover and deposition during PH are still being defined. Main PAs from a fetal sheep PH model exhibited increased mechanical stiffness in the circumferential and axial orientations (62), likely caused by ECM remodeling due to hemodynamic stress. Mathematical modeling and histologic findings showed collagen fibers contributed a greater role to the ECM than elastin. During hypoxia, collagen synthesis has been shown to be upregulated by aldosterone (156), which interacts with mineral corticosteroid receptors to upregulate connective tissue growth factor (CTGF) and collagen III. Inflammatory pathways are also likely to contribute to fibrosis. In a model of antenatal inflammation due to endotoxin exposure, a strong risk factor for subsequent development of PH, pre- and post-natal vitamin D supplementation decreased PA and RV stiffness by restoring the normal organization of ECM collagen and elastin (154), likely due to preserved lung vascular development secondary to the anti-inflammatory properties of vitamin D. Changes PA stiffness were associated with improved oxygen saturations, postnatal survival, and EC function (155).
In addition to increasing afterload via arterial stiffness, collagen metabolism may also play a direct role in RV function. In PAH patients, RV diastolic stiffness was increased and correlated with RV fibrosis and collagen content (195). Similarly, SuHx rats exhibit increased RV fibrosis, collagen deposition and procollagen expression (94). In these animals, the expression of CTGF was also increased, consistent with the effects of mineralocorticoids in the vasculature. Mutant mice in which collagen type 1 turnover was impaired exhibited preserved RV function in response to PH induced by SuHx (93). In these mice RVSP was similar to wild-type SuHx, suggesting that RV-PA coupling was perhaps better maintained. While arterial stiffness was not measured, exposure to CH alone caused reduced arterial stiffening as well as decreased RVSP (176). PKCβII and -δ protein expression and p38 dephosphorylation were increased in cardiac fibroblasts from CH rats with RV fibrosis (46). In vitro application of angiotensin II recapitulated the effects of CH, causing PKCβII and -δ-dependent inactivation of p38 and collagen deposition (46). Whether mineralocorticoids, PKC, or p38 regulate RV collagen accumulation in PAH patients remains to be determined.
Transglutaminase 2.
Calcium signaling has been a recurrent motif in pathways that contribute to altered PASMC function in PH. Transglutaminase 2 (TG2) is a Ca2+-dependent enzyme that catalyzes posttranslational attachment of serotonin to glutamine (Gln) residues. TG2 activity increased in the lungs of hypoxic mice and TG2 inhibition attenuated the development of PH in the SuHx mouse (61). In cultured bovine PASMCs, hypoxia-related elevations in cell proliferation were found to depend on TG2 activity (181). Furthermore, extracellular Ca2+-sensing receptor or the transient receptor potential channel V4 was necessary for hypoxic increases in TG2 activity, whereas HIF-1α was necessary for hypoxic induction of TG2 expression. Thus TG2 activation represents one downstream avenue via which Ca2+ fluxes effect changes in PASMC function in the setting of hypoxia.
In addition to modifications of Gln, TG2 also catalyzes covalent crosslinking of lysine residues in the ECM, providing stability, rigidity, and protection from degradation (148). TG2 has recently been suggested to be responsive to mechanical stress (119) and, when activated, can facilitate arterial stiffening (207) and narrowing of mesenteric arteries in organ culture (15) and in vivo (16, 185). These results suggest the possibility that increased mechanical loads (i.e., increased shear stress or elevated wall strain) during PH could trigger stiffening and/or narrowing via activation of TG2.
Potential Treatments
ET-1 receptor antagonists.
The vasoactive peptide ET-1 has long been recognized as a contributor to the development of many forms of PH. Research in animal models has revealed many of the cellular mechanisms involved in the vascular actions of ET-1, including inhibition of K+ channels, elevation in intracellular calcium, enhancing sensitivity of the contractile apparatus, and production of ROS (184, 220). Widely recognized as one of the most potent vasoconstrictors, ET-1 can also act as a mitogen (281). While primarily derived from ECs, PASMCs and other cells can be a source of ET-1. Indeed, a role for SMC-derived ET-1 in modulating PASMC function has been shown previously, where ET-1 mediated the effects of hypoxia on ion channel expression via upregulation of HIF-1α (184, 265). More recently, SMC-specific loss of ET-1 reduced CH-induced PH and PASMC migration/proliferation (135). Interestingly, inhibition of ETA or ETB receptors did not prevent the effects of hypoxia, suggesting a potential intracellular role for ET-1 or signaling via a non-ETA or ETB receptor. This is in contrast to other studies, which found blockade of either receptor prevented the ET-1-mediated effects of hypoxia (184, 265).
Because ET-1 levels were found to be elevated in almost all forms of PH and preclinical data suggested beneficial actions, endothelin receptor antagonists (ETAs) were rapidly translated to clinical use and the mixed ETA/ETB antagonist bosentan remains one of the frontline therapies for treatment (54). However, recent advances in developing novel chemical analogs of bosentan have provided some new options. In particular, the recently approved drug macitentan appears quite promising. Macitentan differs from other antagonists in its slow receptor dissociation, rendering the compound more pharmacologically active than other ET receptor antagonists (86). Initial clinical studies demonstrated that macitentan significantly improved mortality in PAH patients, with a better safety profile than other ETAs and minimal adverse events (190). Interestingly, unlike most other drugs, macitentan entered clinical trials before being tested in preclinical PH models. It is now known that macitentan is effective at reversing established PH in SuHx (137) and monocrotaline (236) models. In the SuHx model, macitentan was able to partially reverse RVSP and RVH at both early and later time points; in both cases proliferation of cells in occlusive lesions was decreased. Additionally, with early treatment, apoptosis was increased (221). Thus preclinical research continues to improve our understanding of the role of ET-1 in mediating PH and of the effects of novel ETAs that may provide improved ability to target pathways that will allow regression of remodeling with more favorable safety profiles.
KV7 (KCNQ) Channel Activators
K+ channels play a significant role in setting the resting membrane potential. Several lines of evidence indicate that downregulation or inhibition of KV channels is a likely modulator of pulmonary vascular tone and PH. Recent interest has focused on KV7 channels as possible targets for modulation of tone (103). PASMCs have been shown to express several KCNQ family members, including KV7.1, KV7.4, and KV7.5 (131). Evidence is accumulating that these channels, especially KV7.4, play a major role in regulating tone in PAs. For example, in contrast to most other KV channels, KV7 channels are activated at much more negative potentials (−80 to −60 mV), suggesting that these channels are likely open at the resting membrane potential in PASMCs (103).
Inhibitor and activator studies also suggest that KCNQ channels may be contributing to PH. KV7 inhibitors have minimal effect on systemic vessels (131) but cause pulmonary vascular contraction (131, 214) that is lost with exposure to hypoxia (214). On the other hand, flupirtine, an orally active KV7 activator, reduced PPA in rats that were exposed to short-term (5 days) hypoxia (214). Marked downregulation of KV7.4 mRNA expression was observed in this model, but surprisingly, protein levels were not altered. Whether the lack of effect on KV7.4 protein requires longer duration of exposure to become evident is unknown. KV7.5 protein expression was also reduced by hypoxia in PASMCs (147). What remains unclear is the mechanism by which hypoxia might decrease KV7 channel expression; however, KCNQ5 (KV7.5) was found to be a target of miR190 (147). Since hypoxia downregulates KV7.5 and upregulates miR190 (147), it is tempting to speculate that miR190 targeting of KV7.5 might contribute to increased vasoreactivity in CH-induced PH.
Flupirtine also protected against development of PH in CH and SERT+ mice (163). The use of drugs targeting these channels are attractive since the KCNQ activators flupirtine and retigabine have completed clinical trials for pain and seizures and appear to be relatively safe when administered to humans, with minimal side effects (146, 187).
cGMP pathway.
It has long been recognized that impaired NO production and/or responsiveness can contribute to PH. NO, produced primarily from ECs, diffuses to the SMCs, where it activates soluble guanylate cyclase (sGC), leading to production of cGMP, activation of protein kinase G (PKG) and relaxation (253). Phosphodiesterases (PDEs) catalyze cGMP breakdown, thus antagonizing the effects of the NO-cGMP-PKG pathway (89). In PH, ample evidence has accumulated that decreased production of NO and reduced sGC activity, coupled with increased PDE activity, may all facilitate reduced effectiveness of the NO pathway to maintain low vasomotor tone. For example, loss of eNOS (71, 227) or PKG isoform 1 (PKG-1) (284) increases PPA under control conditions. Alternative splicing of the gene encoding PKG-1 results in two isoforms, of which PKG-1α contains a function leucine zipper motif that allows binding to myosin light chain phosphatase and RhoA and mediates relaxation. Selective loss of PKG-1α activity, via a mutation in the leucine zipper domain, increased ROCK activity and resulted in spontaneous development of PH in mice (196).
Reduced NO production has been well documented in PH (132), in some cases due to reduced expression of eNOS (90, 249). In addition, oxidant stress can lead to reduced phosphorylation of eNOS at Ser 1177, which is required for efficient NO production (60). During CH, levels of Ser1177 phosphorylated eNOS are markedly reduced (138) in adult animals. In newborns, oxidative stress also caused persistent downregulation of the NO pathway that resulted in enhanced PA contractility and muscularity and RV hypertrophy that persisted into adulthood (218). Along these lines, lung NOS levels were reduced in an experimental model of congenital diaphragmatic hernia (CDH) in fetal rats (153). Based on observations of high PVR in PPHN, augmentation and/or stimulation of the NO pathway has been an area of intense focus in the treatment of the pediatric population (253). In infants, inhaled NO has been used widely in the clinical setting, and preclinical studies have assessed methods to enhance endogenous NO production. For example, in the hypoxic piglet model, treatment with l-citrulline, the precursor for NO production, helps to recouple eNOS and attenuates PH (77). Similarly, treatment with genestein, a phytoestrogen with tyrosine kinase inhibiting and anti-oxidant properties, restored eNOS phosphorylation, NO production, and cGMP levels and prevented PH (138). The actions of genestein may result from upregulated Akt activity, which has been shown to activate eNOS (60). Finally, antenatal therapy with simvastatin in a CDH model restored NOS levels, normalized pulmonary vessel density and diameter and reduced arteriolar remodeling (153). While the results from the latter study would seem promising, caution should be exercised when interpreting these results, as the effects on PPA or RVH were not assessed and simvastatin has a number of effects unrelated to NOS.
Although inhaled NO remains the first line defense in infants with PH, it should be noted that only ∼60% of patients in this population are NO responsive. The reason for lack of response in some patients is unknown, but may be due to impairments in downstream signaling. For example, in newborn rats the cGMP pathway appears to desensitize, as vasorelaxation induced by activating sGC or application of an NO donor was lost when the vessels were preincubated with agents that increase cGMP activity (70). These results indicate that other modes of activating the sGC/cGMP pathway or alleviating cGMP desensitization will be needed.
In the adult PH population, the PDE5 inhibitors sildenafil and tadalafil have been used to increase cGMP levels with positive impact on pulmonary hemodynamics and exercise capacity (28, 89, 136). A recent study compared the effect of sildenafil with exercise training in the CH mouse model (261), finding that exercise training reduced RVSP and vascular remodeling to a similar extent as sildenafil, with no additive effect. The beneficial effect of exercise training were similar to results reported in PH patients (35, 99), although whether the effects of exercise are due to increased local or systemic production of NO and whether the effects of exercise and sildenafil are synergistic in patients remains to be determined.
While most attention has focused on PDE5, a novel PDE2 inhibitor, BAY 60–7550, ameliorated CH- and bleomycin-induced PH and reduced proliferation in PASMCs from PAH patients (32), suggesting that other PDEs may also be suitable targets. Interestingly, while PDE5 inhibition is most often associated with vasodilation due to reduced cGMP breakdown, it has also been found to interact with the ET-1 pathway, reducing ET-1-induced contraction (285), suggesting that these agents may have multifactorial benefits.
PDE inhibition would be expected to lead to an accumulation of cGMP due to reduced breakdown; however, increasing cGMP synthesis is another potential approach to stimulate the pathway. Interestingly, sGC expression appears to be upregulated during PH (211), suggesting that reduced activity may reflect oxidation and reduced NO responsiveness of the enzyme (202). A number of compounds have been developed to increase sGC activity. Some, such as BAY 58–2667 (cinaciguat) activate sGC when the heme iron is in its oxidized state or missing (202). BAY 58–2667 reduced PPA in fetal lambs after ductus arteriosus ligation (41) and PH in CH mouse and monocrotaline rat models (66). Other methods of increasing sGC activity target sGC that is missing a heme moiety. For example, δ-aminolevulinic acid, the rate-limiting step in heme biosynthesis, targets heme-lacking sGC. In this case, increasing protoporphyrin IX, the iron-free form of heme, activates sGC by allowing protoporphyrin IX to bind to the heme site. Consistent with other methods that activate sGC, δ-aminolevulinic acid prevented PH in mice exposed to CH (8).
Similarly, stimulating sGC with the novel, orally available compound BAY 63–2521 (riociguat), which also increases sensitivity of sGC to NO or stimulates sGC in the absence of bound NO, increased NO-stimulated cGMP production and prevented progression of PH in CH, SuHx, and monocrotaline-treated animals (142, 211). In clinical trials, riociguat improved patient outcomes (88) and has now been approved for the treatment of Group 1 and Group 4 PH.
Cell metabolism.
Several pharmacological agents directed at treating altered metabolism in PH are currently under investigation. Metformin, commonly used in the treatment of type 2 diabetes, activates AMP-activated protein kinase (AMPK), which serves as a key regulator of cellular energy metabolism via a plethora of downstream effects (106). Metformin has been found to prevent PH induced by either monocrotaline or CH (5). More recently, both nitrite and metformin were shown to activate a SIRT3-AMPK-GLUT4 signaling pathway in skeletal muscle, enhancing muscle glucose uptake (140). Additionally, in a rat model of metabolic syndrome with PH associated with heart failure with preserved ejection fraction (PH-HFpEF) induced by a combination of leptin receptor defect and SU5416 injection, treatment with metformin at the time of SU5416 injection prevented increased PPA and vascular remodeling. However, there was no benefit of metformin therapy in older rats with established PH-HFpEF. In the female rat SuHx model of PH, metformin therapy reversed elevated PPA and vascular remodeling (56). In this study, metformin had several effects on SuHx lungs, including increased AMPK activity, decreased aromatase and estrogen levels and decreased PASMC proliferation, suggesting that one of the downstream effects of metformin-induced AMPK activation is to regulate the sex hormone axis via aromatase inhibition. A clinical trial of metformin in Group 1 PH is now ongoing (NCT01884051).
Dichloroacetate (DCA), which inhibits pyruvate dehydrogenase kinase, resulting in activation of pyruvate dehydrogenase and thereby promoting glucose oxidation, has long been of interest in the study of PH. DCA prevents and reverses CH- and monocrotaline-induced PH in rats via upregulation of KV1.5 and increased apoptosis, together with decreased proliferation of PASMCs (160, 161). DCA also improves glucose oxidation in the RV, resulting in better function (183). Results from a phase 1 study of DCA in PAH patients are pending (NCT01083524).
Oxidative stress.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a nuclear transcription factor that regulates expression of multiple genes encoding antioxidant enzymes, including NAD phosphate reduced quinone oxidoreductase (NQO1), glutathione S-transferase, and hemoxygenase-1 (HO-1), as well as genes controlling mitochondrial DNA replication, such as the mitochondrial transcription factor A (TFAM). Nrf2 is known to act in a PAEC signaling pathway whereby BMPR2 regulates TFAM and mitochondrial DNA replication (59). Mice treated with the Nrf-2 activator olitpraz showed an attenuated increase in RVH and pulmonary vascular remodeling but surprisingly showed no change in RVSP (67). A pharmacologic Nrf2 activator, bardoxolone methyl, which also inhibits NF-κB, is currently in a phase 2 clinical trial in patients with World Health Organization (WHO) Group I, III, or V PH (NCT02036970).
ASK1/p38 MAPK.
Targeting the p38 MAP kinase signaling pathway has been of interest in a number of disease states. Unfortunately, p38 inhibitors have been marked by poor side-effect profiles (79). Thus there has been interest in targeting other components of the MAPK signaling cascade. Apoptosis signal-regulating kinase 1 (ASK1) is a MAPK kinase kinase that lies upstream of both p38 MAPK as well as c-Jun NH2-terminal kinase (JNK), another member of the MAPK family. Hypoxia increases JNK, as well as p38 MAPK, activity in rat PASMCs (127), while serotonin-induced enhancement of PASMC proliferation and migration are regulated by JNK, which signals through the Akt pathway (260). Additionally, JNK inhibition decreases proliferation of human PAH PASMCs (268). These findings raise the possibility that ASK1 inhibition could therefore lead to beneficial effects in PH via downstream inhibition of both p38 MAPK and JNK. Indeed, a phase 2 clinical trial of the ASK1 inhibitor GS-4997 is underway in WHO Group I PH patients (NCT02234141). Another potential method of inhibiting MAPKs would be to activate MAPK phosphatases. Genetic loss of MAPK phosphatase-1 (MKP-1) in mice resulted in exaggerated increases in RVSP, RVH, and vascular remodeling in response to CH (as well as exaggerated increases in lung arginase I and II), supporting the idea of MAPK phosphatases as potential therapeutic targets (128). Despite the potential for targeting MAPK signaling as a treatment option, given their multiple physiological roles, it seems likely that distal parts of the signaling cascades, rather than more proximal, will provide better therapeutic options with reduced toxicity and less frequent unwanted side effects.
Sex hormones.
Dehydroepiandrosterone (DHEA), a steroid hormone, exhibited protective effects in preclinical models of PH (9, 25, 64) and low levels of DHEA are associated with the development of PAH in men (246). Although DHEA is a precursor for estrogens, and can bind estrogen receptors, the actions of DHEA are likely to be multifold. For example, DHEA inhibits G6PD (102, 104), a key player in altered cellar metabolism. In PASMCs, DHEA also inhibits Src/STAT3 signaling pathways (180), which are involved in both proliferative and contractile mechanisms, and downregulates ROCK signaling (115). In addition to inhibiting pathways that promote contraction, DHEA also increases the expression of K+ channels (25) and inhibits Ca2+ influx through T-type Ca2+ channels (45), perhaps helping to reduce intracellular Ca2+ levels. With the effects on calcium influx, coupled with upregulation of sGC (175) and activation of PKG (177), the overall effect of DHEA is to facilitate vasorelaxation. Clinical trials of DHEA in PH patients are ongoing, although encouraging results from an initial pilot study of patients with PH secondary to COPD reported beneficial effects on PPA, PVR, and 6-min walk distance (65).
Other studies have been launched to investigate hormone therapy in PAH. A double-blind, placebo-controlled randomized phase II clinical trial using anastrozole in PAH patients (NCT01545336) has recently shown that this therapy was well-tolerated and effectively reduced E2 levels (133). Although limited by the small sample size, preliminary results suggest potential positive benefits on 6 min walk test, laying the foundation for future phase III trials.
Elafin.
Since increased matrix deposition and crosslinking is clearly linked to vascular stiffening, targeting the ECM could provide a potential therapeutic option. The serine elastase inhibitor elafin is endogenously produced primarily in the epithelium and provides protection from inflammatory damage. Mice engineered to genetically overexpress elafin are protected from CH-induced PH (280), with reduced metalloprotease activity. However, recent studies demonstrating that treatment with elafin regressed RVSP and vascular remodeling in the SuHx rat model (167) through a mechanism involving augmentation of endothelial BMPR2 signaling and caveolin-1 interactions indicates a previously unappreciated link to endothelial dysfunction. Similarly, in celiac disease, elafin inhibited TG2 activity (84), suggesting additional cross-over with pathways known to be involved in PH. Clinical trials have been performed or are currently underway in coronary artery bypass graft surgery (6) and esophagectomy and elafin, under the trade name Tiprelestat, has been granted orphan drug status for PAH, with clinical trials likely forthcoming. Given the systemic roles of elastase inhibitors, and potential as yet unidentified actions of elafin, limiting the systemic effects of elafin by lung-directed targeting, perhaps through inhaled versions, deserves consideration.
Circulating Biomarkers
Reliable biomarkers for diagnosis, prognostication, and treatment response would be ideal for the management of PH (80). However, the search for convenient, minimally invasive, specific, and sensitive biomarkers for PH has not revealed any one circulating marker that can be used to accurately diagnose PH or monitor disease progression. While ET-1 and BNP/NH2-terminal-proBNP have proven useful in certain cases, circulating ET-1 levels only represent “spillover” rather than local concentrations and BNP can be affected by demographic factors and may differ with PH etiology (182). As such, only BNP/NT-proBNP are used clinically and the search for additional biomarkers continues.
Circulating biomarkers that have been examined in the pediatric population include NT-proBNP (21, 141) and circulating ECs (144) and fibrocytes (277). In addition, proteomics analysis of plasma derived from children with PH was able to distinguish responders to long-term vasodilators (276). More recently, prospective investigation of the relationship between two markers specific for pulmonary angiogenesis, vascular endothelial growth factor A (VEGFA) and placental growth factor (PLGF), in a cohort of neonates with CDH undergoing standard of care revealed a relationship between a higher VEGFA:PLGF ratio and echocardiographic measures of PH, including RVSP, diastolic ventricular dysfunction, and oxygenation index (178). Most striking, the ratio was elevated at days 3–4 of life and in the second week in patients who died (178). Thus the relationship between VEGFA:PLGF may provide a promising biomarker in the pediatric patient population.
In the adult PH population, novel biomarkers that may identify those patients that respond to vasodilator therapy have been identified in circulating lymphocytes (109). In particular, combined high mRNA levels of RHOQ, which encodes a cytoskeletal protein involved in insulin-mediated signaling, and TPD52, a protein cancer marker, could correctly distinguish patients that were nonresponsive to vasodilators.
While vascular levels of collagen and collagen metabolism have been shown to increase with PH, circulating biomarkers of collagen metabolism have been shown to predict disease severity in PAH patients, with higher levels of NH2-terminal pro-peptide of type III procollagen (PIIINP), COOH-terminal telopeptide of collagen type I, MMP-9, and TIMP1 in PAH patients (205). Moreover, levels of PIIINP significantly correlated with severity of PAH (205) and worse quality of life (206). High circulating levels of collagen XVIII, and its cleavage product, endostatin, have also been shown to be positively correlated with poor outcomes in PAH (53, 113), as has galectin-3 (75), a regulator of matrix deposition and PIIINP. While further testing will be needed, panels of biomarkers based on collagen metabolism appear promising.
Finally, metabolic profiling indicates that several pathways are disturbed in PAH patients. Tracking metabolic abnormalities may provide insight into disease progression and offer clues to therapeutic efficacy. Along these lines, several plasma metabolite biomarkers of PH and associated RV-pulmonary vasculature dysfunction were identified, including metabolites of the indoleamine 2,3-dioxygenase (IDO) pathway (145). The IDO pathway was also upregulated in CH mice. Since these metabolites are thought to function as vasodilators (258), these markers are not believed to be pathogenic but rather to reflect a compensatory mechanism.
Because PH is multifactorial with respect to underlying etiology and pathophysiology, it is difficult to envision a single biomarker that will fit all cases. However, only time and additional testing will tell if any of these proposed biomarkers, alone or in combination, will prove useful for early diagnosis or monitoring disease activity.
Conclusion
PH remains a complex and mysterious disease with often fatal outcome. Tremendous progress has been made over the last few years in elucidating novel cellular mechanisms involved in the pathogenesis of disease, developing new therapeutic strategies to treat the patient population, and identifying potential biomarkers that can facilitate diagnosis, predict prognosis and monitor disease progression (Fig. 4). However, as is often the case, with new insights arise new questions. Which biomarkers will ultimately allow physicians to distinguish patients that will respond to a given therapy? Which drug, or combination of drugs, will prove most effective in slowing progression of disease? In addition, perhaps the holy grail of PH research: will any therapy ultimately lead to reversal of disease (i.e., curative) in the patient population? Given the progress made to date, confidence is high that ongoing and future research will provide additional insight toward answering these pressing issues.
Fig. 4.

Overview of mechanisms contributing to pulmonary hypertension. Pulmonary hypertension is characterized by remodeling of all layers of the pulmonary vasculature, including endothelial cells (ECs), smooth muscle cells (SMCs), and fibroblasts, resulting in vascular wall thickening and occlusion of the lumen, which play roles of varied prominence depending on the etiology of disease. In addition, recruitment of inflammatory cells contributes to the remodeling process. Included here are several of the pathogenic mechanisms that are highlighted in this review of recent progress in the field. AMPK, AMP-activated protein kinase; G6PD, glucose-6-phosphate dehydrogenase; SOD, superoxide dismutase; Nrf2, nuclear factor E2-related factor 2; PPARg, peroxisome proliferator-activated receptor-γ; JNK, c-Jun NH2-terminal kinase; CYP1B1, cytochrome P450 1B1; DHEA, didehydroepiandrosterone; HIMF, hypoxia-induced mitogenic factor; 15-HETE, 15-hydroxyeicosatetraenoic acid.
GRANTS
This work was funded by National Institutes of Health Grants HL-124727, HD-044355, HL-126514, HL-073859, and HL-124930.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.C.H. and L.A.S. prepared figures; J.C.H., K.S., M.B., and L.A.S. drafted manuscript; J.C.H., K.S., M.B., and L.A.S. edited and revised manuscript; J.C.H., K.S., M.B., and L.A.S. approved final version of manuscript.
REFERENCES
- 1.Abe K, Shinoda M, Tanaka M, Kuwabara Y, Yoshida K, Hirooka Y, McMurtry IF, Oka M, Sunagawa K. Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc Res 111: 16–25, 2016. [DOI] [PubMed] [Google Scholar]
- 2.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Abman SH, Shanley PF, Accurso FJ. Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest 83: 1849–1858, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, Pritchard K, Konduri GG. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol 306: L351–L360, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agard C, Rolli-Derkinderen M, Dumas-de-La-Roque E, Rio M, Sagan C, Savineau JP, Loirand G, Pacaud P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol 158: 1285–1294, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alam SR, Lewis SC, Zamvar V, Pessotto R, Dweck MR, Krishan A, Goodman K, Oatey K, Harkess R, Milne L, Thomas S, Mills NM, Moore C, Semple S, Wiedow O, Stirrat C, Mirsadraee S, Newby DE, Henriksen PA. Perioperative elafin for ischaemia-reperfusion injury during coronary artery bypass graft surgery: a randomised-controlled trial. Heart 101: 1639–1645, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albertine KH. Utility of large-animal models of BPD: chronically ventilated preterm lambs. Am J Physiol Lung Cell Mol Physiol 308: L983–L1001, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alhawaj R, Patel D, Kelly MR, Sun D, Wolin MS. Heme biosynthesis modulation via δ-aminolevulinic acid administration attenuates chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L719–L728, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 304: H1708–H1718, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator-activated receptor gamma (PPARγ) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res 92: 1162–1169, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 52: 1390–1400, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA 3rd.. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 34: 1093–1099, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awad KS, Elinoff JM, Wang S, Gairhe S, Ferreyra GA, Cai R, Sun J, Solomon MA, Danner RL. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 310: L187–L201, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 54: S55-66, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res 96: 119–126, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Bakker EN, Pistea A, Spaan JA, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow-dependent remodeling of small arteries in mice deficient for tissue-type transglutaminase: possible compensation by macrophage-derived factor XIII. Circ Res 99: 86–92, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Banasova A, Maxova H, Hampl V, Vizek M, Povysilova V, Novotna J, Vajnerova O, Hnilickova O, Herget J. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration 76: 102–107, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Bauer NR, Moore TM, McMurtry IF. Rodent models of PAH: are we there yet? Am J Physiol Lung Cell Mol Physiol 293: L580–L582, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 61: 599–610, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol 307: L936–L947, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernus A, Wagner BD, Accurso F, Doran A, Kaess H, Ivy DD. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest 135: 745–751, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, Hart CM. Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med 80: 111–120, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blood AB, Terry MH, Merritt TA, Papamatheakis DG, Blood Q, Ross JM, Power GG, Longo LD, Wilson SM. Effect of chronic perinatal hypoxia on the role of Rho-kinase in pulmonary artery contraction in newborn lambs. Am J Physiol Regul Integr Comp Physiol 304: R136–R146, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135: 794–804, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Bonnet S, Dumas-de-La-Roque E, Begueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA 100: 9488–9493, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braun MU, LaRosee P, Simonis G, Borst MM, Strasser RH. Regulation of protein kinase C isozymes in volume overload cardiac hypertrophy. Mol Cell Biochem 262: 135–143, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest 111: 1475–1486, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitling S, Ravindran K, Goldenberg NM, Kuebler WM. The pathophysiology of pulmonary hypertension in left heart disease. Am J Physiol Lung Cell Mol Physiol 309: L924–L941, 2015. [DOI] [PubMed] [Google Scholar]
- 29.Brown RD, Ambler SK, Li M, Sullivan TM, Henry LN, Crossno JT Jr, Long CS, Garrington TP, Stenmark KR. MAP kinase kinase kinase-2 (MEKK2) regulates hypertrophic remodeling of the right ventricle in hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 304: H269–H281, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruns DR, Brown RD, Stenmark KR, Buttrick PM, Walker LA. Mitochondrial integrity in a neonatal bovine model of right ventricular dysfunction. Am J Physiol Lung Cell Mol Physiol 308: L158–L167, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruns DR, Buttrick PM, Walker LA. Genetic ablation of interleukin-18 does not attenuate hypobaric hypoxia-induced right ventricular hypertrophy. Am J Physiol Lung Cell Mol Physiol 310: L542–L550, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bubb KJ, Trinder SL, Baliga RS, Patel J, Clapp LH, MacAllister RJ, Hobbs AJ. Inhibition of phosphodiesterase 2 augments cGMP and cAMP signaling to ameliorate pulmonary hypertension. Circulation 130: 496–507, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Can MM, Kaymaz C, Tanboga IH, Tokgoz HC, Canpolat N, Turkyilmaz E, Sonmez K, Ozdemir N. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin Nucl Med 36: 743–748, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Cero FT, Hillestad V, Sjaastad I, Yndestad A, Aukrust P, Ranheim T, Lunde IG, Olsen MB, Lien E, Zhang L, Haugstad SB, Loberg EM, Christensen G, Larsen KO, Skjonsberg OH. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol 309: L378–L387, 2015. [DOI] [PubMed] [Google Scholar]
- 35.Chan L, Chin LM, Kennedy M, Woolstenhulme JG, Nathan SD, Weinstein AA, Connors G, Weir NA, Drinkard B, Lamberti J, Keyser RE. Benefits of intensive treadmill exercise training on cardiorespiratory function and quality of life in patients with pulmonary hypertension. Chest 143: 333–343, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan MC, Weisman AS, Kang H, Nguyen PH, Hickman T, Mecker SV, Hill NS, Lagna G, Hata A. The amiloride derivative phenamil attenuates pulmonary vascular remodeling by activating NFAT and the bone morphogenetic protein signaling pathway. Mol Cell Biol 31: 517–530, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chava KR, Karpurapu M, Wang D, Bhanoori M, Kundumani-Sridharan V, Zhang Q, Ichiki T, Glasgow WC, Rao GN. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler Thromb Vasc Biol 29: 809–815, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol 297: L1151–L1159, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Chen B, Xue J, Meng X, Slutzky JL, Calvert AE, Chicoine LG. Resveratrol prevents hypoxia-induced arginase II expression and proliferation of human pulmonary artery smooth muscle cells via Akt-dependent signaling. Am J Physiol Lung Cell Mol Physiol 307: L317–L325, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Talati M, Fessel JP, Hemnes AR, Gladson S, French J, Shay S, Trammell A, Phillips JA, Hamid R, Cogan JD, Dawson EP, Womble KE, Hedges LK, Martinez EG, Wheeler LA, Loyd JE, Majka SJ, West J, Austin ED. Estrogen metabolite 16α-hydroxyestrone exacerbates bone morphogenetic protein receptor type II-associated pulmonary arterial hypertension through microRNA-29-mediated modulation of cellular metabolism. Circulation 133: 82–97, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chester M, Seedorf G, Tourneux P, Gien J, Tseng N, Grover T, Wright J, Stasch JP, Abman SH. Cinaciguat, a soluble guanylate cyclase activator, augments cGMP after oxidative stress and causes pulmonary vasodilation in neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 301: L755–L764, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chettimada S, Joshi SR, Alzoubi A, Gebb SA, McMurtry IF, Gupte R, Gupte SA. Glucose-6-phosphate dehydrogenase plays a critical role in hypoxia-induced CD133+ progenitor cells self-renewal and stimulates their accumulation in the lungs of pulmonary hypertensive rats. Am J Physiol Lung Cell Mol Physiol 307: L545–L556, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chevalier M, Gilbert G, Lory P, Marthan R, Quignard JF, Savineau JP. Dehydroepiandrosterone (DHEA) inhibits voltage-gated T-type calcium channels. Biochem Pharmacol 83: 1530–1539, 2012. [DOI] [PubMed] [Google Scholar]
- 46.Chichger H, Vang A, O'Connell KA, Zhang P, Mende U, Harrington EO, Choudhary G. PKC delta and betaII regulate angiotensin II-mediated fibrosis through p38: a mechanism of RV fibrosis in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L827–L836, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Church AC, Martin DH, Wadsworth R, Bryson G, Fisher AJ, Welsh DJ, Peacock AJ. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-α: a potential novel anti-inflammatory strategy in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 309: L333–L347, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 184: 1171–1182, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Cowburn AS, Crosby A, Macias D, Branco C, Colaco RD, Southwood M, Toshner M, Crotty Alexander LE, Morrell NW, Chilvers ER, Johnson RS. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci USA 113: 8801–8806, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crossno JT Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol 292: L885–L897, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, Hu F, Ballabh P, Podlutsky A, Losonczy G, de Cabo R, Mathew R, Wolin MS, Ungvari Z. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 54: 668–675, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dahal BK, Kosanovic D, Kaulen C, Cornitescu T, Savai R, Hoffmann J, Reiss I, Ghofrani HA, Weissmann N, Kuebler WM, Seeger W, Grimminger F, Schermuly RT. Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir Res 12: 60, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Damico R, Kolb TM, Valera L, Wang L, Housten T, Tedford RJ, Kass DA, Rafaels N, Gao L, Barnes KC, Benza RL, Rand JL, Hamid R, Loyd JE, Robbins IM, Hemnes AR, Chung WK, Austin ED, Drummond MB, Mathai SC, Hassoun PM. Serum endostatin is a genetically determined predictor of survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 191: 208–218, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davie NJ, Schermuly RT, Weissmann N, Grimminger F, Ghofrani HA. The science of endothelin-1 and endothelin receptor antagonists in the management of pulmonary arterial hypertension: current understanding and future studies. Eur J Clin Invest 39, Suppl 2: 38–49, 2009. [DOI] [PubMed] [Google Scholar]
- 55.de Raaf MA, Kroeze Y, Middelman A, de Man FS, de Jong H, Vonk-Noordegraaf A, de Korte C, Voelkel NF, Homberg J, Bogaard HJ. Serotonin transporter is not required for the development of severe pulmonary hypertension in the Sugen hypoxia rat model. Am J Physiol Lung Cell Mol Physiol 309: L1164–L1173, 2015. [DOI] [PubMed] [Google Scholar]
- 56.Dean A, Nilsen M, Loughlin L, Salt IP, MacLean MR. Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension 68: 446–454, 2016. [DOI] [PubMed] [Google Scholar]
- 57.Dempsie Y, MacRitchie NA, White K, Morecroft I, Wright AF, Nilsen M, Loughlin L, Mair KM, MacLean MR. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res 99: 24–34, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dib H, Tamby MC, Bussone G, Regent A, Berezne A, Lafine C, Broussard C, Simonneau G, Guillevin L, Witko-Sarsat V, Humbert M, Mouthon L. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Respir J 39: 1405–1414, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, Nakahira K, Alcazar MA, Hopper RK, Ji L, Feldman BJ, Rabinovitch M. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 21: 596–608, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399: 601–605, 1999. [DOI] [PubMed] [Google Scholar]
- 61.DiRaimondo TR, Klock C, Warburton R, Herrera Z, Penumatsa K, Toksoz D, Hill N, Khosla C, Fanburg B. Elevated transglutaminase 2 activity is associated with hypoxia-induced experimental pulmonary hypertension in mice. ACS Chem Biol 9: 266–275, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dodson RB, Morgan MR, Galambos C, Hunter KS, Abman SH. Chronic intrauterine pulmonary hypertension increases main pulmonary artery stiffness and adventitial remodeling in fetal sheep. Am J Physiol Lung Cell Mol Physiol 307: L822–L828, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res 113: 126–136, 2013. [DOI] [PubMed] [Google Scholar]
- 64.Dumas de La Roque E, Bellance N, Rossignol R, Begueret H, Billaud M, dos Santos P, Ducret T, Marthan R, Dahan D, Ramos-Barbon D, Amor-Carro O, Savineau JP, Fayon M. Dehydroepiandrosterone reverses chronic hypoxia/reoxygenation-induced right ventricular dysfunction in rats. Eur Respir J 40: 1420–1429, 2012. [DOI] [PubMed] [Google Scholar]
- 65.Dumas de La Roque E, Savineau JP, Metivier AC, Billes MA, Kraemer JP, Doutreleau S, Jougon J, Marthan R, Moore N, Fayon M, Baulieu EE, Dromer C. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): a pilot study. Ann Endocrinol (Paris) 73: 20–25, 2012. [DOI] [PubMed] [Google Scholar]
- 66.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 113: 286–295, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Eba S, Hoshikawa Y, Moriguchi T, Mitsuishi Y, Satoh H, Ishida K, Watanabe T, Shimizu T, Shimokawa H, Okada Y, Yamamoto M, Kondo T. The nuclear factor erythroid 2-related factor 2 activator oltipraz attenuates chronic hypoxia-induced cardiopulmonary alterations in mice. Am J Respir Cell Mol Biol 49: 324–333, 2013. [DOI] [PubMed] [Google Scholar]
- 68.El Chami H, Hassoun PM. Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog Cardiovasc Dis 55: 218–228, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.El Chami H, Hassoun PM. Inflammatory mechanisms in the pathogenesis of pulmonary arterial hypertension. Compr Physiol 1: 1929–1941, 2011. [DOI] [PubMed] [Google Scholar]
- 70.Enomoto M, Jain A, Pan J, Shifrin Y, Van Vliet T, McNamara PJ, Jankov RP, Belik J. Newborn rat response to single vs. combined cGMP-dependent pulmonary vasodilators. Am J Physiol Lung Cell Mol Physiol 306: L207–L215, 2014. [DOI] [PubMed] [Google Scholar]
- 71.Fagan KA, Tyler RC, Sato K, Fouty BW, Morris KG Jr, Huang PL, McMurtry IF, Rodman DM. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am J Physiol Lung Cell Mol Physiol 277: L472–L478, 1999. [DOI] [PubMed] [Google Scholar]
- 72.Falkenham A, de Antueno R, Rosin N, Betsch D, Lee TD, Duncan R, Legare JF. Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol 185: 927–942, 2015. [DOI] [PubMed] [Google Scholar]
- 73.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112: 1419–1428, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fediuk J, Sikarwar AS, Nolette N, Dakshinamurti S. Thromboxane-induced actin polymerization in hypoxic neonatal pulmonary arterial myocytes involves Cdc42 signaling. Am J Physiol Lung Cell Mol Physiol 307: L877–L887, 2014. [DOI] [PubMed] [Google Scholar]
- 75.Fenster BE, Lasalvia L, Schroeder JD, Smyser J, Silveira LJ, Buckner JK, Brown KK. Galectin-3 levels are associated with right ventricular functional and morphologic changes in pulmonary arterial hypertension. Heart Vessels 31: 939–946, 2016. [DOI] [PubMed] [Google Scholar]
- 76.Fessel JP, West JD. Redox biology in pulmonary arterial hypertension (2013 Grover Conference Series). Pulm Circ 5: 599–609, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fike CD, Dikalova A, Kaplowitz MR, Cunningham G, Summar M, Aschner JL. Rescue treatment with l-citrulline inhibits hypoxia-induced pulmonary hypertension in newborn pigs. Am J Respir Cell Mol Biol 53: 255–264, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fike CD, Kaplowitz MR. Effect of chronic hypoxia on pulmonary vascular pressures in isolated lungs of newborn pigs. J Appl Physiol (1985) 77: 2853–2862, 1994. [DOI] [PubMed] [Google Scholar]
- 79.Fisk M, Gajendragadkar PR, Maki-Petaja KM, Wilkinson IB, Cheriyan J. Therapeutic potential of p38 MAP kinase inhibition in the management of cardiovascular disease. Am J Cardiovasc Drugs 14: 155–165, 2014. [DOI] [PubMed] [Google Scholar]
- 80.Foris V, Kovacs G, Tscherner M, Olschewski A, Olschewski H. Biomarkers in pulmonary hypertension: what do we know? Chest 144: 274–283, 2013. [DOI] [PubMed] [Google Scholar]
- 81.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol 168: 659–669, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, Fierst J, Whitson J, Cucci AR, Brown MB, Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol 308: L873–L890, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Aboyans V, Vaz Carneiro A, Achenbach S, Agewall S, Allanore Y, Asteggiano R, Paolo Badano L, Albert Barbera J, Bouvaist H, Bueno H, Byrne RA, Carerj S, Castro G, Erol C, Falk V, Funck-Brentano C, Gorenflo M, Granton J, Iung B, Kiely DG, Kirchhof P, Kjellstrom B, Landmesser U, Lekakis J, Lionis C, Lip GY, Orfanos SE, Park MH, Piepoli MF, Ponikowski P, Revel MP, Rigau D, Rosenkranz S, Voller H, Luis Zamorano J. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and The European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37: 67–119, 2016. [DOI] [PubMed] [Google Scholar]
- 84.Galipeau HJ, Wiepjes M, Motta JP, Schulz JD, Jury J, Natividad JM, Pinto-Sanchez I, Sinclair D, Rousset P, Martin-Rosique R, Bermudez-Humaran L, Leroux JC, Murray JA, Smecuol E, Bai JC, Vergnolle N, Langella P, Verdu EF. Novel role of the serine protease inhibitor elafin in gluten-related disorders. Am J Gastroenterol 109: 748–756, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 132: 1906–1912, 2007. [DOI] [PubMed] [Google Scholar]
- 86.Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One 7: e47662, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gharaee-Kermani M, McCullumsmith RE, Charo IF, Kunkel SL, Phan SH. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine 24: 266–276, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Ghofrani HA, Humbert M, Langleben D, Schermuly R, Stasch JP, Wilkins MR, Klinger JR. Riociguat: mode of action and clinical development in pulmonary hypertension. Chest 2016. [DOI] [PubMed]
- 89.Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov 5: 689–702, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221, 1995. [DOI] [PubMed] [Google Scholar]
- 91.Gien J, Tseng N, Seedorf G, Roe G, Abman SH. Peroxisome proliferator activated receptor-γ-Rho-kinase interactions contribute to vascular remodeling after chronic intrauterine pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306: L299–L308, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest 128: 572S–573S, 2005. [DOI] [PubMed] [Google Scholar]
- 93.Golob MJ, Wang Z, Prostrollo AJ, Hacker TA, Chesler NC. Limiting collagen turnover via collagenase-resistance attenuates right ventricular dysfunction and fibrosis in pulmonary arterial hypertension. Physiol Rep 4: e12815, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gomez-Arroyo J, Sakagami M, Syed AA, Farkas L, Van Tassell B, Kraskauskas D, Mizuno S, Abbate A, Bogaard HJ, Byron PR, Voelkel NF. Iloprost reverses established fibrosis in experimental right ventricular failure. Eur Respir J 45: 449–462, 2015. [DOI] [PubMed] [Google Scholar]
- 95.Goss KN, Cucci AR, Fisher AJ, Albrecht M, Frump A, Tursunova R, Gao Y, Brown MB, Petrache I, Tepper RS, Ahlfeld SK, Lahm T. Neonatal hyperoxic lung injury favorably alters adult right ventricular remodeling response to chronic hypoxia exposure. Am J Physiol Lung Cell Mol Physiol 308: L797–L806, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Graham BB, Kumar R, Mickael C, Sanders L, Gebreab L, Huber KM, Perez M, Smith-Jones P, Serkova NJ, Tuder RM. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am J Physiol Lung Cell Mol Physiol 309: L435–L440, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grasemann H, Dhaliwal R, Ivanovska J, Kantores C, McNamara PJ, Scott JA, Belik J, Jankov RP. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am J Physiol Lung Cell Mol Physiol 308: L503–L510, 2015. [DOI] [PubMed] [Google Scholar]
- 98.Green DE, Murphy TC, Kang BY, Searles CD, Hart CM. PPARγ ligands attenuate hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of microRNA-21. PLoS One 10: e0133391, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grunig E, Lichtblau M, Ehlken N, Ghofrani HA, Reichenberger F, Staehler G, Halank M, Fischer C, Seyfarth HJ, Klose H, Meyer A, Sorichter S, Wilkens H, Rosenkranz S, Opitz C, Leuchte H, Karger G, Speich R, Nagel C. Safety and efficacy of exercise training in various forms of pulmonary hypertension. Eur Respir J 40: 84–92, 2012. [DOI] [PubMed] [Google Scholar]
- 100.Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, Rabinovitch M. Tie2-mediated loss of peroxisome proliferator-activated receptor-γ in mice causes PDGF receptor-β-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol 297: L1082–L1090, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guignabert C, Izikki M, Tu LI, Li Z, Zadigue P, Barlier-Mur AM, Hanoun N, Rodman D, Hamon M, Adnot S, Eddahibi S. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res 98: 1323–1330, 2006. [DOI] [PubMed] [Google Scholar]
- 102.Gupte SA, Li KX, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther 301: 299–305, 2002. [DOI] [PubMed] [Google Scholar]
- 103.Gurney AM, Joshi S, Manoury B. KCNQ potassium channels: new targets for pulmonary vasodilator drugs? Adv Exp Med Biol 661: 405–417, 2010. [DOI] [PubMed] [Google Scholar]
- 104.Hamilton NM, Dawson M, Fairweather EE, Hamilton NS, Hitchin JR, James DI, Jones SD, Jordan AM, Lyons AJ, Small HF, Thomson GJ, Waddell ID, Ogilvie DJ. Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. J Med Chem 55: 4431–4445, 2012. [DOI] [PubMed] [Google Scholar]
- 105.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 118: 1846–1857, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hartwig SM, Holman KM, Varga SM. Depletion of alveolar macrophages ameliorates virus-induced disease following a pulmonary coronavirus infection. PLoS One 9: e90720, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, Minami M, Inagaki T, Miyagawa S, Sawa Y, Murakami M, Kumanogoh A, Yamauchi-Takihara K, Okumura M, Kishimoto T, Komuro I, Shirai M, Sakata Y, Nakaoka Y. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci USA 112: E2677-2686, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hemnes AR, Trammell AW, Archer SL, Rich S, Yu C, Nian H, Penner N, Funke M, Wheeler L, Robbins IM, Austin ED, Newman JH, West J. Peripheral blood signature of vasodilator-responsive pulmonary arterial hypertension. Circulation 131: 401–409; discussion 409, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Heymann F, Trautwein C, Tacke F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm Allergy Drug Targets 8: 307–318, 2009. [DOI] [PubMed] [Google Scholar]
- 111.Hilgendorff A, Parai K, Ertsey R, Navarro E, Jain N, Carandang F, Peterson J, Mokres L, Milla C, Preuss S, Alcazar MA, Khan S, Masumi J, Ferreira-Tojais N, Mujahid S, Starcher B, Rabinovitch M, Bland R. Lung matrix and vascular remodeling in mechanically ventilated elastin haploinsufficient newborn mice. Am J Physiol Lung Cell Mol Physiol 308: L464–L478, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hiram R, Rizcallah E, Marouan S, Sirois C, Sirois M, Morin C, Fortin S, Rousseau E. Resolvin E1 normalizes contractility, Ca2+ sensitivity and smooth muscle cell migration rate in TNF-α- and IL-6-pretreated human pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 309: L776–L788, 2015. [DOI] [PubMed] [Google Scholar]
- 113.Hoffmann J, Marsh LM, Pieper M, Stacher E, Ghanim B, Kovacs G, Konig P, Wilkens H, Haitchi HM, Hoefler G, Klepetko W, Olschewski H, Olschewski A, Kwapiszewska G. Compartment-specific expression of collagens and their processing enzymes in intrapulmonary arteries of IPAH patients. Am J Physiol Lung Cell Mol Physiol 308: L1002–L1013, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoffmann J, Yin J, Kukucka M, Yin N, Saarikko I, Sterner-Kock A, Fujii H, Leong-Poi H, Kuppe H, Schermuly RT, Kuebler WM. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J 37: 1400–1410, 2011. [DOI] [PubMed] [Google Scholar]
- 115.Homma N, Nagaoka T, Karoor V, Imamura M, Taraseviciene-Stewart L, Walker LA, Fagan KA, McMurtry IF, Oka M. Involvement of RhoA/Rho kinase signaling in protection against monocrotaline-induced pulmonary hypertension in pneumonectomized rats by dehydroepiandrosterone. Am J Physiol Lung Cell Mol Physiol 295: L71–L78, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547: 133–145, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hsia CC, Hyde DM, Ochs M, Weibel ER; ATS/ ERS. Joint Task Force on Quantitative Assessment of Lung Structure. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 181: 394–418, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hsu JH, Oishi P, Wiseman DA, Hou Y, Chikovani O, Datar S, Sajti E, Johengen MJ, Harmon C, Black SM, Fineman JR. Nitric oxide alterations following acute ductal constriction in the fetal lamb: a role for superoxide. Am J Physiol Lung Cell Mol Physiol 298: L880–L887, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huelsz-Prince G, Belkin AM, VanBavel E, Bakker EN. Activation of extracellular transglutaminase 2 by mechanical force in the arterial wall. J Vasc Res 50: 383–395, 2013. [DOI] [PubMed] [Google Scholar]
- 120.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151: 1628–1631, 1995. [DOI] [PubMed] [Google Scholar]
- 121.Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A, Vonk Noordegraaf A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 145: 1230–1236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jain A, McNamara PJ. Persistent pulmonary hypertension of the newborn: Advances in diagnosis and treatment. Semin Fetal Neonatal Med 20: 262–271, 2015. [DOI] [PubMed] [Google Scholar]
- 123.Jankov RP, Luo X, Demin P, Aslam R, Hannam V, Tanswell AK, Pace-Asciak CR. Hepoxilin analogs inhibit bleomycin-induced pulmonary fibrosis in the mouse. J Pharmacol Exp Ther 301: 435–440, 2002. [DOI] [PubMed] [Google Scholar]
- 124.Jasiewicz M, Knapp M, Waszkiewicz E, Ptaszynska-Kopczynska K, Szpakowicz A, Sobkowicz B, Musial WJ, Kaminski KA. Enhanced IL-6 trans-signaling in pulmonary arterial hypertension and its potential role in disease-related systemic damage. Cytokine 76: 187–192, 2015. [DOI] [PubMed] [Google Scholar]
- 125.Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW, Stewart DJ. Marked strain-specific differences in the su5416 rat model of severe pulmonary arterial hypertension. Am J Respir Cell Mol Biol 54: 461–468, 2016. [DOI] [PubMed] [Google Scholar]
- 126.Jie W, Guo J, Shen Z, Wang X, Zheng S, Wang G, Ao Q. Contribution of myocardin in the hypoxia-induced phenotypic switching of rat pulmonary arterial smooth muscle cells. Exp Mol Pathol 89: 301–306, 2010. [DOI] [PubMed] [Google Scholar]
- 127.Jin N, Hatton N, Swartz DR, Xia X, Harrington MA, Larsen SH, Rhoades RA. Hypoxia activates jun-N-terminal kinase, extracellular signal-regulated protein kinase, and p38 kinase in pulmonary arteries. Am J Respir Cell Mol Biol 23: 593–601, 2000. [DOI] [PubMed] [Google Scholar]
- 128.Jin Y, Calvert TJ, Chen B, Chicoine LG, Joshi M, Bauer JA, Liu Y, Nelin LD. Mice deficient in Mkp-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am J Physiol Heart Circ Physiol 298: H1518–H1528, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jin Y, Jin Y, Chen B, Tipple TE, Nelin LD. Arginase II is a target of miR-17-5p and regulates miR-17-5p expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 307: L197–L204, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Johansen AK, Dean A, Morecroft I, Hood K, Nilsen M, Loughlin L, Anagnostopoulou A, Touyz RM, White K, MacLean MR. The serotonin transporter promotes a pathological estrogen metabolic pathway in pulmonary hypertension via cytochrome P450 1B1. Pulm Circ 6: 82–92, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther 329: 368–376, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kaneko FT, Arroliga AC, Dweik RA, Comhair SA, Laskowski D, Oppedisano R, Thomassen MJ, Erzurum SC. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am J Respir Crit Care Med 158: 917–923, 1998. [DOI] [PubMed] [Google Scholar]
- 133.Kawut S, Archer-Chicko C, DeMichele A, Dubow D, Fritz JS, Ky B, Palevsky H, Palmisciano AJ, Patel M, Pinder D, Propert K, Smith K, Stanczyk F, Vaidya A, Ventetuolo C. A randomized clinical trial of anastrozole in pulmonary arterial hypertension. Am J Respir Crit Care Med 193: A6312, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kawut SM, Al-Naamani N, Agerstrand C, Rosenzweig EB, Rowan C, Barst RJ, Bergmann S, Horn EM. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest 135: 752–759, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim FY, Barnes EA, Ying L, Chen C, Lee L, Alvira CM, Cornfield DN. Pulmonary artery smooth muscle cell endothelin-1 expression modulates the pulmonary vascular response to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 308: L368–L377, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Klinger JR. Tadalafil for the treatment of pulmonary arterial hypertension. Expert Rev Respir Med 5: 315–328, 2011. [DOI] [PubMed] [Google Scholar]
- 137.Kunita-Takanezawa M, Abe K, Hirooka Y, Kuwabara Y, Hirano K, Oka M, Sunagawa K. Novel dual endothelin receptor antagonist macitentan reverses severe pulmonary arterial hypertension in rats. J Cardiovasc Pharmacol 64: 473–480, 2014. [DOI] [PubMed] [Google Scholar]
- 138.Kuriyama S, Morio Y, Toba M, Nagaoka T, Takahashi F, Iwakami S, Seyama K, Takahashi K. Genistein attenuates hypoxic pulmonary hypertension via enhanced nitric oxide signaling and the erythropoietin system. Am J Physiol Lung Cell Mol Physiol 306: L996–L1005, 2014. [DOI] [PubMed] [Google Scholar]
- 139.Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L7–L26, 2014. [DOI] [PubMed] [Google Scholar]
- 140.Lai YC, Tabima DM, Dube JJ, Hughan KS, Vanderpool RR, Goncharov DA, St Croix CM, Garcia-Ocana A, Goncharova EA, Tofovic SP, Mora AL, Gladwin MT. SIRT3-AMP-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation 133: 717–731, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lammers AE, Hislop AA, Haworth SG. Prognostic value of B-type natriuretic peptide in children with pulmonary hypertension. Int J Cardiol 135: 21–26, 2009. [DOI] [PubMed] [Google Scholar]
- 142.Lang M, Kojonazarov B, Tian X, Kalymbetov A, Weissmann N, Grimminger F, Kretschmer A, Stasch JP, Seeger W, Ghofrani HA, Schermuly RT. The soluble guanylate cyclase stimulator riociguat ameliorates pulmonary hypertension induced by hypoxia and SU5416 in rats. PLoS One 7: e43433, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lemler MS, Bies RD, Frid MG, Sastravaha A, Zisman LS, Bohlmeyer T, Gerdes AM, Reeves JT, Stenmark KR. Myocyte cytoskeletal disorganization and right heart failure in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Heart Circ Physiol 279: H1365–H1376, 2000. [DOI] [PubMed] [Google Scholar]
- 144.Levy M, Bonnet D, Mauge L, Celermajer DS, Gaussem P, Smadja DM. Circulating endothelial cells in refractory pulmonary hypertension in children: markers of treatment efficacy and clinical worsening. PLoS One 8: e65114, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lewis GD, Ngo D, Hemnes AR, Farrell L, Domos C, Pappagianopoulos PP, Dhakal BP, Souza A, Shi X, Pugh ME, Beloiartsev A, Sinha S, Clish CB, Gerszten RE. Metabolic profiling of right ventricular-pulmonary vascular function reveals circulating biomarkers of pulmonary hypertension. J Am Coll Cardiol 67: 174–189, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li C, Ni J, Wang Z, Li M, Gasparic M, Terhaag B, Uberall MA. Analgesic efficacy and tolerability of flupirtine vs. tramadol in patients with subacute low back pain: a double-blind multicentre trial. Curr Med Res Opin 24: 3523–3530, 2008. [DOI] [PubMed] [Google Scholar]
- 147.Li SS, Ran YJ, Zhang DD, Li SZ, Zhu D. MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K+ channel in arterial smooth muscle cells. J Cell Biochem 115: 1196–1205, 2014. [DOI] [PubMed] [Google Scholar]
- 148.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4: 140–156, 2003. [DOI] [PubMed] [Google Scholar]
- 149.Lu X, Murphy TC, Nanes MS, Hart CM. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am J Physiol Lung Cell Mol Physiol 299: L559–L566, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, Asosingh K, Comhair SA, DiFilippo FP, Neumann DR, Davis L, Graham BB, Tuder RM, Dostanic I, Erzurum SC. Fasting 2-deoxy-2-[18F]fluoro-d-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc 10: 1–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maclean MR, Dempsie Y. The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol 661: 309–322, 2010. [DOI] [PubMed] [Google Scholar]
- 152.MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, Colston J, Loughlin L, Nilsen M, Dempsie Y, Harmar A. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation 109: 2150–2155, 2004. [DOI] [PubMed] [Google Scholar]
- 153.Makanga M, Maruyama H, Dewachter C, Da Costa AM, Hupkens E, de Medina G, Naeije R, Dewachter L. Prevention of pulmonary hypoplasia and pulmonary vascular remodeling by antenatal simvastatin treatment in nitrofen-induced congenital diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 308: L672–L682, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mandell E, Powers KN, Harral JW, Seedorf GJ, Hunter KS, Abman SH, Dodson RB. Intrauterine endotoxin-induced impairs pulmonary vascular function and right ventricular performance in infant rats and improvement with early vitamin D therapy. Am J Physiol Lung Cell Mol Physiol 309: L1438–L1446, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mandell E, Seedorf G, Gien J, Abman SH. Vitamin D treatment improves survival and infant lung structure after intra-amniotic endotoxin exposure in rats: potential role for the prevention of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 306: L420–L428, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, Loscalzo J, Leopold JA. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 130: 168–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Maruyama H, Dewachter C, Belhaj A, Rondelet B, Sakai S, Remmelink M, Vachiery JL, Naeije R, Dewachter L. Endothelin-Bone morphogenetic protein type 2 receptor interaction induces pulmonary artery smooth muscle cell hyperplasia in pulmonary arterial hypertension. J Heart Lung Transplant 34: 468–478, 2015. [DOI] [PubMed] [Google Scholar]
- 158.Mathie SA, Dixon KL, Walker SA, Tyrrell V, Mondhe M, O'Donnell VB, Gregory LG, Lloyd CM. Alveolar macrophages are sentinels of murine pulmonary homeostasis following inhaled antigen challenge. Allergy 70: 80–89, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.McMurtry IF, Frith CH, Will DH. Cardiopulmonary responses of male and female swine to simulated high altitude. J Appl Physiol 35: 459–462, 1973. [DOI] [PubMed] [Google Scholar]
- 160.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95: 830–840, 2004. [DOI] [PubMed] [Google Scholar]
- 161.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 105: 244–250, 2002. [DOI] [PubMed] [Google Scholar]
- 162.Mizuno S, Farkas L, Al Husseini A, Farkas D, Gomez-Arroyo J, Kraskauskas D, Nicolls MR, Cool CD, Bogaard HJ, Voelkel NF. Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am J Respir Cell Mol Biol 47: 679–687, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Morecroft I, Murray A, Nilsen M, Gurney AM, MacLean MR. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br J Pharmacol 157: 1241–1249, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Morisawa D, Hirotani S, Oboshi M, Nishimura K, Sawada H, Eguchi A, Okuhara Y, Iwasaku T, Naito Y, Mano T, Okamura H, Masuyama T. Interleukin-18 disruption suppresses hypoxia-induced pulmonary artery hypertension in mice. Int J Cardiol 202: 522–524, 2016. [DOI] [PubMed] [Google Scholar]
- 165.Mortimer HJ, Peacock AJ, Kirk A, Welsh DJ.. p38 MAP kinase: essential role in hypoxia-mediated human pulmonary artery fibroblast proliferation. Pulm Pharmacol Ther 20: 718–725, 2007. [DOI] [PubMed] [Google Scholar]
- 166.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med 171: 494–499, 2005. [DOI] [PubMed] [Google Scholar]
- 167.Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, Hennigs JK, Kim KY, Miyagawa K, Wang L, Cao A, Sa S, Jiang X, Stockstill RW, Nicolls MR, Zamanian RT, Bland RD, Rabinovitch M. Elafin reverses pulmonary hypertension via caveolin-1-dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med 191: 1273–1286, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez-Arroyo JG, Voelkel NF, Bogaard HJ. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ 2: 434–442, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nijmeh H, Balasubramaniam V, Burns N, Ahmad A, Stenmark KR, Gerasimovskaya EV. High proliferative potential endothelial colony-forming cells contribute to hypoxia-induced pulmonary artery vasa vasorum neovascularization. Am J Physiol Lung Cell Mol Physiol 306: L661–L671, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol 42: 482–490, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nozik-Grayck E, Woods C, Stearman RS, Venkataraman S, Ferguson BS, Swain K, Bowler RP, Geraci MW, Ihida-Stansbury K, Stenmark KR, McKinsey TA, Domann FE. Histone deacetylation contributes to low extracellular superoxide dismutase expression in human idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 311: L124–L134, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nozik-Grayck E, Woods C, Taylor JM, Benninger RK, Johnson RD, Villegas LR, Stenmark KR, Harrison DG, Majka SM, Irwin D, Farrow KN. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L868–L876, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, Limbird J, Imamura M, Gebb SA, Fagan KA, McMurtry IF. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res 74: 377–387, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ooi CY, Wang Z, Tabima DM, Eickhoff JC, Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 299: H1823–H1831, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Patel D, Kandhi S, Kelly M, Neo BH, Wolin MS. Dehydroepiandrosterone promotes pulmonary artery relaxation by NADPH oxidation-elicited subunit dimerization of protein kinase G-1α. Am J Physiol Lung Cell Mol Physiol 306: L383–L391, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Patel N, Moenkemeyer F, Germano S, Cheung MM. Plasma vascular endothelial growth factor A and placental growth factor: novel biomarkers of pulmonary hypertension in congenital diaphragmatic hernia. Am J Physiol Lung Cell Mol Physiol 308: L378–L383, 2015. [DOI] [PubMed] [Google Scholar]
- 179.Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, Michelakis ED. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab 20: 827–839, 2014. [DOI] [PubMed] [Google Scholar]
- 180.Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 301: H1798–H1809, 2011. [DOI] [PubMed] [Google Scholar]
- 181.Penumatsa KC, Toksoz D, Warburton RR, Hilmer AJ, Liu T, Khosla C, Comhair SA, Fanburg BL. Role of hypoxia-induced transglutaminase 2 in pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 307: L576–L585, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pezzuto B, Badagliacca R, Poscia R, Ghio S, D'Alto M, Vitulo P, Mule M, Albera C, Volterrani M, Fedele F, Vizza CD. Circulating biomarkers in pulmonary arterial hypertension: update and future direction. J Heart Lung Transplant 34: 282–305, 2015. [DOI] [PubMed] [Google Scholar]
- 183.Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, Archer SL. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med 91: 333–346, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 304: L549–L561, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pistea A, Bakker EN, Spaan JA, Hardeman MR, van Rooijen N, VanBavel E. Small artery remodeling and erythrocyte deformability in l-NAME-induced hypertension: role of transglutaminases. J Vasc Res 45: 10–18, 2008. [DOI] [PubMed] [Google Scholar]
- 186.Plomaritas DR, Herbert LM, Yellowhair TR, Resta TC, Gonzalez Bosc LV, Walker BR, Jernigan NL. Chronic hypoxia limits H2O2-induced inhibition of ASIC1-dependent store-operated calcium entry in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 307: L419–L430, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Porter RJ, Burdette DE, Gil-Nagel A, Hall ST, White R, Shaikh S, DeRossett SE. Retigabine as adjunctive therapy in adults with partial-onset seizures: integrated analysis of three pivotal controlled trials. Epilepsy Res 101: 103–112, 2012. [DOI] [PubMed] [Google Scholar]
- 188.Price LC, Wort SJ, Perros F, Dorfmuller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest 141: 210–221, 2012. [DOI] [PubMed] [Google Scholar]
- 189.Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol 308: L229–L252, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G SERAPHIN. Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 369: 809–818, 2013. [DOI] [PubMed] [Google Scholar]
- 191.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rabinovitch M, Chesler N, Molthen RC. Point:Counterpoint: Chronic hypoxia-induced pulmonary hypertension does/does not lead to loss of pulmonary vasculature. J Appl Physiol (1985) 103: 1449–1451, 2007. [DOI] [PubMed] [Google Scholar]
- 193.Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol Heart Circ Physiol 240: H62–H72, 1981. [DOI] [PubMed] [Google Scholar]
- 194.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 115: 165–175, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmuller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 128: 2016–2025, 2013. [DOI] [PubMed] [Google Scholar]
- 196.Ramchandran R, Raghavan A, Geenen D, Sun M, Bach L, Yang Q, Raj JU. PKG-1α leucine zipper domain defect increases pulmonary vascular tone: implications in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L537–L544, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol 280: L88–L97, 2001. [DOI] [PubMed] [Google Scholar]
- 198.Rhodes CJ, Im H, Cao A, Hennigs JK, Wang L, Sa S, Chen PI, Nickel NP, Miyagawa K, Hopper RK, Tojais NF, Li CG, Gu M, Spiekerkoetter E, Xian Z, Chen R, Zhao M, Kaschwich M, Del Rosario PA, Bernstein D, Zamanian RT, Wu JC, Snyder MP, Rabinovitch M. RNA Sequencing analysis detection of a novel pathway of endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 192: 356–366, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rich S, Kieras K, Hart K, Groves BM, Stobo JD, Brundage BH. Antinuclear antibodies in primary pulmonary hypertension. J Am Coll Cardiol 8: 1307–1311, 1986. [DOI] [PubMed] [Google Scholar]
- 200.Rose F, Grimminger F, Appel J, Heller M, Pies V, Weissmann N, Fink L, Schmidt S, Krick S, Camenisch G, Gassmann M, Seeger W, Hanze J. Hypoxic pulmonary artery fibroblasts trigger proliferation of vascular smooth muscle cells: role of hypoxia-inducible transcription factors. FASEB J 16: 1660–1661, 2002. [DOI] [PubMed] [Google Scholar]
- 201.Ross DJ, Strieter RM, Fishbein MC, Ardehali A, Belperio JA. Type I immune response cytokine-chemokine cascade is associated with pulmonary arterial hypertension. J Heart Lung Transplant 31: 865–873, 2012. [DOI] [PubMed] [Google Scholar]
- 202.Roy B, Mo E, Vernon J, Garthwaite J. Probing the presence of the ligand-binding haem in cellular nitric oxide receptors. Br J Pharmacol 153: 1495–1504, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 131: 1691–1702, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol 24: 1333–1347, 1992. [DOI] [PubMed] [Google Scholar]
- 205.Safdar Z, Tamez E, Chan W, Arya B, Ge Y, Deswal A, Bozkurt B, Frost A, Entman M. Circulating collagen biomarkers as indicators of disease severity in pulmonary arterial hypertension. JACC Heart Fail 2: 412–421, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Safdar Z, Tamez E, Frost A, Guffey D, Minard CG, Entman ML. Collagen metabolism biomarkers and health related quality of life in pulmonary arterial hypertension. Int J Cardiovasc Res 4: 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM, Berkowitz DE. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ Res 107: 117–125, 2010. [DOI] [PubMed] [Google Scholar]
- 208.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, Klussmann S, Ghofrani HA, Weissmann N, Klepetko W, Banat GA, Seeger W, Grimminger F, Schermuly RT. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 897–908, 2012. [DOI] [PubMed] [Google Scholar]
- 209.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res 10: 6, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schafer M, Myers C, Brown RD, Frid MG, Tan W, Hunter K, Stenmark KR. Pulmonary arterial stiffness: toward a new paradigm in pulmonary arterial hypertension pathophysiology and assessment. Curr Hypertens Rep 18: 4, 2016. [DOI] [PubMed] [Google Scholar]
- 211.Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D, Schluter KD, Dingendorf A, Hackemack S, Kolosionek E, Kaulen C, Dumitrascu R, Weissmann N, Mittendorf J, Klepetko W, Seeger W, Ghofrani HA, Grimminger F. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur Respir J 32: 881–891, 2008. [DOI] [PubMed] [Google Scholar]
- 212.Schneider JP, Ochs M. Alterations of mouse lung tissue dimensions during processing for morphometry: a comparison of methods. Am J Physiol Lung Cell Mol Physiol 306: L341–L350, 2014. [DOI] [PubMed] [Google Scholar]
- 213.Schreier D, Hacker T, Song G, Chesler N. The role of collagen synthesis in ventricular and vascular adaptation to hypoxic pulmonary hypertension. J Biomech Eng 135: 021018, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sedivy V, Joshi S, Ghaly Y, Mizera R, Zaloudikova M, Brennan S, Novotna J, Herget J, Gurney AM. Role of Kv7 channels in responses of the pulmonary circulation to hypoxia. Am J Physiol Lung Cell Mol Physiol 308: L48–L57, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 32: 2361–2367, 2000. [DOI] [PubMed] [Google Scholar]
- 216.Shen T, Shi J, Wang N, Yu X, Zhang C, Li J, Wei L, Ma C, Zhao X, Lian M, Jiang C, Zhu D. 15-Lipoxygenase and 15-hydroxyeicosatetraenoic acid regulate intravascular thrombosis in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 309: L449–L462, 2015. [DOI] [PubMed] [Google Scholar]
- 217.Shifren A, Durmowicz AG, Knutsen RH, Faury G, Mecham RP. Elastin insufficiency predisposes to elevated pulmonary circulatory pressures through changes in elastic artery structure. J Appl Physiol (1985) 105: 1610–1619, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shifrin Y, Sadeghi S, Pan J, Jain A, Fajardo AF, McNamara PJ, Belik J. Maternal-pup interaction disturbances induce long-lasting changes in the newborn rat pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol 309: L1186–L1198, 2015. [DOI] [PubMed] [Google Scholar]
- 219.Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med 91: 297–309, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Shimoda LA, Sham JS, Liu Q, Sylvester JT. Acute and chronic hypoxic pulmonary vasoconstriction: a central role for endothelin-1? Respir Physiol Neurobiol 132: 93–106, 2002. [DOI] [PubMed] [Google Scholar]
- 221.Shinohara T, Sawada H, Otsuki S, Yodoya N, Kato T, Ohashi H, Zhang E, Saitoh S, Shimpo H, Maruyama K, Komada Y, Mitani Y. Macitentan reverses early obstructive pulmonary vasculopathy in rats: early intervention in overcoming the survivin-mediated resistance to apoptosis. Am J Physiol Lung Cell Mol Physiol 308: L523–L538, 2015. [DOI] [PubMed] [Google Scholar]
- 222.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093–1129, 2005. [DOI] [PubMed] [Google Scholar]
- 223.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104: 236–244, 228p following 244, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. [DOI] [PubMed] [Google Scholar]
- 225.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 226.Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol (1985) 119: 1164–1172, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res 81: 34–41, 1997. [DOI] [PubMed] [Google Scholar]
- 228.Suresh K, Shimoda LA. Lung circulation. Compr Physiol 6: 897–943, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Talati M, Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ 5: 269–278, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Pena-Lefebvre P, Brunet S, Servettaz A, Weill B, Simonneau G, Guillevin L, Boissier MC, Mouthon L. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial. hypertension. Thorax 60: 765–772, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tamby MC, Humbert M, Guilpain P, Servettaz A, Dupin N, Christner JJ, Simonneau G, Fermanian J, Weill B, Guillevin L, Mouthon L. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur Respir J 28: 799–807, 2006. [DOI] [PubMed] [Google Scholar]
- 232.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. [DOI] [PubMed] [Google Scholar]
- 235.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med 175: 1280–1289, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Temple IP, Monfredi O, Quigley G, Schneider H, Zi M, Cartwright EJ, Boyett MR, Mahadevan VS, Hart G. Macitentan treatment retards the progression of established pulmonary arterial hypertension in an animal model. Int J Cardiol 177: 423–428, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Thenappan T, Goel A, Marsboom G, Fang YH, Toth PT, Zhang HJ, Kajimoto H, Hong Z, Paul J, Wietholt C, Pogoriler J, Piao L, Rehman J, Archer SL. A central role for CD68(+) macrophages in hepatopulmonary syndrome. Reversal by macrophage depletion. Am J Respir Crit Care Med 183: 1080–1091, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Thenappan T, Prins KW, Pritzker MR, Scandurra J, Volmers K, Weir EK. The critical role of pulmonary arterial compliance in pulmonary hypertension. Ann Am Thorac Soc 13: 276–284, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med 5: 200ra117, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Toba M, Alzoubi A, O'Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 306: H243–H250, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Tozzi CA, Christiansen DL, Poiani GJ, Riley DJ. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am J Respir Crit Care Med 149: 1317–1326, 1994. [DOI] [PubMed] [Google Scholar]
- 242.Tuder RM. How do we measure pathology in PAH (lung and RV) and what does it tell us about the disease. Drug Discov Today 19: 1257–1263, 2014. [DOI] [PubMed] [Google Scholar]
- 243.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 144: 275–285, 1994. [PMC free article] [PubMed] [Google Scholar]
- 244.Uenoyama M, Ogata S, Nakanishi K, Kanazawa F, Hiroi S, Tominaga S, Seo A, Matsui T, Kawai T, Suzuki S. Protein kinase C mRNA and protein expressions in hypobaric hypoxia-induced cardiac hypertrophy in rats. Acta Physiol (Oxf) 198: 431–440, 2010. [DOI] [PubMed] [Google Scholar]
- 245.van Loenhout RB, Tibboel D, Post M, Keijzer R. Congenital diaphragmatic hernia: comparison of animal models and relevance to the human situation. Neonatology 96: 137–149, 2009. [DOI] [PubMed] [Google Scholar]
- 246.Ventetuolo CE, Baird GL, Barr RG, Bluemke DA, Fritz JS, Hill NS, Klinger JR, Lima JA, Ouyang P, Palevsky HI, Palmisciano AJ, Krishnan I, Pinder D, Preston IR, Roberts KE, Kawut SM. Higher Estradiol and Lower Dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med 193: 1168–1175, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ventetuolo CE, Lima JA, Barr RG, Bristow MR, Bagiella E, Chahal H, Kizer JR, Lederer DJ, Bluemke DA, Kawut SM. The renin-angiotensin system and right ventricular structure and function: The MESA-Right Ventricle Study. Pulm Circ 2: 379–386, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123: 1986–1995, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Villanueva ME, Zaher FM, Svinarich DM, Konduri GG. Decreased gene expression of endothelial nitric oxide synthase in newborns with persistent pulmonary hypertension. Pediatr Res 44: 338–343, 1998. [DOI] [PubMed] [Google Scholar]
- 250.Villegas LR, Kluck D, Field C, Oberley-Deegan RE, Woods C, Yeager ME, El Kasmi KC, Savani RC, Bowler RP, Nozik-Grayck E. Superoxide dismutase mimetic, MnTE-2-PyP, attenuates chronic hypoxia-induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid Redox Signal 18: 1753–1764, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, Kourembanas S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: long-term follow-up. Pulm Circ 4: 619–629, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Voelkel NF, Tamosiuniene R, Nicolls MR. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert Rev Cardiovasc Ther 14: 939–951, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wagenaar GT, Hiemstra PS, Gosens R. Therapeutic potential of soluble guanylate cyclase modulators in neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol 309: L1037–L1040, 2015. [DOI] [PubMed] [Google Scholar]
- 254.Wagenaar GT, Laghmani el H, Fidder M, Sengers RM, de Visser YP, de Vries L, Rink R, Roks AJ, Folkerts G, Walther FJ. Agonists of MAS oncogene and angiotensin II type 2 receptors attenuate cardiopulmonary disease in rats with neonatal hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol 305: L341–L351, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Wagenaar GT, Sengers RM, Laghmani el H, Chen X, Lindeboom MP, Roks AJ, Folkerts G, Walther FJ. Angiotensin II type 2 receptor ligand PD123319 attenuates hyperoxia-induced lung and heart injury at a low dose in newborn rats. Am J Physiol Lung Cell Mol Physiol 307: L261–L272, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol 289: H1209–H1217, 2005. [DOI] [PubMed] [Google Scholar]
- 257.Walker LA, Walker JS, Glazier A, Brown DR, Stenmark KR, Buttrick PM. Biochemical and myofilament responses of the right ventricle to severe pulmonary hypertension. Am J Physiol Heart Circ Physiol 301: H832–H840, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, Changsirivathanathamrong D, Wu BJ, Ball HJ, Thomas SR, Kapoor V, Celermajer DS, Mellor AL, Keaney JF Jr, Hunt NH, Stocker R. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med 16: 279–285, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wedgwood S, Lakshminrusimha S, Schumacker PT, Steinhorn RH. Cyclic stretch stimulates mitochondrial reactive oxygen species and Nox4 signaling in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 309: L196–L203, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wei L, Liu Y, Kaneto H, Fanburg BL. JNK regulates serotonin-mediated proliferation and migration of pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 298: L863–L869, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Weissmann N, Peters DM, Klopping C, Kruger K, Pilat C, Katta S, Seimetz M, Ghofrani HA, Schermuly RT, Witzenrath M, Seeger W, Grimminger F, Mooren FC. Structural and functional prevention of hypoxia-induced pulmonary hypertension by individualized exercise training in mice. Am J Physiol Lung Cell Mol Physiol 306: L986–L995, 2014. [DOI] [PubMed] [Google Scholar]
- 262.Wen Y, Gu J, Chakrabarti SK, Aylor K, Marshall J, Takahashi Y, Yoshimoto T, Nadler JL. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-α in macrophages. Endocrinology 148: 1313–1322, 2007. [DOI] [PubMed] [Google Scholar]
- 263.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 90: 373–382, 2011. [DOI] [PubMed] [Google Scholar]
- 264.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, Morecroft I, Loughlin L, McClure JD, Thomas M, Mair KM, MacLean MR. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 126: 1087–1098, 2012. [DOI] [PubMed] [Google Scholar]
- 265.Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 294: L309–L318, 2008. [DOI] [PubMed] [Google Scholar]
- 266.Willfuhr A, Brandenberger C, Piatkowski T, Grothausmann R, Nyengaard JR, Ochs M, Muhlfeld C. Estimation of the number of alveolar capillaries by the Euler number (Euler-Poincare characteristic). Am J Physiol Lung Cell Mol Physiol 309: L1286–L1293, 2015. [DOI] [PubMed] [Google Scholar]
- 267.Wilson JL, Rupasinghe C, Usheva A, Warburton R, Kaplan C, Taylor L, Hill N, Mierke DF, Polgar P. Modulating the dysregulated migration of pulmonary arterial hypertensive smooth muscle cells with motif mimicking cell permeable peptides. Curr Top Pept Protein Res 16: 1–17, 2015. [PMC free article] [PubMed] [Google Scholar]
- 268.Wilson JL, Yu J, Taylor L, Polgar P. Hyperplastic growth of pulmonary artery smooth muscle cells from subjects with pulmonary arterial hypertension is activated through JNK and p38 MAPK. PLoS One 10: e0123662, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Wohrley JD, Frid MG, Moiseeva EP, Orton EC, Belknap JK, Stenmark KR. Hypoxia selectively induces proliferation in a specific subpopulation of smooth muscle cells in the bovine neonatal pulmonary arterial media. J Clin Invest 96: 273–281, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Wolf D, Tseng N, Seedorf G, Roe G, Abman SH, Gien J. Endothelin-1 decreases endothelial PPARgamma signaling and impairs angiogenesis after chronic intrauterine pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306: L361–L371, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X, Xu Y, Tang Q, Hu D, Somani A, Geurts AM, Ostertag E, Bache RJ, Weir EK, Chen Y. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension 58: 303–309, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 18: 1746–1748, 2004. [DOI] [PubMed] [Google Scholar]
- 273.Yamaji-Kegan K, Takimoto E, Zhang A, Weiner NC, Meuchel LW, Berger AE, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (FIZZ1/RELMa) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306: L1090–L1103, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU. Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. Am J Physiol Lung Cell Mol Physiol 303: L1001–L1010, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Yang T, Wang L, Xiong CM, He JG, Zhang Y, Gu Q, Zhao ZH, Ni XH, Fang W, Liu ZH. The ratio of (18)F-FDG activity uptake between the right and left ventricle in patients with pulmonary hypertension correlates with the right ventricular function. Clin Nucl Med 39: 426–430, 2014. [DOI] [PubMed] [Google Scholar]
- 276.Yeager ME, Colvin KL, Everett AD, Stenmark KR, Ivy DD. Plasma proteomics of differential outcome to long-term therapy in children with idiopathic pulmonary arterial hypertension. Proteomics Clin Appl 6: 257–267, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Yeager ME, Nguyen CM, Belchenko DD, Colvin KL, Takatsuki S, Ivy DD, Stenmark KR. Circulating fibrocytes are increased in children and young adults with pulmonary hypertension. Eur Respir J 39: 104–111, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O'Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol 178: 2601–2610, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Young KC, Torres E, Hehre D, Wu S, Suguihara C, Hare JM. Antagonism of stem cell factor/c-kit signaling attenuates neonatal chronic hypoxia-induced pulmonary vascular remodeling. Pediatr Res 79: 637–646, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 105: 516–521, 2002. [DOI] [PubMed] [Google Scholar]
- 281.Zamora MA, Dempsey EC, Walchak SJ, Stelzner TJ. BQ123, an ETA receptor antagonist, inhibits endothelin-1-mediated proliferation of human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 9: 429–433, 1993. [DOI] [PubMed] [Google Scholar]
- 282.Zhang R, Shi L, Zhou L, Zhang G, Wu X, Shao F, Ma G, Ying K. Transgelin as a therapeutic target to prevent hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306: L574–L583, 2014. [DOI] [PubMed] [Google Scholar]
- 283.Zhang R, Zhou L, Li Q, Liu J, Yao W, Wan H. Up-regulation of two actin-associated proteins prompts pulmonary artery smooth muscle cell migration under hypoxia. Am J Respir Cell Mol Biol 41: 467–475, 2009. [DOI] [PubMed] [Google Scholar]
- 284.Zhao YD, Cai L, Mirza MK, Huang X, Geenen DL, Hofmann F, Yuan JX, Zhao YY. Protein kinase G-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation. Am J Pathol 180: 2268–2275, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Zhou Z, de Beer VJ, de Wijs-Meijler D, Bender SB, Hoekstra M, Laughlin MH, Duncker DJ, Merkus D. Pulmonary vasoconstrictor influence of endothelin in exercising swine depends critically on phosphodiesterase 5 activity. Am J Physiol Lung Cell Mol Physiol 306: L442–L452, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]