

Abstract
Background
Cutis tricolor is a skin abnormality consisting in a combination of congenital hyper- and hypopigmented skin lesions (in the form of paired macules, patches or streaks) in close proximity to each other in a background of normal skin. It is currently regarded as a twin-spotting (mosaic) phenomenon and today is clear that not all cases of cutis tricolor represent one single entity. This phenomenon has been reported so far either: (I) as an purely cutaneous trait; (II) as a part of a complex malformation phenotype (Ruggieri-Happle syndrome, RHS) including distinct facial features, eye (cataract), skeletal (skull and vertebral defects, and long bones dysplasia), nervous system (corpus callosum, cerebellar and white matter anomalies, cavum vergae and holoprosencephaly) and systemic abnormalities; (III) as a distinct type with multiple, disseminated smaller skin macules (cutis tricolor parvimaculata); and (IV) in association with other skin disturbances [e.g., cutis marmorata telangectasica congenita (phacomatosis achromico-melano-marmorata)] or in the context of other skin (e.g., ataxia-telangiectasia and phacomatosis pigmentovascularis, PPV) or complex malformation phenotypes (e.g., microcephaly and dwarfism).
Methods
(I) Review of the existing literature; and (II) information on our personal experience (clinical, laboratory and imaging data) on new cases with cutis tricolor seen and followed-up at our institutions during years 2010–2016.
Results
The existing literature revealed 19 previous studies (35 cases) with pure cutaneous or syndromic cutis tricolor phenomena. Our personal experience included 5 unpublished patients (3 boys; 2 girls; currently aged 2 to 14 years) seen and followed-up at our Institutions in Italy who had: (I) skin manifestations of the cutis tricolor type (N=5); (II) skeletal abnormalities including small skull (n=2), obtuse angle of mandible (n=3), mild to moderate scoliosis (n=3), vertebral defects (n=3), and long bones bowing (n=3); mild psychomotor delay (n=3); epilepsy (n=2); anomalies of the corpus callosum (n=3); and cavum vergae (n =2).
Conclusions
This study further confirms and expands the overall phenotype of cutis tricolor. By literature review and personal experience we conclude that the skin abnormalities of the cutis tricolor type are stable over time; the skeletal defects are mild to moderate and do not progress or cause relevant orthopaedic complications; the neurological/behavioural phenotype does not progress and the paroxysmal events (when present) tend to decrease over time; there is a typical facial phenotype in some patients (long, elongated face, thick and brushy eyebrows, hypertelorism, deep nasal bridge with large bulbous nose and anteverted nostrils), which characterizes a somewhat distinct syndromic phenotype; some patients may develop early onset cataracts. The allelic dydymotic hypothesis of post-zygotic mutations likely involving the same gene loci could well explain the overall skin, bone, lens and nervous system phenomena of migration of different streaks of clones in the different tissues.
Keywords: Cutis tricolor, Ruggieri-Happle syndrome (RHS), mosic, mosaicism, skin, neurological, imaging, magnetic resonance imaging (MRI)
Introduction
The term cutis tricolor describes the combination of congenital hyper- and hypopigmented lesions (in the form of macules, patches or streaks), in close proximity to each other, in a background of normal skin (1-20). This phenomenon has been reported so far either (18): (I) as an purely cutaneous trait (3,5,7,11,17,18); (II) as a part of a complex malformation phenotype (Ruggieri-Happle syndrome, RHS) (18,21-23) including distinct facial (a peculiar facial phenotype consisting in coarse, asymmetric and later in adolescence elongated face, thick and brushy eyebrows, hypertelorism, deep nasal bridge with large bulbous nose and anteverted nostrils, low-set ears, large phyltrum), eye (congenital cataract), skeletal (small skull, dystrophic vertebrae, and mildly bowed long bones), nervous system [corpus callosum anomalies, white matter abnormalities (i.e., delayed myelination), holoprosencephaly and cerebellar anomalies] and systemic abnormalities (2,3,6,8,11-14,16,18,19); (III) as a distinct type with multiple, disseminated smaller skin macules (cutis tricolor parvimaculata) (9,15,18) [different from the autosomal dominant congenital hypo- and hyperpigmented macules (Westerhof syndrome; MIM # 154000)]; and (IV) in association with other skin disturbances [e.g., cutis marmorata telangectasica congenita (phacomatosis achromico-melano-marmorata)] or in the context of other skin disorders (e.g., ataxia-telangiectasia and phacomatosis pigmentovascularis, PPV) or neurocutaneous phenotypes (4,10,13,20).
Cutis tricolor has been postulated to represent a twin-spotting phenomenon (18,24-26). Paradominant inheritance has been postulated as a possible mechanism (22) to explain familial occurrence as in the case of Baba et al. (5) who reported a pure skin phenotype of the “cutis tricolor” type in two sisters.
In 1997 Happle and co-workers coined the term “cutis tricolor” for the unusual combination of three degrees of skin pigmentation in close proximity to each other. In their 17-year-old boy they described congenital hyper- and hypopigmented macules confined to circumscribed body segments on a background of normal intermediate skin complexion in association with multiple birth defects (2). Ruggieri (3) subsequently expanded the cutaneous phenotype by including cases with abnormal paired pigmentation in the form of large patches (in the form of a yet unclassified archetypical pattern of cutaneous mosaicism) (1,23,26-28) or streaks (sash-like pattern of cutaneous mosaicism) (1,23) diffusely involving the body associated (case 2) (3) or not (case 1) (3) to systemic defects. The spectrum of extra-cutaneous manifestations further expanded in recent years including specific facial dysmorphic, skeletal and brain abnormalities (6,11-14,16,19).
Pure cutaneous traits (3,5,7,11,17) and variant forms have been also reported (9,15) and the skin anomaly has been recorded within the context of well-known and less well-known phenotypes (4,10,20).
Suggested eponyms for the complex malformation syndrome with cutis tricolor have been cutis tricolor syndrome (1,21) or RHS (1,18,21-23).
Methods
Patients
The Unit of Rare Diseases of the Nervous System in Childhood at the University of Catania, Italy, has been established in 2016 (formerly—since 1992—Unit of Paediatric Neurology) and caters to children and their families with different neurologic disorders in the paediatric age group (including neurocutaneous diseases), which are referred for diagnosis, management advice, and genetic counselling from the southern, central, and eastern Sicilian region (i.e., the towns and provinces of Catania, Siracusa, Ragusa, and Enna, with an approximate population of 1.9 million inhabitants out of the 5.8 million inhabitants of Sicily). Subjects are referred from all the city hospitals, the general paediatricians, and general practitioners, the regional lay group associations, and the consultants in dermatology, paediatrics, neurology, ophthalmology, radiology, and genetics in the regions. The hospitals, the lay groups, and the practitioners and consultants are contacted annually via letters for their needs of referrals.
Following our reports of 2010–2011 (13,14)—between 2010 and 2016—patients with cutis tricolour were diagnosed and followed-up at our institution in Catania. All these patients underwent a full clinical evaluation and laboratory investigation at the time of the initial diagnostic work-up and re-investigated at the follow-up visits.
The diagnosis of cutis tricolour was based on the following: the presence of hyper- and hypo-pigmented skin abnormalities in the form of macules, patches, or streaks in close proximity to each other on a background of normal skin.
The general and neurologic status was assessed during first referral and follow-up visits by means of standard clinical evaluations and neuropsychological testing.
Magnetic resonance imaging (MRI) studies were performed on a 1.5 T Philips Gyroscan and electroencephalography (standard EEG and video-EEG) was also performed in the presence of a clear or suspect neurologic anomaly at diagnosis and during follow-up.
In all patients, a standard and array-CGH analysis was performed.
Literature review
Studies published in the literature (Medline, Scopus, WOS and bibliographies) reporting on cutis tricolor and/or conditions associated to skin phenomena of the cutis tricolor type in association or not with systemic/neurological involvement were reviewed (all years). Literature search was performed by using the following key words: “cutis tricolor”, “twin spotting”, “skeletal”, “bone”, “neurological”, “nervous system”, and “MRI”: were considered studies reporting data in English, German, French and Spanish.
Results
Patients
Our personal experience included five unpublished patients (3 boys; 2 girls; currently aged 2 to 14 years) first seen and followed-up at our Institutions by means of the above methods.
The clinical, laboratory, and imaging findings of these five patients studied are summarised in Tables 1 and 2.
Table 1. Clinical findings in five cases with cutis tricolor syndrome.
| Main features | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Sex | Male | Female | Female | Male | Male |
| Age | 2 | 4 | 12 | 13 | 14 |
| Clinical manifestations | |||||
| Height | 25th | 50th | 95th | 75th | 50th |
| Weight | 50th | 50th | 97th | 97th | 50th |
| OFC | 25th | 50th | 98th | 25th | 50th |
| Cutis tricolor (pattern) | L, P | P | L, P | P | M |
| Face | + | − | − | + | − |
| Trunk | + | + | + | + | + |
| Upper limbs | − | + | + | − | − |
| Lower limbs | + | + | + | + | − |
| Coarse face | + | − | + | + | − |
| Facial asymmetry | + | − | + | + | − |
| Dolichocephalism | + | − | − | + | |
| Frontal bossing | + | − | + | + | − |
| Orbital bossing | + | − | + | + | − |
| Brushy eyebrows | + | + | + | + | + |
| Hypertelorism | + | − | + | + | − |
| Epicanthus | + | − | − | + | − |
| Deep nasal bridge | + | − | + | + | − |
| Large/bulbous nose | + | − | + | + | − |
| Large/anteverted nostrils | + | − | − | + | − |
| Low set ears | + | − | − | + | − |
| Posteriorly angulated ears | + | − | − | + | − |
| Wide philtrum | + | − | + | + | − |
| Prominent/thick lips | + | − | − | + | − |
| Prominent chin | + | + | + | + | − |
| Clinodactyly | + | + | + | − | − |
| Short neck | + | + | + | + | − |
| Pectus excavatum | + | + | + | + | + |
| Developmental milestoned | D | N | D | D | N |
| Hypotonia | + | − | + | + | − |
| Poor co-ordination | + | − | − | − | − |
| Language | D | N | D | D | N |
| Epilepsy | − | − | − | + | + |
| Onset | NA | NA | NA | 6y | 2y |
| Seizure type | NA | NA | NA | GTCS | GTCS |
| Outcome/age ceased | NA | NA | NA | 9y | 11y |
| Behavioural anomalies | + | − | − | + | + |
| Mental retardation | Mo | − | Mi | Mo | Mi |
| Other | − | − | Cataract | − | VSD |
OFC, occipitofrontal circumference; L, linear; P, patches; M, multiple maculae; +, present; −, absent; N, normal; D, delayed; NA, not applicable; y, years; GTCS, generalised tonic clonic seizures; Mi, mild; Mo, moderate; Se, severe; VSD, ventricular septal defect.
Table 2. Laboratory and imaging findings in five cases with cutis tricolor syndrome.
| Main features | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Sex | M | F | F | M | M |
| Age | 2 | 4 | 12 | 13 | 14 |
| Radiographic feature | |||||
| Small skull | + | − | − | + | − |
| Prognathism | + | − | + | − | − |
| Obtuse angle of mandible | + | − | + | + | − |
| Absent posterior arch atlas | + | − | − | − | − |
| Scoliosis | + | − | + | + | − |
| Cervical | + | − | − | − | − |
| Thoracic | + | − | + | + | − |
| Lumbar | − | − | + | + | − |
| Kyphosis | + | − | + | + | − |
| Lordosis | + | − | + | + | − |
| Vertebral scalloping | + | − | + | + | − |
| Increased distance pedicles | + | − | + | + | − |
| Altered pedicles | + | − | + | + | − |
| Metaphyseal osteosclerosis | − | − | − | + | − |
| Rib abnormalities | + | − | + | + | − |
| Bowing of long bones | + | − | + | + | − |
| Upper limbs | − | − | − | − | − |
| Lower limbs | + | − | + | + | − |
| Leg length discrepancy | + | − | + | + | − |
| Neuroimaging features | |||||
| Asymmetric ventricles | + | − | + | − | − |
| White matter anomalies | + | − | − | + | − |
| Corpus callosum anomalies | + | − | + | + | − |
| Laboratory findings | |||||
| EEG abnormalities | + | − | + | − | − |
| Cytogenetic analysis | |||||
| Array-CGH (anomalies) | − | − | − | − | − |
M, male; F, female; +, present; −, absent; NP, not performed; EEG, electroencephalography; CGH, comparative genomic hybridization.
Literature review
The existing literature revealed 19 previous studies (2-20) for a total number of 35 cases with purely cutaneous traits (3,5,7,11,17,18) and with a combination of paired skin phenomena of the cutis tricolor type and associated cutaneous/systemic/neurological manifestations (2-4,6,8-14,15-23).
Discussion
By literature review (2-20) and personal experience (Tables 1,2) we have recorded 40 cases so far with the following demographic, clinical, laboratory and imaging features and natural history.
Incidence and prevalence
In the recorded cases [(2-20); and Tables 1,2] there were no significant sex and race differences (18), even though the figures are too low to draw demographic conclusions.
Clinical manifestations
The hallmarks of disease are the paired skin anomalies. Here below we summarises the main cutaneous and extra-cutaneous manifestations including imaging abnormalities.
Skin manifestations
These consist in the combination of hypopigmented and hyperpigmented anomalies in close proximity one to each other in a background of normal complexion (Figure 1). There is a wide variety of juxtaposed cutaneous lesions including macules, patches, and streaks, arranged in different archetypical (e.g., patchy, linear, sash-like, diffuse) (Figure 1A) (1,23,26) or less-well defined (unclassifiable) (Figure 1B) patterns (1,23,26) over confined or large body areas. The hallmark of the skin anomaly is: (I) the close spatial proximity of the two different birthmarks (Figure 1); and (II) the coexistence of three different complexions, which reflects the twin-spotting phenomenon (Figure 1A,B) (see below). Lesions usually manifest at birth or in the first months of life. The degree and (more rarely) the extent of the pigmentary anomaly can increase in the first years of life: then they stabilise and could also revert to normal or near-normal colour in some areas.
Figure 1.
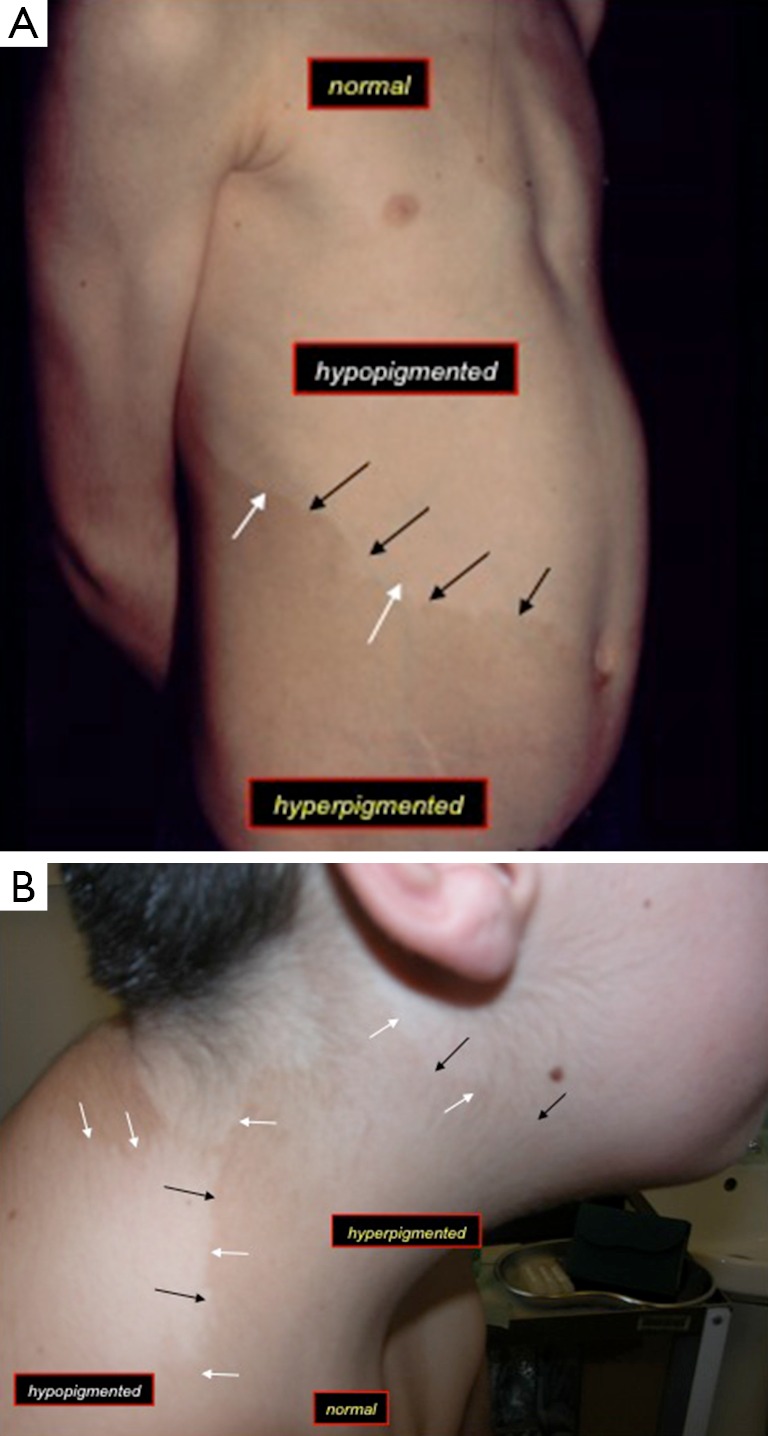
Close view-up of the skin in two children with cutis tricolor: as a pure trait (A) in an archetypical sash-like (spiral) pattern of mosaic distribution [case 2 in (3)]; and (B) as a complex malformation phenotype in a less-well defined/unclassifiable mosaic pattern of distribution [case 1 in (3)]. Note the hyperpigmented (the “hyperpigmentation” is pointed by the black arrows) and hypopigmented (the “hypopigmentation” is pointed by the white arrows) areas in close proximity one to each other in a background of normal complexion (labelled as “normal”).
Facial phenotype
In the most severely affected cases (1,3,6,16) the face is coarse and mildly asymmetric with dolichocephaly and prominent metopic suture; there is orbital bossing, thick and brushy eyebrows, hypertelorism, mild epicanthus, deep nasal bridge with large, bulbous nose and anteverted nostrils; low set and anteriorly rotated ears; large phyltrum and thick lips; the palate is narrow with small and spaced teeth; the chin is prominent. In our experience [(3,6,16), Table 1], at birth—in the cases with facial dysmorphisms—the face could present only with a minor (minimal) degree of dysmorphisms: then, over the years, mostly across the pre-pubertal and pubertal period, the face tends to elongate and to thin, and the nasal features become more prominent (3,6,16).
Eye findings
In one study (12), three children with syndromic cutis tricolor developed moderately to large sized, dense (with thick fibrosis under the anterior capsule), cortical-nuclear and subcapsular, monocular to binocular cataracts. Their ages at diagnosis of cataracts was comprised between 11 and 14 to 15 years. They did not develop other manifestations in the anterior or posterior segments. All were successfully operated with normal postoperative eye examinations. At gross pathology and histopathological examination there were eosiphilic large fibres arranged in two to multiple streaks with vacuolisation of superficial cortical fibres and extracellular clefts (12). The cataracts were classified as syndermatotic anterior polar cataracts, which usually are associated to dermatologic conditions (12). In the present study, two additional patients (cases 3 and 4; Table 1) had cataracts with similar features to those previously recorded (12).
Skeletal abnormalities
The radiographic features in the syndromic patients so far analysed (2,3,6,8,11-14,16,18,19), include [reviewed in (14)]: a small skull (not microcephalic) with prognathism; the pituitary fossa can be “J” shaped and abnormally shaped (Figure 2A) with implanted teeth sprouts and numerous dental cysts; absent posterior arch of the atlas; obtuse angle of the mandible; scoliosis of different degrees and location (with additional lordosis and kyphosis: the abnormal curvature of the spine can be dystrophic in appearance, right-sided lower thoracic (T10–12) and upper lumbar (L1–3) with asymmetric peduncles and sloping ribs (n=6) or left- and right-sided upper thoracic (T4–T7) and lower lumbar (L1–L4) (Figure 2B); metaphyseal osteosclerosis of the femurs; mild to moderate bowing of long bones (Figure 2C). There is also clinodactyly of the fifth fingers.
Figure 2.

Radiographic abnormalities in three children with syndromic cutis tricolor: lateral view of the skull (A) showing a small skull (not microcephaly) with prognathism and “J” shaped pituitary fossa; antero-posterior view of the spine (B) showing an abnormal (dystrophic) curvature of the spine; antero-posterior view of the lower limbs (C) revealing mild bowing of the tibiae.
Notably, the skeletal dysplasia (29) observed in syndromic patients with skin manifestations of the cutis tricolor type was reminiscent of neurofibromatosis type 1 (29), even though to a lesser degree: the scoliotic changes were milder than that encountered in NF1, the mandibular thinning and vertebral scalloping was not so pronounced, and the tibiae were bowed to a lesser extent than that usually seen in NF1 (29).
Bone age is usually mildly to moderately increased.
Neurological phenotype
In syndromic individuals [(2,3,6,8,11-14,16,18,19); and also Table 1], it consisted in [reviewed in (13)]:
Psychomotor delay with mild to moderate or (less frequently) severe cognitive delay [mean full-scale IQ in the syndromic cutis tricolor population (i.e., RHS) was 82.3 (range, 50–110): i.e., lower than the general population (P<0.05)] (3,6,13,16);
Specific expressive language disabilities (non-progressive delay in production of speech sounds, communication milestones, and expressive language disabilities: improvement was attained after placement in special and early pre-school intervention programs) (3,6,13,16);
Epilepsy (i.e., generalised tonic-clonic or partial types first occurring between 18 months and 11 years of age);
EEG abnormalities consisted in non-specific spike and wave activity in the centro-temporal and fronto-centro-temporal regions; occipital P and PO waves; and a Rasmussen-like pattern in one case [(13): who showed at age 2 years an initial diffuse paroxysmal activity with diffuse slowing and later developed hemispheric slowing activity with a typical Rasmussen type recording];
Altered behaviour [e.g., aggressive, violent and opposite behaviour, ADHD, obsessive-compulsive and autistic-spectrum like disorders: there was a recognisable behavioural phenotype in one study (13), consisting in an early (<5 years of age) phenotype of normal social interaction and eye-to-eye contact with temper tantrum episodes and frequent intense biting of objects, which evolved into obsessive-compulsive disorder or ADHD coupled with aggressive, opposite and near violent behaviour in toddler ages and aggressive behaviour with autistic spectrum disorder-like abnormalities after puberty and during adolescence into young adulthood];
Mood disorders.
Brain imaging abnormalities
Brain MRI studies revealed so far (2,3,6,8,11-14,16,18,19), multifocal, diffuse, confluent, bright signal abnormalities in the posterior periventricular (and subcortical) white matter on T2-weighted images (Figure 3). Other isolated imaging abnormalities were dilatation of the cerebral lateral ventricles and dysmorphisms/dysplasia of the corpus callosum (Figure 4A) with cavum vergae (Figure 4B,C), holoprosencephaly and brain teratoma (19). Studies of the spinal cord yielded normal findings in all the 14 subjects in one study (13).
Figure 3.
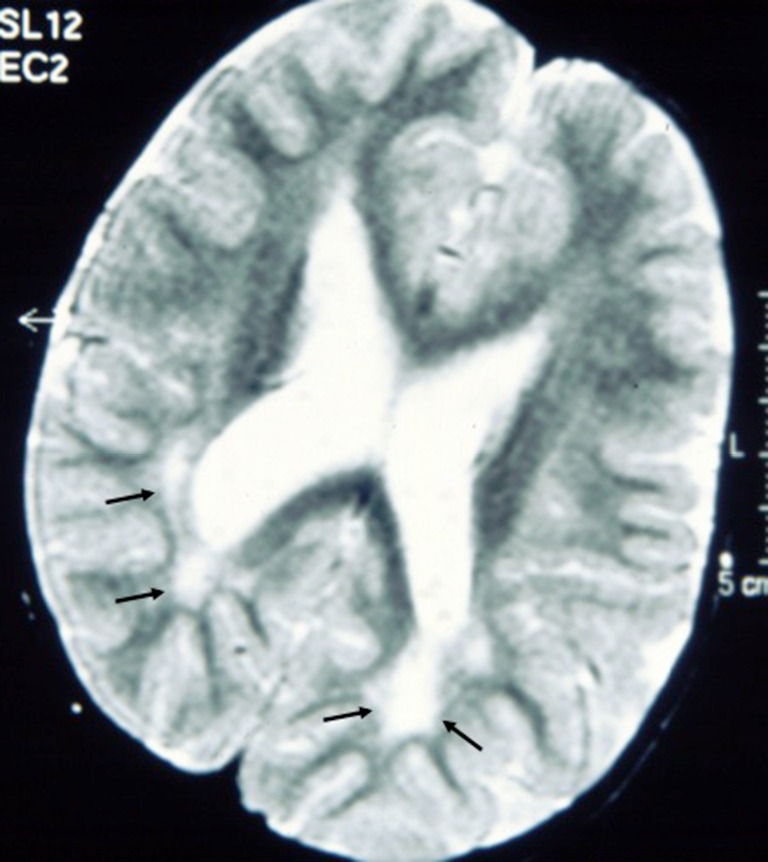
Axial T2-weighted magnetic resonance images of the brain in a child with syndromic cutis tricolor showing high signal lesions in the posterior (peritrigonal) white matter (black arrows): thes hyperintensities were recorded at age 6 years and regarded as delayed myelination.
Figure 4.

Sagittal T2-weighted (A) magnetic resonance imges of the brain in a child with syndromic cutis tricolor reveals a dysmorphic corpus callosum; axial T1- (B) and T2-weighted (C) magnetic resonance images of the brain in a child with syndromic cutis tricolor showing a cavum vergae.
Pathology
Punch biopsies collected from the involved areas showed histologically (2) an increase in melanin content of the basal layer, but no dermal melanin or melanophages in the hyperpigmented areas and a decrease in the melanin content and in the number of melanocytes in the hypopigmented lesions. A normal epidermal and dermal architecture was present in the normally intermediate pigmented areas. Electron microscopy showed (11): (I) in the hypopigmented area basal keratinocytes containing isolate melanosomes, some of them displaying an incomplete maturation; and (II) in the hyperpigmented area basal keratinocytes filled of mature melanosomes.
Molecular genetics and pathogenesis
A tentative genetic explanation of this postulated new phenotype could be a post zygotic somatic mutation (Figure 5) (1,22,23): loss of heterozygosity for the underlying mutation at an early developmental stage would give rise to a complex malformation mosaic pattern (e.g., generalised skin manifestations of the cutis tricolor type in association to extra-cutaneous anomalies) as in the original case of Happle et al. (1) and in the subsequent studies reporting on syndromic phenotypes (2,3,6,8,11-14,16,18,19); post zygotic recombination occurring later during embryogenesis would give rise to pure cutaneous traits (i.e., manifestations confined to the skin only) (3,5,7,11,17,18) or to the distinct type of cutis tricolor parvimaculata (9,15,18). Non-allelic didymosis (22,23) would explain the coexistence of segmental lesions of pigmentary (e.g., paired hypo- and hyper-pigmented macules) and vascular (e.g., cutis marmorata telangiectatica) disturbance (4,10,13,20). Paradominant inheritance has been postulated (1,2,23) as a possible mechanism to explain familial occurrence as in the case of Baba et al. (5) who reported an isolated skin phenotype of the “cutis tricolor” type in two sistrs.
Figure 5.
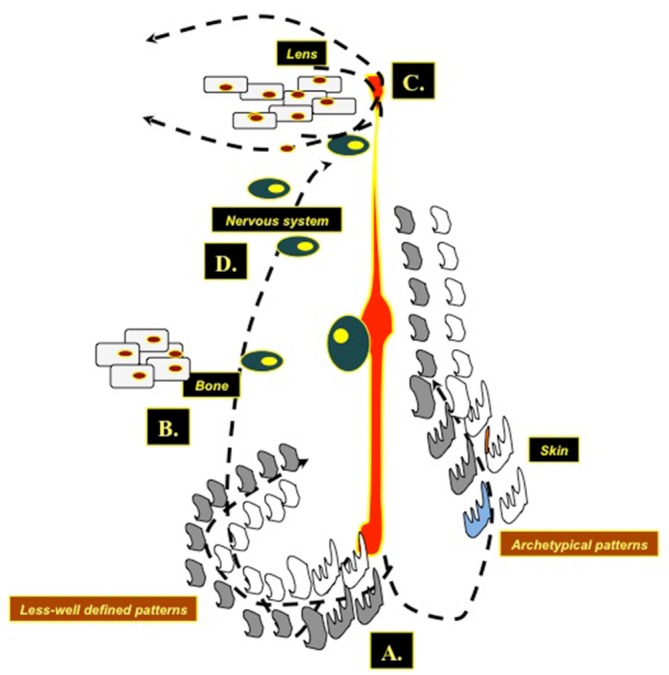
Drawing illustrating the unifying (allelic) dydymotic theory as a pathogenic explanation for the cutis tricolor phenotypes: (A) a post zygotic somatic mutation leading to heterozygosity for the underlying yet undiscovered gene responsible for the skin anomaly at an early developmental stage would give rise to a complex malformation mosaic pattern (e.g., syndromic cutis tricolor) whilst, at a later embryonic stage would give rise to pure cutaneous traits (i.e., manifestations confined to the skin only) or to the distinct type of cutis tricolor parvimaculata. These heterozygous cutaneous cell populations (grey and white cells in the figure) can generate in the skin (A) either archetypical or less-well defined mosaic patterns. Similar phenomena occur in the bone (skeletal abnormalities) (B), in the lenses (C) [note the different populations of cortical-subcortical lens cells migrating in parallel streaks [as found at histopathology in the cataracts of children with cutis tricolor, see (12)], and in the nervous system (D), generating layers disarraying in the subcortical white matter leading to delayed myelination or to clones of disordered neuronal cells.
Conclusions
This study further confirms and expands the overall phenotype of cutis tricolor. The skin abnormalities of the cutis tricolor type are usually stable over time and do not benefit pf sophisticated laser therapeutic techniques; the skeletal defects are mild to moderate and do not progress or cause relevant orthopaedic complications; the neurological/behavioural phenotype and the paroxysmal events (when present) tend to stabilise over time [seizures can be treated similarly to those in patients without cutis tricolor phenomena: some patients with intractable seizures benefited of combination of antiepileptic drugs]; problems at school may need special help and stimulants drugs associated with antiepileptic treatment. notably, a typical facial phenotype in some patients could help the diagnostic work-up as it characterizes a somewhat distinct syndromic phenotype; some patients may develop childhood cataracts.
Paired skin phenomena of the cutis tricolor type are likely more frequent than previously thought and should be investigated (when dictated by clinical findings) by means of: (I) brain and spinal cord MRI studies; (II) skeletal X-ray studies; (III) systemic (i.e., heart, abdominal and pelvic) ultrasound studies; (IV) neurophysiologic studies (EEG); (V) psychomotor testing; (VI) and routine laboratory investigation.
Acknowledgements
None.
Ethical Statement: This study was approved by the local ethics committee (Comitato Etico 1, University of Catania). Patients’ consent was obtained (and/or waived due to the non-interventional retrospective analysis nature of this study).
Footnotes
Conflicts of Interest: The authors have no conflict of interest to declare.
References
- 1.Happle R. Mosaicism in Human Skin. Understanding Nevi, Nevoid Skin Disorders, and Cutaneous Neoplasia. Berlin/Heidlber: Springer-Verlag, 2014. [Google Scholar]
- 2.Happle R, Barbi G, Eckert D, Kennerknecht I. "Cutis tricolor": congenital hyper- and hypopigmented macules associated with a sporadic multisystem birth defect: an unusual example of twin spotting? J Med Genet 1997;34:676-8. 10.1136/jmg.34.8.676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruggieri M. Cutis tricolor: congenital hyper- and hypopigmented lesions in a background of normal skin with and without associated systemic features: further expansion of the phenotype. Eur J Pediatr 2000;159:745-9. 10.1007/PL00008339 [DOI] [PubMed] [Google Scholar]
- 4.Khumalo NP, Joss DV, Huson SM, Burge S. Pigmentary anomalies in ataxia--telangiectasia: a clue to diagnosis and an example of twin spotting. Br J Dermatol 2001;144:369-71. 10.1046/j.1365-2133.2001.04029.x [DOI] [PubMed] [Google Scholar]
- 5.Baba M, Seçkin D, Akçali C, Happle R. Familial cutis tricolor: a possible example of paradominant inheritance. Eur J Dermatol 2003;13:343-5. [PubMed] [Google Scholar]
- 6.Ruggieri M, Iannetti P, Pavone L. Delineation of a newly recognized neurocutaneous malformation syndrome with "cutis tricolor". Am J Med Genet A 2003;120A:110-6. 10.1002/ajmg.a.20011 [DOI] [PubMed] [Google Scholar]
- 7.Seraslan G, Atik E. Cutis tricolor: two case reports. Case Rep Clin Pract Rev 2005; 6:317-9. [Google Scholar]
- 8.Niessen RC, Jonkman MF, Muis N, Hordijk R, van Essen AJ. Pigmentary mosaicism following the lines of Blaschko in a girl with a double aneuploidy mosaicism: (47,XX,+7/45,X). Am J Med Genet A 2005;137A:313-22. 10.1002/ajmg.a.30876 [DOI] [PubMed] [Google Scholar]
- 9.Larralde M, Happle R. Cutis tricolor parvimaculata: a distinct neurocutaneous syndrome? Dermatology 2005;211:149-51. 10.1159/000086446 [DOI] [PubMed] [Google Scholar]
- 10.Boente Mdel C, Obeid R, Asial RA, Bibas-Bonet H, Coronel AM, Happle R. Cutis tricolor coexistent with cutis marmorata telangiectatica congenita: "phacomatosis achromico-melano-marmorata". Eur J Dermatol 2008;18:394-6. [DOI] [PubMed] [Google Scholar]
- 11.Ruggieri M, Roggini M, Kennerknecht I, Schepis C, Iannetti P. Cutis tricolor (Ruggieri-Happle syndrome). In: Ruggieri M, Pascual-Castroviejo I, Di Rocco C. editors. Neurocutaneous Disorders. Phakomatoses & Hamartoneoplastic Syndromes. Wien/New York: Springer-Verlag, 2008:461-71. [Google Scholar]
- 12.Ruggieri M, Iannetti F, Polizzi A, Puzzo L, Di Pietro M, Caltabiano R, Iannetti L, Magro G, Iannetti P. Cataracts in three children with a newly recognised neurocutaneous malformation phenotype with "cutis tricolor". Br J Ophthalmol 2009;93:127-8. 10.1136/bjo.2008.140749 [DOI] [PubMed] [Google Scholar]
- 13.Lionetti E, Pavone P, Kennerknecht I, Failla G, Schepis C, De Pasquale R, Pavone L, Ruggieri M. Neurological manifestations in individuals with pure cutaneous or syndromic (Ruggieri-Happle syndrome) phenotypes with "cutis tricolor": a study of 14 cases. Neuropediatrics 2010;41:60-5. 10.1055/s-0030-1261919 [DOI] [PubMed] [Google Scholar]
- 14.Ruggieri M, Roggini M, Kennerknecht I, Polizzi A, Distefano A, Pavone V. Spectrum of skeletal abnormalities in a complex malformation syndrome with "cutis tricolor" (Ruggieri-Happle syndrome). Acta Paediatr 2011;100:121-7. 10.1111/j.1651-2227.2010.01970.x [DOI] [PubMed] [Google Scholar]
- 15.Boente Mdel C, Bazan C, Montanari D. Cutis tricolor parvimaculata in two patients with ring chromosome 15 syndrome. Pediatr Dermatol 2011;28:670-3. 10.1111/j.1525-1470.2011.01470.x [DOI] [PubMed] [Google Scholar]
- 16.Nicita F, Spalice A, Roggini M, Papetti L, Ursitti F, Tarani L, Ruggieri M. Complex malformation (Ruggieri-Happle) phenotype with "cutis tricolor" in a 10-year-old girl. Brain Dev 2012;34:869-72. 10.1016/j.braindev.2012.01.015 [DOI] [PubMed] [Google Scholar]
- 17.Oiso N, Matsuda H, Kawada A. Cutis tricolor of pure cutaneous trait as leukoderma and nevus spilus. J Dermatol 2013;40:490-1. 10.1111/1346-8138.12155 [DOI] [PubMed] [Google Scholar]
- 18.Torchia D, Schachner LA, Izakovic J. Cutis tricolor. Cutis 2013;91:11-6. [PubMed] [Google Scholar]
- 19.Tekin B, Yucelten AD, Bayri Y. A novel association of an uncommon pigmentation pattern: coexistence of cutis tricolor with intracranial teratoma and holoprosencephaly. Dermatol Online J 2014;20. [PubMed] [Google Scholar]
- 20.Pavone P, Praticò AD, Gentile G, Falsaperla R, Iemmolo R, Guarnaccia M, Cavallaro S, Ruggieri M. A neurocutaneous phenotype with paired hypo- and hyperpigmented macules, microcephaly and stunted growth as prominent features. Eur J Med Genet 2016;59:283-9. 10.1016/j.ejmg.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 21.POSSUM - Pictures of Standard Syndromes and Undiagnosed Malformations. Melbourne: Murdoch Children Research Institute, 2016 [POSSUM web]. Available online: https://www.possum.net.au
- 22.Happle R. Didymotic Skin Disorders. In: Happle R. editor. Mosaicism in Human Skin. Understanding Nevi, Nevoid Skin Disorders, and Cutaneous Neoplasia. Berlin/Heidlber: Springer-Verlag, 2014:109-14. [Google Scholar]
- 23.Ruggieri M, Praticò AD. Mosaic Neurocutaneous Disorders and Their Causes. Semin Pediatr Neurol 2015;22:207-33. 10.1016/j.spen.2015.11.001 [DOI] [PubMed] [Google Scholar]
- 24.Jinnette VA, Courter AM, Lowry JW, Buckley NH, White KT, Hoegerman SF. Mechanisms of twin spotting. Lancet 1990;336:61. 10.1016/0140-6736(90)91581-T [DOI] [PubMed] [Google Scholar]
- 25.Koopman RJ. Concept of twin spotting. Am J Med Genet 1999;85:355-8. [DOI] [PubMed] [Google Scholar]
- 26.Happle R. The categories of cutaneous mosaicism: A proposed classification. Am J Med Genet A 2016;170A:452-9. 10.1002/ajmg.a.37439 [DOI] [PubMed] [Google Scholar]
- 27.Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch Dermatol 1993;129:1460-70. 10.1001/archderm.1993.01680320094012 [DOI] [PubMed] [Google Scholar]
- 28.Happle R. Pigmentary patterns associated with human mosaicism: A proposed classification. Eur J Dermatol 1993;3:170-4. [Google Scholar]
- 29.Taybi H, Lachman RS. Radiology of Syndromes, Metabolic Disorders and Skeletal Dysplasia. 4th ed. St. Louis: Mosby, 1996. [Google Scholar]