

Abstract
Physical inactivity and disuse result in skeletal muscle metabolic disruption, including insulin resistance and mitochondrial dysfunction. The role of the Toll-like receptor 4 (TLR4) signaling pathway in contributing to metabolic decline with muscle disuse is unknown. Therefore, our goal was to determine whether TLR4 is an underlying mechanism of insulin resistance, mitochondrial dysfunction, and skeletal muscle ceramide accumulation following muscle disuse in mice. To address this hypothesis, we subjected (n = 6–8/group) male WT and TLR4−/− mice to 2 wk of hindlimb unloading (HU), while a second group of mice served as ambulatory wild-type controls (WT CON, TLR4−/− CON). Mice were assessed for insulin resistance [homeostatic model assessment-insulin resistance (HOMA-IR), glucose tolerance], and hindlimb muscles (soleus and gastrocnemius) were used to assess muscle sphingolipid abundance, mitochondrial respiration [respiratory control ratio (RCR)], and NF-κB signaling. The primary finding was that HU resulted in insulin resistance, increased total ceramides, specifically Cer18:0 and Cer20:0, and decreased skeletal muscle mitochondrial respiration. Importantly, TLR4−/− HU mice were protected from insulin resistance and altered NF-κB signaling and were partly resistant to muscle atrophy, ceramide accumulation, and decreased RCR. Skeletal muscle ceramides and RCR were correlated with insulin resistance. We conclude that TLR4 is an upstream regulator of insulin sensitivity, while partly upregulating muscle ceramides and worsening mitochondrial respiration during 2 wk of HU.
Keywords: lipids, insulin resistance, physical inactivity, inflammation, sphingolipids
it is well understood that physical inactivity and muscle disuse precipitate metabolic decline and muscle atrophy in rodents and humans (9, 17, 30, 31, 34). Because skeletal muscle is a major site of glucose regulation, it is not surprising that inactivity in the form of muscle disuse can significantly disrupt skeletal muscle glucose handling and induce insulin resistance and mitochondrial dysfunction (6, 10, 31, 34, 47). Further understanding of the mechanisms associated with skeletal muscle metabolic disruption are needed to develop therapies to effectively treat muscle disuse-induced metabolic disorders.
Toll-like receptor 4 (TLR4) is a critical cell surface receptor involved in innate immunity. Skeletal muscle TLR4 abundance has been associated with insulin resistance in diabetic and obese humans (37) and is elevated in aged muscle (22) and in older adults during bed rest (18). In response to specific agonists (e.g., LPS), TLR4 activates NF-κB, thereby, upregulating classical proinflammatory cytokines such as IL-6. In addition to inflammation, skeletal muscle TLR4 signaling can transcriptionally upregulate rate-limiting enzymes involved in de novo ceramide synthesis (25). TLR4 and ceramides are well appreciated to be regulators of insulin resistance (12), particularly in human and mouse models of diabetes, obesity, and lipid overload (2, 13–15, 24, 26, 27, 35, 44–46, 48). However, there is much less mechanistic understanding of TLR4 and muscle ceramides in response to disuse (3, 5, 31). To date, we have shown that TLR4/NF-κB signaling is elevated in mouse and human skeletal muscle after a period of disuse (18, 31). Furthermore, 1 wk of hindlimb unloading was shown to increase specific muscle ceramide species (38, 39). However, the regulatory role of TLR4 on muscle ceramides during hindlimb unloading has not been investigated.
Evidence suggests that TLR4 signaling is also closely tied to altered mitochondrial function. Recent work from the Hulver laboratory showed that TLR4 activation (via LPS) decreased mitochondrial respiration in isolated mitochondria from rodent skeletal muscle (21). In a follow-up study, McMillian et al. (32) reported that overactivation of skeletal muscle TLR4 in mice after 16 wk of high-fat feeding resulted in greater weight gain, further impairment of glucose tolerance, and inability to increase fatty acid oxidation compared with WT controls. Therefore, it is logical to surmise that TLR4 signaling may not only regulate the accumulation of ceramide in muscle but also impact mitochondrial respiration during hindlimb unloading (47).
We previously reported that myeloid differentiation primary response gene 88 (MyD88) mediated skeletal muscle insulin resistance, inflammation, and upregulated ceramide biosynthetic enzymes during 2 wk of hindlimb unloading (31). Since MyD88 is a critical adaptor protein for several TLRs, our next step was to move upstream to a specific Toll receptor, namely, TLR4. Our purpose was to narrow our focus to the specific pathway responsible for disuse-induced insulin resistance, while concurrently addressing the regulatory role of TLR4 on intramyocellular ceramides and mitochondrial respiration. We hypothesized that mice deficient of TLR4 would be protected from insulin resistance, skeletal muscle ceramide accumulation, and a decline in mitochondrial respiration during hindlimb unloading.
MATERIALS AND METHODS
Animals.
Ten-week-old male wild-type (WT) and TLR4−/− mice with a C57BL/6J genetic background (Jackson Laboratories) were housed in a conventional vivarium and maintained on a 12:12 h light-dark cycle and temperature-controlled environment (22–23°C). Animals were acclimated to the current environment approximately 1 wk before beginning the experiments. The Institutional Animal Care and Use Committee of the University of Utah approved these experiments.
Experimental design.
Animals were assigned to one of four experimental groups (n = 6–8/group): 1) WT control (WT CON; n = 7), 2) WT hindlimb unloading (WT HU; n = 6), 3) TRL4−/− control (TLR4−/− CON; n = 7), and 4) TLR4−/− hindlimb unloading (TLR4−/− HU; n = 8). Control animals (WT CON, TLR4−/− CON) were able to freely ambulate in their cage (2 or 3 animals/cage) and have ad libitum access to food (standard chow) and water during the 14-day experimental period. Animals in the HU group (WT HU, TLR4−/− HU) underwent hindlimb suspension (two animals/cage), as we have done previously (31). Briefly, a sterile surgical steel suture was inserted into the animals' tail while the animal was anesthetized. The steel suture was then shaped into a ring for later suspension onto a steel bar with a swivel. After a 5-day recovery period, the animals were suspended off the ground by their hindlimbs (30°) for 14-days. Animals had access to a 360° perimeter within the cage and able to reach food and water. All mice were monitored at least once daily for behavior and to make sure that they could reach their food and water. Body weight was recorded every other day. At day 13, CON and HU mice were fasted for 6 h and underwent a glucose tolerance test. Blood glucose levels (Bayer Contour) via a tail vein were determined immediately before (0) and 5, 15, 30, 60, and 120 min following the glucose injection (1 g/kg body wt). On day 14, mice were fasted for 6 h and anesthetized with isoflurane, and hindlimb muscles (soleus and gastrocnemius) were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C for later analysis. At the same time, a sample of blood was collected for measurement of insulin (Insulin ELISA kit; Crystal Chem, Chicago, IL). Animals were euthanized with CO2 immediately after tissue and blood collection.
Immunoblotting.
Soleus muscles were homogenized 1:12 (wt/vol) using a glass tube and mechanically driven pestle grinder in an ice-cold buffer containing 50 mM Tris (pH 7.5), 250 mM mannitol, 40 mM NaF, 5 mM pyrophosphate, 1 mM EDTA, 1 mM EGTA, and 1% Triton X-100 with a protease inhibitor cocktail. Protein concentration was determined using the Bradford technique. Thirty micrograms of homogenate were separated via polyacrylamide gel electrophoresis, transferred onto a polyvinylidene difluoride membrane, and incubated with primary and secondary antibodies directed against the proteins of interest. Membranes were imaged on a ChemiDoc XRS (Bio-Rad, Hercules, CA) and were quantified with Image lab software (Bio-Rad). The specific antibodies used to detect target proteins were phospho-Akt (Ser-473), total Akt, phospho-nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα, Ser-32/36), total IkBα, IL-6, and microtubule-associated protein 1 light chain 3 (LC3). All antibodies were used at a dilution of 1:500–1:1,000 and were purchased from Cell Signaling Technologies (Danvers, MA). Secondary antibodies (1:6,000) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Data were expressed as phosphorylated relative to total, LC3 was expressed as LC3II/LC3I, and IL-6 protein was normalized to GAPDH.
Mitochondrial respiration.
Freshly dissected soleus samples were placed in ice-cold buffer X containing (in mM) 60 K-MES, 35 KCl, 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 0.5 DTT, 20 taurine, 5.7 ATP, 15 phosphocreatine, and 6.56 MgCl2 (pH 7.1) (4). Muscle fibers were gently pulled apart along the longitudinal axis to create small bundles of <7 fibers. Fiber bundles were then incubated in buffer X with 125 μg/ml saponin while being mixed via mild inversion on a vertical rotator for 30 min at 4°C (4). After incubation in saponin, fiber bundles were washed 3 times for 15 min at 4°C in buffer Z containing (in mM) 110 K-MES, 35 KCl, 1 EGTA, 5 K2HPO4, 3 MgCl2, 0.5 mg/ml BSA, 0.005 glutamate, and 0.002 malate (pH 7.4) before analysis.
Mitochondrial respiration was assessed with a Clark-type high-resolution oxygraph respirometer (Oxytherm, Hansatech Instruments, Kings Lynn, UK) maintained at 37°C (33). Following calibration of the Clark oxygen electrode, permeabilized muscle fibers were incubated in the respirometer with 2 ml of buffer Z containing 20 mM creatine to saturate creatine kinases, while being continuously stirred at 37°C. Oxygen flux through complex I was measured using 5 mM glutamate and 2 mM malate. The ADP-stimulated respiration (state 3) was initiated by adding 0.25 mM ADP to the respiration chamber. Basal respiration (state 4) was determined in the presence of 10 μg/ml oligomycin to inhibit ATP synthesis. The respiratory control ratio (RCR) was calculated by dividing oxygen consumption during state 3 respiration by the oxygen consumption during state 4 respiration.
Sphingolipid analysis.
Sphingolipid analysis was performed as described previously (19). Briefly, ∼20 mg of gastrocnemius tissue was extracted using a modified Folch procedure with the addition of C17 ceramide (d18:1/17:0) as an internal standard (Avanti Polar Lipids, Alabaster AL). Following extraction, the lipid pellets were solubilized in 200 μl of methanol, transferred to LC-MS vials, and analyzed by UPLC-ESI-MS. An Agilent 1290 UPLC system fit with an Acquity UPLC CSH C18 1.7 μm 2.1 × 50 mm column (Waters, Beverly MA) was employed to fractionate the lipid mixture An Agilent 6490 triple quadrupole mass spectrometer operated in the positive mode with the transitions listed in the Table 1 was used for detection. Mobile phase A consisted of acetonitrile in H2O (60% vol/vol) and mobile phase B consists of isopropanol in H2O (90% vol/vol), both containing 10 mM ammonium formate and 0.1% formic acid. The gradient initiated at 60% B, held for 1 min, and then ramped to 99% B over 9 min and maintained for 10 min. Flow rate was 0.2 ml/min, and the injection volume was 3 μl. The source gas temperature was set to 150°C, with a drying gas flow of 14 l/min and a nebulizer pressure of 20 psi. Sheath gas temperature was 350°C, sheath gas flow was 11 l/min, capillary voltage of 3,000 V, high pressure radio frequency (RF) was 150 V, and low pressure RF was 60 V. All collision energies were 29, and cell accelerator voltages were 4. Data analysis was performed using Mass Hunter Quant B.07.00 (Agilent Technologies, Santa Clara CA).
Table 1.
Animal muscle and body weight characteristics from control and hindlimb unloaded wild-type and TLR4−/− mice
| WT CON | WT HU | TLR4−/− CON | TLR4−/− HU | |
|---|---|---|---|---|
| Body weight, g | 24.0 ± 1.2 | 22.9 ± 1.4 | 23.7 ± 1.5 | 21.5 ± 1.1* |
| Soleus, mg | 7.3 ± 1.5 | 3.0 ± 0.7*# | 7.5 ± 0.7 | 4.8 ± 0.7* |
| Gastrocnemius, mg | 134.0 ± 11.2 | 91.9 ± 16.7*# | 128.8 ± 12.3 | 112.9 ± 18.5 |
Data are expressed as means ± SD (6–8/group).
Significantly different from respective control (CON), P < 0.05. #Significantly different from TLR4−/− hindlimb unloaded (HU), P < 0.05.
Statistical analysis.
Data are expressed as means ± SD. Statistical analyses were conducted using Graph Pad Prism (version 7; San Diego, CA). Statistical analyses for animal experiments were conducted using a two-way ANOVA [Treatment (CON, HU) × Group (WT, TLR4)]. When significant interactions occurred, Sidak post hoc tests were performed to determine specific differences. Area under the curve (AUC) was calculated using the trapezoid rule. Pearson correlations were used to determine relationships between ceramides, homeostatic model assessment-insulin resistance (HOMA-IR) glucose AUC, and mitochondrial function. Statistical significance was set at P ≤ 0.05.
RESULTS
TLR4−/− mice maintained insulin sensitivity and partly retained muscle size after 2 wk of hindlimb unloading.
There was a treatment effect for body weight with HU (P = 0.002) (Table 1 and Fig. 1), but there was no group × treatment interaction (P = 0.2401). Body weight decreased by 9% only in TLR4−/− mice after HU (P = 0.0104). There was a group × treatment interaction for soleus and gastrocnemius weight (P = 0.0329 and P = 0.0362, respectively). Soleus weight decreased after HU in both the WT and TLR4−/− groups (P < 0.0001). However, the reduction in soleus weight after HU in the TLR4−/− group was less compared with WT HU (P = 0.003). Gastrocnemius weight also decreased in WT mice (P < 0.0001) and tended to decrease in TLR4−/− mice (P = 0.06). However, the reduction in gastrocnemius weight was attenuated in the TLR4−/− group (P = 0.03). There was a group × treatment interaction for HOMA-IR (Fig. 1A) (P = 0.0140), such that HOMA-IR significantly increased in the WT group after HU (P < 0.0001) and that this increase was significantly greater than the TLR4−/− group after HU (P = 0.0012). Finally, there was a group × treatment interaction for glucose AUC (Fig. 1B) (P = 0.0096). Glucose AUC was significantly higher only in the WT HU group compared with WT CON (P < 0.0001) and was significantly greater than the TLR4−/− group after HU (P = 0.0004).
Fig. 1.
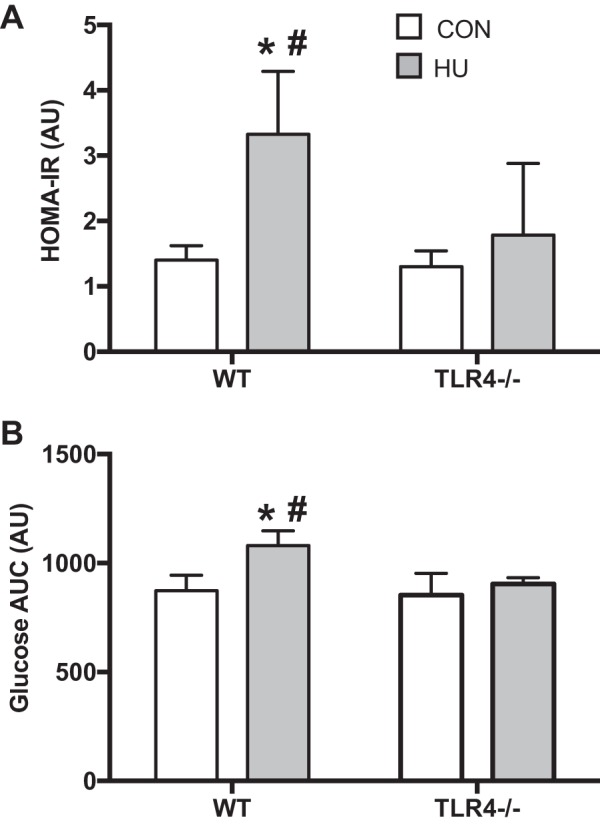
Insulin sensitivity and glucose tolerance. Data (means ± SD; n = 6–8/group) are from ambulatory control (CON; white bars) and after 14 days of hindlimb unloading (HU; gray bars) in WT and TLR4−/− mice. A: homeostatic model assessment-insulin resistance (HOMA-IR) was calculated from serum insulin and glucose values after a 5-h fast. B: glucose AUC was determined from a 2-h glucose tolerance test (1 mg/kg). *Significantly different from respective CON (P < 0.05). #Significantly different from TLR4−/− HU (P < 0.05).
TLR4−/− mice maintained Akt and NF-κB signaling after 2 wk of hindlimb unloading.
There was a group × treatment interaction for Akt phosphorylation at Thr-473 (Fig. 2A) (P = 0.0536). Akt phosphorylation decreased by ∼70% after HU in WT group (P = 0.0046) but was not different from the TLR4−/− group after HU (P = 0.1067). There was a group × treatment interaction for IκBα phosphorylation at Ser-32/36 (Fig. 2B) (P < 0.0001). IκBα phosphorylation significantly increased by ∼70% after HU only in the WT group (P < 0.0001), and this response was higher than the TLR4−/− group after HU (P < 0.0001). There was a group × treatment interaction for IL-6 protein expression (Fig. 2C) (P = 0.0016). IL-6 protein abundance significantly increased by ∼400% after HU in the WT group (P < 0.0001) and ∼150% in TLR4−/− group after HU compared with the respective control groups (P = 0.0062). However, IL-6 protein abundance after HU in the WT group was significantly higher than the TLR4−/− HU group (P < 0.0001). Finally, there was a group × treatment interaction for LC3II/LC3I protein expression (Fig. 2D) (P < 0.0001). LC3II/LC3I expression increased only in the WT group after HU by ∼100% (P < 0.0001) and was significantly different than the TLR4−/− HU group (P < 0.0001).
Fig. 2.

Skeletal muscle protein expression. Data (means ± SD; n = 6–8/group) in the figure are soleus samples from ambulatory control (CON; white bars) and after 14 days of HU (gray bar) in WT and TLR4−/− mice. Muscle homogenates were assessed by immunoblotting for Akt Thr-473 (relative to total Akt; A), IκBα Ser-32/36 (relative to total IκBα) (B), IL-6 protein expression (relative to GAPDH) (C), and microtubule-associated protein 1 light chain 3 (LC3)II/LC3I (D). E: representative Western blot images for each of the target proteins listed above. *Significantly different from respective CON (P < 0.05). #Significantly different from TLR4−/− HU (P < 0.05).
TLR4−/− mice partly maintained mitochondrial respiration after 2 wk of hindlimb unloading.
There was a group × treatment interaction for RCR (Fig. 3A) (P < 0.0001). Respiratory control ratio was significantly decreased by ∼75% after HU in WT group (P < 0.0001) and similarly decreased in TLR4−/− group after HU by ∼30% (P = 0.0005). The decrease in RCR after HU was attenuated in the TLR4−/− group compared with the WT group (P < 0.0001). There was a group × treatment interaction for ADP-stimulated state 3 respiration (Fig. 3B) (P = 0.0518). There was only a trend for state 3 respiration to be depressed in the WT group after HU (P = 0.0711). Finally, there was a group × treatment interaction for oligo-stimulated state 4 respiration (Fig. 3C) (P = 0.0082). State 4 respiration was significantly increased after HU only in the WT group (P < 0.0001), and this response was different than the TLR4−/− group (P < 0.0001).
Fig. 3.
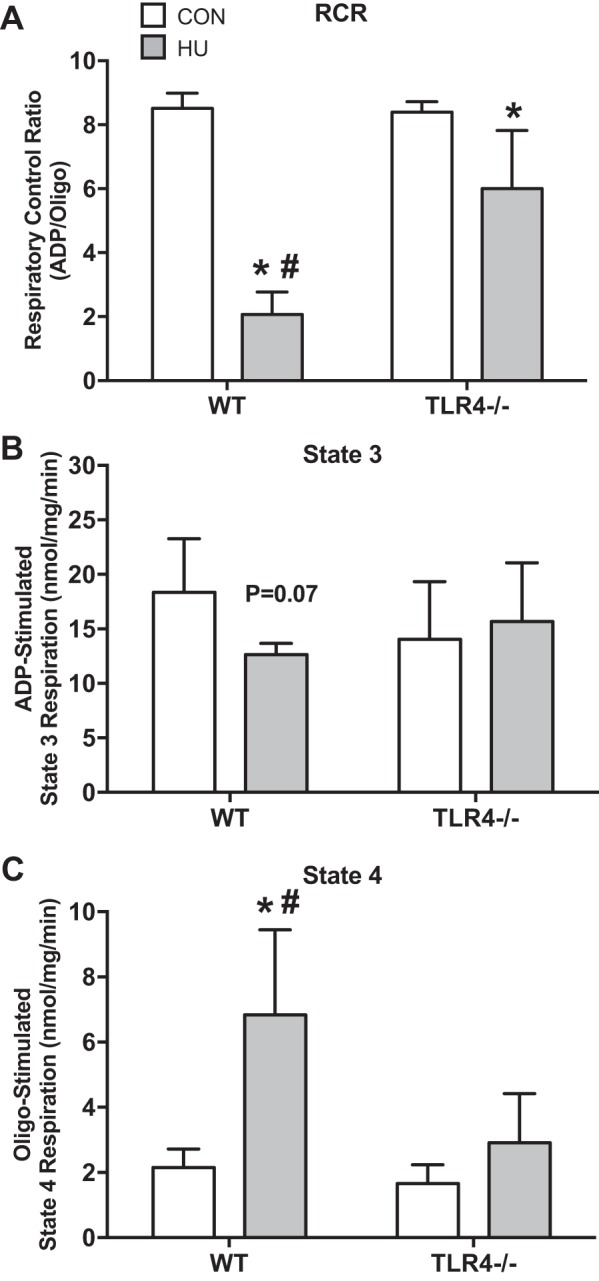
Skeletal muscle mitochondrial respiration. Data (means ± SD; n = 6–8/group) in figure are soleus samples from ambulatory control (CON; white bars) and after 14 days of HU (gray bar) in WT and TLR4−/− mice. Muscle was assessed for mitochondrial respiration. Data are respiratory control ratio (RCR; ADP/Oligo; A), ADP-stimulated state 3 respiration (nmol/mg/min) (B), and Oligo-stimulated state 4 respiration (nmol/mg/min) (C). *Significantly different from respective CON (P < 0.05). #Significantly different from TLR4−/− HU (P < 0.05).
TLR4−/− mice were partly resistant to the accumulation of skeletal muscle ceramides after 2 wk of hindlimb unloading.
The total abundance of skeletal muscle phosphoceramides (Fig. 4A) and sphingomyelin (Fig. 4B) was not altered as result of HU, nor were these different between WT and TLR4−/− groups. Alternately, there was a treatment effect for skeletal muscle glucosylceramides (Fig. 4C) (P = 0.0053) but no interaction (P = 0.6080). Glucosylceramide skeletal muscle abundance was increased by ∼100% after HU in the WT group (P = 0.0436) but not in the TLR4−/− group (P = 0.1376). Finally, there was a group × treatment interaction for total ceramide abundance (Fig. 4D) (P = 0.0488). Total ceramide abundance in skeletal muscle was increased after HU only in the WT group (P = 0.0006). There were no differences between groups as a result of HU (P = 0.2204).
Fig. 4.

Skeletal muscle sphingolipids. Data (means ± SD; n = 6–8/group) in figure are gastrocnemius samples from ambulatory control (CON; white bars) and after 14 days of HU (gray bar) in WT and TLR4−/− mice. Muscle was assessed for total abundance of select sphingolipid pools. Data are total phosphoceramides (pmol/mg) (A), sphingomyelin (pmol/mg) (B), glucosylceramides (pmol/mg) (C), and ceramides (pmol/mg) (D). *Significantly different from respective CON (P < 0.05).
TLR4−/− mice were protected from the accumulation of skeletal muscle Cer20:0 but not Cer18:0 after hindlimb unloading.
There was a treatment effect for Cer18:0 (P = 0.0011). Cer18:0 significantly increased by ∼30% as a result of HU in both WT and TLR4−/− groups (WT: P = 0.0202; TLR4−/−: P = 0.0227). There was a tendency for a group × treatment interaction for Cer20:0 (P = 0.0634). Cer20:0 increased ∼20% only in WT group as a result of HU (P = 0.0145), but there was no detected difference between the WT and TLR4−/− group as a result of HU (P = 0.3073) (Fig. 5). No other ceramide species were altered as a result of the treatment or between groups.
Fig. 5.

Specific ceramide species. Data (means ± SD; n = 6–8/group) in figure are gastrocnemius samples from WT ambulatory control (CON; white bars) and WT HU (gray bars) mice and from TLR4−/− ambulatory control (CON; hatched bar), and TLR4−/− HU (black bar) mice. Lipids isolated from muscle were assessed for detectable individual ceramide species ordered from low- to high-fatty acid chain length. *Significantly different from respective CON (P < 0.05).
Skeletal muscle ceramides and mitochondrial respiration are related to insulin sensitivity and glucose tolerance.
There was a positive correlation between total ceramides (Fig. 6A) (P = 0.0104), Cer18:0 (Fig. 6B) (P = 0.0336), and Cer20:0 (Fig. 6C) (P = 0.0304) vs. glucose AUC. HOMA-IR was positively correlated with total ceramides (Fig. 6D) (P = 0.0504) and inversely correlated with RCR (Fig. 6E) (P < 0.0001). There was no relationship between HOMA-IR with Cer18:0 or Cer20:0 (data not shown).
Fig. 6.

Relationships between muscle ceramides, insulin sensitivity, and mitochondrial respiration. Data (means ± SD) represent Pearson correlation tests from data collected from WT and TLR4−/− mice: total ceramides (pmol/mg) vs. glucose AUC (A), Cer18:0 (pmol/mg) vs. glucose AUC (B), Cer20:0 (pmol/mg) vs. glucose AUC (C), total ceramides (pmol/mg) vs. HOMA-IR (AU) (D), and respiratory control ratio (ADP/Oligo) vs. HOMA-IR (AU) (E). Pearson r and P values are displayed in the upper left corner.
DISCUSSION
The purpose of this study was to determine the regulatory role of TLR4 on muscle and metabolic disruption that occurs with physical inactivity and/or muscle disuse. The primary finding was that TLR4-deficient mice were protected from insulin resistance and impaired muscle NF-κB signaling caused by HU. Moreover, TLR4 deficiency partly prevented the HU-induced accumulation of muscle ceramides and the decrease in mitochondrial respiration. Finally, intramyocellular ceramides and mitochondrial respiration were related to insulin resistance and glucose tolerance. These data provide evidence that TLR4 plays an important role in metabolic decline with muscle disuse. Disrupting activated TLR4 signaling during periods of inactivity/muscle disuse may serve as an important therapeutic strategy at reducing metabolic disease development.
In our prior study, we showed that MyD88, a downstream adaptor protein of TLR signaling, was a potent mediator of insulin resistance, glucose intolerance, impaired Akt and NF-κB signaling, and partially induced muscle atrophy during 2 wk of HU (31). We add to these findings and report for the first time that TLR4 similarly regulates insulin sensitivity, aberrant muscle cell signaling, and muscle size with HU. It is reasonable to speculate that modest weight loss observed in the TLR4−/− mice may have partially contributed to improved insulin sensitivity during HU since weight loss after a high-fat diet in mice improve glucose homeostasis (29). Although this may be the case, we contend that muscle and metabolic adaptations to HU are also due to TLR4 since TLR4 signaling is well documented to robustly influence metabolic dysfunction during high-fat feeding (25, 41, 43). Moreover, we noted striking muscle and metabolic similarities in the current study compared with MyD88−/− mice after HU (31). This is in light of MyD88−/− mice not losing body weight during HU (31). MyD88 is also an essential adaptor protein for TLR4. At this stage, it is unclear what cells (muscle, fat, liver) were responsible for muscle atrophy and metabolic decline with HU since we used a whole body TLR4 knockout animal in these experiments. It is quite possible that TLR4 signaling in one or more cell types could be driving the muscle and metabolic phenotype observed with HU. For example, Jia et al. (28) noted that hepatocyte-specific, but not myeloid-specific, TLR4-deficient mice were protected from obesity and inflammation after a 12-wk high-fat diet. In another study, mice with overexpressed skeletal muscle-specific TLR4 gained more weight and were more glucose intolerant on a high-fat diet than WT controls (32). Nonetheless, our current and past data support that TLR4, likely through a MyD88-dependent pathway (31), is a primary mechanism that causes insulin resistance and partly underlies muscle atrophy during HU. Studies evaluating tissue-specific effects of TLR4 during HU are warranted.
A major finding of this study was that 2 wk of HU in mice increased the sphingolipid intermediate, ceramide by ∼34% within skeletal muscle tissue, and this response was partly ablated in TLR4−/− mice. In contrast, Salaun et al. (39) did not observe any change in total muscle ceramides following 1 wk of HU in rats. The discordant findings could simply be a result of a shorter duration of HU compared with our study. However, when looking at the individual ceramide species, Salaun et al. (39) and our current study showed an increase in the Cer18:0 moiety following HU. Cer18:0, the most abundant skeletal muscle ceramide species, has been shown to be elevated in mouse skeletal muscle following a high-fat diet (7) and in myotubes of patients with Type 2 diabetes vs. physically active adults (5). We also observed an increase in the ceramide species, Cer20:0, and, like Cer18:0, it was positively correlated to glucose AUC. The specific metabolic role (if any) of Cer20:0 is unknown. It is important to point out that long-chained ceramides (e.g., Cer22:0, Cer24:0) have been linked with insulin resistance in liver (36), perhaps suggesting Cer20:0 may also play a similar role in skeletal muscle with HU. Interestingly, total muscle ceramide and Cer20:0 accumulation were modestly prevented in TLR4−/− mice after HU. This finding suggests that ceramide accumulation partly stems from TLR4 signaling, but additional regulators of ceramide metabolism, such as other TLRs (e.g., TLR2) or alterations in ceramide flux (hydrolysis, salvage pathway) could play a part in ceramide accumulation with HU. Future investigations using myriocin and/or animals deficient in ceramide turnover would help address whether muscle ceramides are directly responsible for metabolic dysfunction with disuse.
Another interesting observation was that HU increased the total skeletal muscle abundance of glucosylceramides. The Summers laboratory (11) has elegantly showed that glucosylceramides impaired insulin sensitivity by accumulating in adipose but not muscle cells in mice fed a high-fat diet. In contrast, we found that glycosylceramides increased in gastrocnemius skeletal muscle after HU. We were limited in identifying the specific cell source of glucosylceramide accumulation in our study, since whole muscle, in addition to other muscle-resident cells such as adipose, immune and fibroblasts, were pooled during lipid isolation and lipidomic analysis. Regardless, heightened glucosylceramides in muscle-derived adipocytes could have secondary effects in skeletal muscle, such as altered insulin signaling (1). The specific role of glucosylceramides in muscle will need to be further investigated to determine whether these sphingolipids regulate insulin sensitivity as a result of HU.
One of the other major goals of this study was to determine whether TLR4 regulates mitochondrial function since mitochondrial respiration has been reported to be impaired following HU in rodents (47). Indeed, we are the first to show that under TLR4-null conditions following HU, mitochondrial respiration (RCR) was partially preserved, and this was mainly driven by maintenance of oligo-stimulated state 4 respiration. These data are complementary with those of Frisard et al. (21), who showed that LPS, a stimulator of TLR4 signaling, reduced RCR in isolated mouse skeletal muscle mitochondria by ∼70% (21). In another study by this group, TLR4-mutant mice were protected from LPS-induced dysregulation in glucose and fatty acid oxidation (20). We add to the body of TLR4 literature that TLR4 partly regulates mitochondrial respiration during HU, raising the investigation that additional mechanisms are involved in reducing mitochondrial respiration. For instance, decreased mitochondrial respiration may be regulated by ceramides since muscle ceramides remained partly elevated in TLR4−/− mice after HU. Emerging evidence suggest a possible regulatory role of ceramides on mitochondrial function (8, 16, 23, 40, 42, 45). For example, Ussher et al. (45) showed that malonyl CoA decarboxylase KO mice (animals with deficient fatty acid oxidation) were protected from 12 wk of diet-induced insulin resistance and muscle ceramide accumulation. In this same study, the authors showed that myriocin not only protected against high-fat diet-induced insulin resistance and obesity, but also preserved oxygen consumption rates and treadmill exercise exhaustion time and distance (45). However, caution is warranted in identifying a relationship between ceramides and mitochondrial respiration in our study, since these endpoints were examined in different muscles (soleus vs. gastrocnemius) because of limited tissue quantities. Together, our data support that TLR4 alters mitochondrial respiration with HU. Future experiments are needed to test whether ceramides mediate mitochondrial dysfunction with muscle disuse.
Perspectives and Significance
Our findings are the first to show that TLR4 mediates insulin resistance and partially regulates muscle ceramide accumulation, mitochondrial respiration, and muscle atrophy during HU. We also found that intramyocellular ceramides and mitochondrial respiration were modestly related to insulin sensitivity. Inhibiting molecules along the TLR4 axis may prove useful in preventing or reversing the development of metabolic disorders caused by muscle disuse.
GRANTS
This study was supported by the University of Utah's Diabetes and Metabolism Center and by the National Institutes of Health (Grants R56AG050781 and R01AG050781). Metabolomics Core Facility was supported by National Center for Research Resources Shared Instrumentation Grant 1 S10 OD016232-01.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
O.S.K., D.S.N., K.M.B., and M.J.D. performed experiments; O.S.K. and M.J.D. analyzed data; O.S.K., D.S.N., K.M.B., R.M.O., and M.J.D. edited and revised manuscript; O.S.K., D.S.N., K.M.B., R.M.O., and M.J.D. approved final version of manuscript; R.M.O. and M.J.D. conception and design of research; M.J.D. interpreted results of experiments; M.J.D. prepared figures; M.J.D. drafted manuscript.
REFERENCES
- 1.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409: 729–733, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM 2nd Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53: 25–31, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Amati F, Dube JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, Switzer GE, Bickel PE, Stefanovic-Racic M, Toledo FG, Goodpaster BH. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60: 2588–2597, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bajpeyi S, Myrland CK, Covington JD, Obanda D, Cefalu WT, Smith SR, Rustan AC, Ravussin E. Lipid in skeletal muscle myotubes is associated to the donors' insulin sensitivity and physical activity phenotypes. Obesity (Silver Spring) 22: 426–434, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bienso RS, Ringholm S, Kiilerich K, Aachmann-Andersen NJ, Krogh-Madsen R, Guerra B, Plomgaard P, van Hall G, Treebak JT, Saltin B, Lundby C, Calbet JA, Pilegaard H, Wojtaszewski JF. GLUT4 and glycogen synthase are key players in bed rest-induced insulin resistance. Diabetes 61: 1090–1099, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bikman BT, Guan Y, Shui G, Siddique MM, Holland WL, Kim JY, Fabrias G, Wenk MR, Summers SA. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J Biol Chem 287: 17,426–17,437, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest 121: 4222–4230, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Booth FW, Roberts CK, Laye MJ. Lack of exercise is a major cause of chronic diseases. Compr Physiol 2: 1143–1211, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA. The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. J Physiol 593: 1981–1995, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chavez JA, Siddique MM, Wang ST, Ching J, Shayman JA, Summers SA. Ceramides and glucosylceramides are independent antagonists of insulin signaling. J Biol Chem 289: 723–734, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 15: 585–594, 2012. [DOI] [PubMed] [Google Scholar]
- 13.Coen PM, Hames KC, Leachman EM, DeLany JP, Ritov VB, Menshikova EV, Dube JJ, Stefanovic-Racic M, Toledo FG, Goodpaster BH. Reduced skeletal muscle oxidative capacity and elevated ceramide but not diacylglycerol content in severe obesity. Obesity (Silver Spring) 21: 2362–2371, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 33: 861–868, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekker MJ, Baker C, Naples M, Samsoondar J, Zhang R, Qiu W, Sacco J, Adeli K. Inhibition of sphingolipid synthesis improves dyslipidemia in the diet-induced hamster model of insulin resistance: evidence for the role of sphingosine and sphinganine in hepatic VLDL-apoB100 overproduction. Atherosclerosis 228: 98–109, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Di Paola M, Cocco T, Lorusso M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 39: 6660–6668, 2000. [DOI] [PubMed] [Google Scholar]
- 17.Dirks ML, Wall BT, van de Valk B, Holloway TM, Holloway GP, Chabowski A, Goossens GH, van Loon LJ. One week of bed rest leads to substantial muscle atrophy and induces whole-body insulin resistance in the absence of skeletal muscle lipid accumulation. Diabetes db15 1661, 2016. [DOI] [PubMed] [Google Scholar]
- 18.Drummond MJ, Timmerman KL, Markofski MM, Walker DK, Dickinson JM, Jamaluddin M, Brasier AR, Rasmussen BB, Volpi E. Short-term bed rest increases TLR4 and IL-6 expression in skeletal muscle of older adults. Am J Physiol Regul Integr Comp Physiol 305: R216–R223, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson KA, Smith ME, Anthonymuthu TS, Evanson MJ, Brassfield ES, Hodson AE, Bressler MA, Tucker BJ, Thatcher MO, Prince JT, Hancock CR, Bikman BT. AICAR inhibits ceramide biosynthesis in skeletal muscle. Diabetol Metab Syndr 4: 45, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, Heilbronn L, Haynie K, Muoio B, Li L, Hulver MW. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab 298: E988–E998, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisard MI, Wu Y, McMillan RP, Voelker KA, Wahlberg KA, Anderson AS, Boutagy N, Resendes K, Ravussin E, Hulver MW. Low levels of lipopolysaccharide modulate mitochondrial oxygen consumption in skeletal muscle. Metab Clin Exp 64: 416–427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh S, Lertwattanarak R, Garduno JD, Galeana JJ, Li J, Zamarripa F, Lancaster JL, Mohan S, Hussey S, Musi N. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J Gerontol A Biol Sci Med Sci 70: 232–246, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem 272: 24,154–24,158, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, Kirwan JP. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58: 337–343, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE, Summers SA. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121: 1858–1870, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5: 167–179, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Hussey SE, Lum H, Alvarez A, Cipriani Y, Garduno-Garcia J, Anaya L, Dube J, Musi N. A sustained increase in plasma NEFA upregulates the Toll-like receptor network in human muscle. Diabetologia 57: 582–591, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, Lee S, Scherer PE, Elmquist JK. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun 5: 3878, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung DY, Ko HJ, Lichtman EI, Lee E, Lawton E, Ong H, Yu K, Azuma Y, Friedline RH, Lee KW, Kim JK. Short-term weight loss attenuates local tissue inflammation and improves insulin sensitivity without affecting adipose inflammation in obese mice. Am J Physiol Endocrinol Metab 304: E964–E976, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kump DS, Booth FW. Alterations in insulin receptor signalling in the rat epitrochlearis muscle upon cessation of voluntary exercise. J Physiol 562: 829–838, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon OS, Tanner RE, Barrows KM, Runtsch M, Symons JD, Jalili T, Bikman BT, McClain DA, O'Connell RM, Drummond MJ. MyD88 regulates physical inactivity-induced skeletal muscle inflammation, ceramide biosynthesis signaling, and glucose intolerance. Am J Physiol Endocrinol Metab 309: E11–E21, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMillan RP, Wu Y, Voelker K, Fundaro G, Kavanaugh J, Stevens JR, Shabrokh E, Ali M, Harvey M, Anderson AS, Boutagy NE, Mynatt RL, Frisard MI, Hulver MW. Selective overexpression of Toll-like receptor-4 in skeletal muscle impairs metabolic adaptation to high-fat feeding. Am J Physiol Regul Integr Comp Physiol 309: R304–R313, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol (1985) 111: 1459–1466, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen RH, Krogh-Madsen R, Thomsen C, Booth FW, Pedersen BK. Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA 299: 1261–1263, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Pierre N, Deldicque L, Barbe C, Naslain D, Cani PD, Francaux M. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PloS One 8: e65061, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Ohman MK, Takeda K, Sugii S, Pewzner-Jung Y, Futerman AH, Summers SA. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 20: 687–695, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A, Musi N. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57: 2595–2602, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salaun E, Gratas-Delamarche A, Derbre F. De novo ceramides synthesis is not involved in skeletal muscle atrophy induced by short-term mechanical unloading. Free Rad Biol Med 75, Suppl 1: S28, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Salaun E, Lefeuvre-Orfila L, Cavey T, Martin B, Turlin B, Ropert M, Loreal O, Derbre F. Myriocin prevents muscle ceramide accumulation but not muscle fiber atrophy during short-term mechanical unloading. J Appl Physiol (1985) 120: 178–187, 2016. [DOI] [PubMed] [Google Scholar]
- 40.Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, Bielawski J, Ogretmen B. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol 8: 831–838, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–3025, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith ME, Tippetts TS, Brassfield ES, Tucker BJ, Ockey A, Swensen AC, Anthonymuthu TS, Washburn TD, Kane DA, Prince JT, Bikman BT. Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem J 456: 427–439, 2013. [DOI] [PubMed] [Google Scholar]
- 43.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56: 1986–1998, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, Xu E, Hammerschmidt P, Bronneke HS, Trifunovic A, LoSasso G, Wunderlich FT, Kornfeld JW, Bluher M, Kronke M, Bruning JC. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20: 678–686, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, Lopaschuk DG, Proctor SD, Keung W, Muoio DM, Lopaschuk GD. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 59: 2453–2464, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem J 417: 791–801, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Yajid F, Mercier JG, Mercier BM, Dubouchaud H, Prefaut C. Effects of 4 wk of hindlimb suspension on skeletal muscle mitochondrial respiration in rats. J Appl Physiol (1985) 84: 479–485, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab 297: E211–E224, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]