

Abstract
Autonomic neural activation of intracellular Ca2+ release in parotid acinar cells induces the secretion of the fluid and protein components of primary saliva critical for maintaining overall oral homeostasis. In the current study, we profiled the role of acidic organelles in shaping the Ca2+ signals of parotid acini using a variety of imaging and pharmacological approaches. Results demonstrate that zymogen granules predominate as an apically polarized population of acidic organelles that contributes to the initial Ca2+ release. Moreover, we provide evidence that indicates a role for the intracellular messenger NAADP in the release of Ca2+ from acidic organelles following elevation of cAMP. Our data are consistent with the “trigger” hypothesis where localized release of Ca2+ sensitizes canonical intracellular Ca2+ channels to enhance signals from the endoplasmic reticulum. Release from acidic stores may be important for initiating saliva secretion at low levels of stimulation and a potential therapeutic target to augment secretory activity in hypofunctioning salivary glands.
Keywords: calcium signaling, NAADP, acidic organelles, parotid salivary gland, cAMP
the parotid gland is one of three major paired salivary glands (along with the submandibular and sublingual) that produces, elaborates, and transports saliva in response to neural input arising from the autonomic nervous system (3, 24, 58). In contrast to the canonical “gas and brake” relationship between autonomic inputs, sympathetically driven β-adrenergic receptors (β-AR) can act concurrently with parasympathetic activation of muscarinic M3 receptors to evoke or potentiate secretion of the protein and fluid components of saliva (2, 25, 52) via inositol trisphosphate (IP3)-mediated Ca2+ release from the endoplasmic reticulum (ER). However, recent literature has demonstrated that intracellular acidic organellular compartments can also act as releasable Ca2+ stores. Ca2+ signals derived from acidic organelles have been shown to regulate a wide variety of cellular functions from fertilization in starfish oocytes (39) to glucose-induced insulin release from β-cells (42) and neurite outgrowth (10). Importantly, a large content of acidic Ca2+ stores situated in the granular region are participants in Ca2+ signaling events important for fluid and digestive enzyme release from pancreatic acini (27, 50, 66). Although one particular study using submandibular acini did not find evidence of acidic organelle Ca2+ release (31), differences are known to exist between the major paired salivary glands. The current study examined the acidic store content of parotid acini and interrogated whether acidic organelles contribute to agonist-evoked Ca2+ release.
To characterize whether acidic Ca2+ stores contributed to agonist-evoked Ca2+ signals in the parotid salivary gland, we used enzymatically dissociated cultures of acinar cells in combination with immunofluorescence, live-cell fluorescence imaging, and pharmacological approaches. Our work revealed that acidic organelles are largely localized to the apical region of the acini and are predominantly comprised of secretory granules. The data indicate that acidic Ca2+ stores are functionally recruited following cyclic adenosine monophosphate (cAMP) elevation and suggest that nicotinic acid adenine dinucleotide phosphate (NAADP) is the second messenger that releases Ca2+ from secretory granules. Due to the crucial nature of Ca2+ signals in regulating saliva production, this pathway may have importance for understanding or treating diseases that produce salivary gland hypofunction.
MATERIALS AND METHODS
Animal use.
Male C57BL/6J mice ∼6 mo in age were obtained from The Jackson Laboratory (Bar Harbor, ME) and euthanized using CO2 asphyxiation and subsequently punctured through the heart. All protocols involving animals were approved by the University of Toledo Institutional Animal Care and Use Committee and conformed to Guide for the Care and Use of Laboratory Animals. Access to food and water was ad libitum.
Preparation of parotid acinar clusters.
The parotid glands were quickly removed by dissection and placed in 2 ml of an enzymatic digestion solution containing 20 ml Eagle's minimal essential medium, 0.5–0.6 mg of collagenase P, 200 μl l-glutamine (2 mM), and 200 mg bovine serum albumin (1%). Glands were cleaned of fat and connective tissue, minced for several minutes until homogenous, and then placed in a 25-ml vented flask of enzymatic digest solution. Cells were incubated for 20 min with continuous shaking and gassed with CO2 at 37°C in a water bath. The tissue was transferred to a tube and centrifuged for 1 min at 162 relative centrifugal force (rcf) with the supernatant subsequently discarded. Cells were transferred to a freshly gassed 25-ml vented flask containing the enzymatic digest solution and triturated using a 10 ml serological pipette fitted with a 200-μl tip. Tissue underwent successive 15-min digestions with shaking and gassing followed by trituration breaks using flame-polished glass pipettes with progressively smaller ends (1.19–0.6 mm). When digested to clusters containing ∼16 acini, cells were spun down for 1 min at 76 rcf, the supernatant was discarded, and the digest was halted by addition of a rinse solution containing BSA-free basal medium. After being washed, the pellet was centrifuged at 76 rcf for 1 min, the supernatant was discarded, and the pellet was resuspended in a 1:1 solution containing the rinse solution and rinse solution with 200 μl l-glutamine (2 mM) and 400 μl Penn/Strep. Parotid acinar clusters were plated on autoclaved, ethanol-wiped glass coverslips (25CIR-1), and allowed to adhere for 30 min.
Live-cell imaging.
All Ca2+ measurements were made at room temperature using a Nikon Eclipse TE2000S as previously described (7). Briefly, the TE2000 with DIC optics was coupled to a Polychrome IV monochromator-based high-speed digital imaging system (TILL Photonics, Gräfelfing, Germany) ported to a fiber optic guide and epifluorescence condenser. Acinar clusters were loaded with fura-2 AM, a cell-permeable ratiometric fluorescent Ca2+ indicator, and illuminated with dual wavelength light (340 and 380 nm) focused onto the image plane via a DM400 dichroic mirror and Nikon SuperFluor ×40 oil-immersion objective, and fluorescence was obtained through a 525 ± 25 nm band-pass filter (Chroma Technologies, Brattleboro, VT).
Immunofluorescence.
Prepared clusters of parotid acini were adhered to autoclaved glass coverslips (25CIR-1) using Cell-Tak. The acini were fixed using 4% paraformaldehyde for 15–20 min and washed three times with PBS. Permeabilization was achieved using 0.3% Triton-X-0.1% BSA for 15 min and acini were blocked to prevent nonspecific binding with 5% nonfat dry milk and 0.1% Triton-X (washing buffer) at 1 h. Antibodies for lysosomal-associated membrane protein 1 (LAMP1), vesicular-associated membrane protein 8 (VAMP8), two-pore channel 2 (TPC2), and early endosome antigen 1 (EEA1) were used at a dilution of 1:50 in washing buffer and left overnight in a humidified chamber at 4°C with mild shaking. Following incubation with the primary, the acini were washed three times in the washing buffer and Alexa Fluor secondary antibodies were prepared in the washing buffer at a dilution of 1:500 or Alexa Fluor 555-conjugated phalloidin at 1:200. Secondary antibodies were incubated at room temperature for 1 h with mild shaking. Cells were washed twice with washing buffer and once with PBS before being attached to slides on top of the antifade agent Vectashield (Burlingame, CA). After drying, slides were sealed with nail polish and imaged at room temperature on a TCS SP5 multiphoton laser scanning confocal microscope with a ×40 oil-immersion objective (Leica Microsystems).
Time-differentiated imaging.
Time-differentiated imaging was performed as described previously (7). Briefly, brightfield images were acquired every 500 ms using a high-speed Uniblitz VS35 optical shutter (Vincent Associates, Rochester, NY) placed in the tungsten lamp illumination path during recording. Images were subtracted from each previous one and exocytotic events visualized as the resultant changes in optical density.
Texas Red dextran labeling.
Isolated parotid acini plated on coverslips were incubated with 500 μl Texas red dextran solution (1 mg/ml) for 5 min. Exocytosis was then induced by adding agonist pipetted to the Texas Red solution for 10 min at 37°C. Following the stimulation period, cells were immediately fixed with 4% paraformaldehyde for 30 min. The paraformaldehyde was exchanged three times to ensure successful removal of excess Texas Red. Coverslips were washed three times at 5 min with PBS to remove paraformaldehyde and immediately imaged using confocal microscopy.
Amylase collection and Western blotting.
Parotid acinar clusters were prepared as previously described and adhered to glass coverslips. A small volume of ∼200 to 300 μl of saline was layered over the acini and amylase release was induced by addition of agonist for 10 min on ice. Following stimulation, the saline containing released amylase was collected, centrifuged for 1 min, and then frozen. The acini on the coverslips were then lysed with 200–300 μl of a buffer containing 25 mM Tris·HCl, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS to determine the remaining amylase content. Samples were loaded by concentration and resolved using 12% Bis-Tris gel electrophoresis. Western blotting was performed using a primary antibody for α-amylase (1:500) overnight with mild shaking at 4°C. Following incubation with the primary antibody, the membrane was incubated for 1 h at room temperature with a donkey anti-goat horseradish peroxidase-conjugated secondary antibody (1:10,000) and the blot was imaged used an ECL detection system.
Materials.
Collagenase-P used for cell preparations was bought from Roche Applied Science (Indianapolis, IN). Isoprenaline hydrochloride, carbamoylcholine chloride (carbachol), forskolin, 3-isobutyl-1-methylxanthine, and TPCN-2 primary antibody were obtained from Sigma Aldrich (St. Louis, MO). VAMP8 was acquired from Synaptic Systems (Göttingen, Germany), α-amylase, and glycyl-l-phenylalanine-β-napthylamide from Santa Cruz Biotechnology (Santa Cruz, CA), and EEA1 from Cell Signaling (Danvers, MA). LysoTracker red, Texas Red dextran, calcein green, and all secondary antibodies were purchased from Life Technologies (Eugene, OR). TefLabs (Austin, TX) supplied fura-2 AM. Bafilomyin A1 was bought from LCLabs (Woburn, MA) and NED19 acquired from R&D Systems (Minneapolis, MN).
Data analysis and image processing:.
As controls and experimental treatments were performed on different acini clusters, unpaired t-tests were used to test for statistical significance between groups with GraphPad Prism 3 (GraphPad Software, La Jolla, CA). Values represent means ± SE, with P < 0.05 taken as statistically significant. Welch's correction was used when the variances were significantly different. Bar graphs were normalized to control values for ease of presentation. All Ca2+ traces and subsequent analyses were done using Igor Pro software (WaveMetrics, Lake Oswego, OR). The rate of the initial Ca2+ rise was determined by a linear fit to the rising phase of the Ca2+ response. Adjustments of brightness and contrast was achieved through ImageJ (W. S. Rasband, National Institutes of Health, Bethesda, MD), and these adjustments were applied to every pixel in the image and did not obscure, eliminate, or misrepresent any information present in the original. Immunoblot data analysis was also performed using ImageJ. The relative intensity of the bands was determined and secreted amylase is expressed as percentage of total amylase.
RESULTS
Parotid acinar cells contain abundant acidic organelles with distinct distributions.
In an initial set of experiments, the acidic organelle content of parotid acini was probed using 300 nM LysoTracker red, a vital fluorescent dye that preferentially accumulates in organelles with low pH. In addition, 1 μM calcein green was used to label the cytoplasm. After 30 min of dye loading, acini were imaged using confocal microscopy. Figure 1A shows a confocal image of an acinar cell cluster with an intense red apical signal, indicating abundant acidic organelles were present at the apicoluminal pole of the acini. Occasionally ductal fragments were also observed. The pattern of LysoTracker labeling in ductal cells was less intense and typically localized to a perinuclear region (Fig. 1A, asterisk). This expression pattern indicated that there were differences in the identity of acidic organelles in acinar vs. ductal cells. Moreover, acidic organelle expression pattern varied by exocrine gland. Submandibular acini had a diffuse pattern of labeling (Fig. 1B) while lacrimal acini had abundant and polarized acidic organelles (Fig. 1C).
Fig. 1.

Acidic organelle localization and identification. LysoTracker (red, acidic organelles) and calcein (green, cytoplasm) fluorescent dyes were used for live-cell imaging (A–E) to localize acidic organelles in parotid acini (A) and a ductal fragment (A, *), submandibular acini (B), and lacrimal acini (C) and in parotid acini following treatment with BAF (D) or GPN and cathepsin inhibitor (E). The secretory granules (F) were immunolabeled using the VAMP8 antibody (green) and the subluminal F-actin network by Alexa Fluor 555-conjugated phalloidin (red). Lysosomes in acinar cells (G) and a ductal fragment (H) were visualized using LAMP1(G and H, green) and costained with the F-actin network (red). Endosomes were labeled by EEA1 antisera (I, green) with actin visualized by fluorescent phalloidin (red). All scale bars are 10 μm.
Treatment with bafilomycin A1 (BAF) to reversibly inhibit the vacuolar H+-ATPase and disrupt the pH gradient of acidic organelles confirmed that the labeled organelles were acidic and validated BAF as a pharmacological tool for subsequent functional studies. In these experiments, acini were incubated in 1 μM BAF for 1 h, loaded with LysoTracker and calcein, washed with fresh saline, and imaged using confocal microscopy. As shown in Fig. 1D, there was nearly a complete abolishment of the red fluorescent labeling compared with control cells treated with vehicle only. However, the identity of these organelles was not known. To further characterize parotid acinar acidic organelles, we treated cells with the lysosomal selective disruptor glycyl-l-phenylalanine-β-napthylamide (GPN) with inclusion of cathepsin inhibitor 1 to prevent off target cleavage of GPN. GPN is a substrate preferentially cleaved by the lysosomal specific protease cathepsin C to induce osmotic rupture of lysosomes. Parotid acini were treated with GPN and inhibitor for 10 min, loaded with LysoTracker and calcein, washed with saline, and imaged by confocal microscopy. Acini treated with GPN and inhibitor still displayed a robust albeit somewhat more spatially restricted LysoTracker signal (Fig. 1E). We determined the area of the cell stained by LysoTracker as a percentage of the total area of the cell stained for both control and treated acini. On average, 49% of the cell was stained by LysoTracker in control (n = 5) while 47% of the cell was stained by LysoTracker in GPN-treated acini (n = 5). This was not significantly different (P = 0.62). The overall apical pattern was generally maintained and likely represented secretory granules.
Maintenance of the LysoTracker signal following GPN treatment demonstrates that secretory granules were the more prominent acidic store in parotid acini. Accordingly, we used immunofluorescence and selective antibodies to localize a variety of acidic organelles including secretory granules, lysosomes/late endosomes, and early endosomes to ascertain the relative cell content of different acidic organelles. The vesicular-associated membrane protein 8 (VAMP8) was used as a marker for secretory granules. Figure 1F shows that acini clusters displayed immunopositive signals for VAMP8 concentrated along apicolateral surfaces, in agreement with a previous study that tested VAMP8 in parotid gland slices (62). This pattern of staining was reminiscent of the aforementioned live-cell LysoTracker images. Lysosomal stores and late endosomes were localized using lysosomal associated membrane protein 1 (LAMP1). Parotid acini displayed a pattern of LAMP1 immunoreactivity along the apical and lateral boarders of acinar cells in addition to closely apposed to the actin network (Fig. 1G). This staining mirrored our VAMP8 distribution; however, experiments assessing colocalization could not be performed as VAMP8/LAMP1 antibodies were derived from the same host. Interestingly, a ductal fragment isolated alongside the acini displayed a different LAMP1 immunoreactive pattern (Fig. 1H). This fragment displayed a more perinuclear distribution that mirrored our ductal LysoTracker image from Fig. 1A suggesting that different acidic organelles predominate between acinar and ductal cells. Although our LAMP1 studies suggests an extensive population of lysosomes, the LysoTracker fluorescent signal was largely unaffected following selective elimination by GPN treatment (Fig. 1E). This suggested the possibility that LAMP1 was on secretory granules, consistent with a previously described relationship between granules-lysosomes (8, 66). A final set of confocal images assessed the acinar endosomal distribution using the early endosome antigen 1 (EEA1) antibody (Fig. 1I). Early endosomes had a more diffuse staining pattern, with immunoreactivity in the apicolateral regions as well as some basal localization. Together, the confocal data demonstrate there are likely distinct acidic stores in acinar cells with the secretory granules comprising the majority of acidic organelles distributed apicolaterally, lysosomes/late endosomes sharing a similar distribution as well as being in close proximity to the actin network, and a more homogenous distribution of early endosomes.
Functional assessment of acidic store Ca2+ signaling.
Although the predominant Ca2+ release reservoir in most cell types is the ER (5, 28, 33) acidic organelles such as lysosomes, secretory granules, and endosomes can also function as releasable Ca2+ stores (49, 51). However, it is not well elucidated how these acidic stores participate in or are mobilized for global Ca2+ signals. Because parotid acini contained abundant acidic organelles, we began to assess the role of these organelles in agonist-evoked Ca2+ signals. Cells were loaded with fura-2 AM and stimulated with carbachol (CCh), an acetylcholine agonist, at concentrations we determined experimentally to evoke a threshold (50–100 nM CCh) or maximal (2 μM CCh) Ca2+ release (Fig. 2). It was especially important to empirically determine the threshold for CCh-evoked responses as day-to-day variations in enzymatic digestion of the tissue likely resulted in differences to cell surface receptor content between preparations. Next, the role of acidic organelles in CCh-evoked Ca2+ release was assessed following treatment with 1 μM BAF. Figure 3, A and B, shows that BAF-treated cells showed no significant difference in the average peak amplitude of the Ca2+ response at both threshold and maximal cholinergic stimulation compared with controls. Moreover, under our conditions no significant differences were observed in the average initial rate of Ca2+ increase or number of oscillations between control and treated cells (Table 1).
Fig. 2.
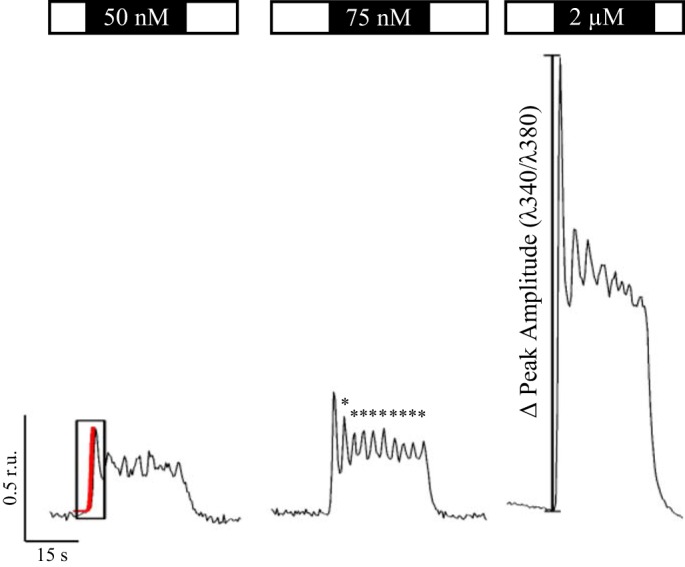
Representative Ca2+ traces following stimulation with varying concentrations of CCh. Ca2+ signals were recorded in parotid acinar cells following stimulation with the acetylcholine agonist CCh at threshold and maximal simulating concentrations (open box is saline, and black box represents concentration of CCh). Because the enzymatically dispersed cell preparations varied in their sensitivity to agonist stimulation, the threshold concentrations used to evoke Ca2+ signals was empirically determined. Threshold was defined as the minimum stimulus needed to typically evoke a clear initial peak release and produce oscillations (*) whose amplitudes were approximately three times the baseline noise and lasted for 2 s. Depending on the day, a threshold CCh concentration usually ranged between 50 and 75 nM. Complex Ca2+ waveforms were analyzed for the number of oscillations, the peak amplitude of initial Ca2+ rise (in ratio units or r.u), and rate of the initial Ca2+ rise via the slope (ratio units per second) estimated from a linear function fitted to the Ca2+ trace (red).
Fig. 3.

cAMP-dependent recruitment of acidic organelle Ca2+ release. Open box represents saline, and black box indicates duration and concentration of CCh stimulus. Acini were stimulated at threshold and maximal concentrations of CCh in the absence (A, black trace) or presence of BAF pretreatment (A, red trace). Compared with CCh alone responses [0.24 ± 0.034 r.u., n = 17], there was no change in the peak amplitude of Ca2+ release following BAF treatment (0.17 ± 0.031 r.u., n = 17, P = 0.16) at a threshold level of stimulation (B). Additionally, there was no significant change in peak amplitudes of CCh alone responses (1.3 ± 0.13 r.u., n = 17) compared with BAF + CCh peak amplitudes (1.2 ± 0.14 r.u., n = 17, P = 0.55) at maximal stimulation (B). However, when FORS-treated acini were stimulated at threshold and maximal concentrations of CCh with (C, red trace) or without (C, black trace) BAF pretreatment, there was a significant diminishment (D) in the threshold peak amplitude of initial Ca2+ release (0.11 ± 0.016 r.u., n = 6, ***P = 0.0004) compared with FORS alone responses (0.31 ± 0.035 r.u., n = 6). This was also true for maximal stimulation (D), with FORS alone peak amplitudes (2.2 ± 0.17 r.u., n = 6) significantly reduced following BAF treatment (1.6 ± 0.096 r.u., n = 6, *P = 0.013).
Table 1.
Functional assessment of acidic organelle Ca2+ release
| Treatment | Maximal Peak Amplitude, r.u. | Threshold Rate, r.u./s | Maximal Rate, r.u./s | Threshold Oscillations | Maximal Oscillations |
|---|---|---|---|---|---|
| CCha | N/A | 0.11 ± 0.027 (n = 16) | 0.80 ± 0.11 (n = 16) | 4.9 ± 0.59 (n = 17) | 7.0 ± 0.49 (n = 17) |
| BAF + CCha | N/A | 0.12 ± 0.042 (n = 17) | 0.69 ± 0.11 (n = 17) | 4.5 ± 0.81 (n = 17) | 5.8 ± 0.61 (n = 17) |
| FORS + CChb | N/A | 0.12 ± 0.023 (n = 6) | 1.5 ± 0.17 (n = 6) | 4.0 ± 0.63 (n = 6) | 6.3 ± 0.71 (n = 6) |
| FORS + BAF + CChb | N/A | 0.041 ± 0.012* (n = 6) | 1.2 ± 0.066 (n = 6) | 5.8 ± 0.65 (n = 6) | 6.8 ± 0.40 (n = 6) |
| ISOP/IBMX + CChc | 1.5 ± 0.13 (n = 13) | 0.083 ± 0.023 (n = 13) | 0.76 ± 0.11 (n = 13) | 5.0 ± 0.74 (n = 13) | 4.5 ± 0.61 (n = 13) |
| SOP/IBMX + BAF + CChc | 1.4 ± 0.22 (n = 10) | 0.025 ± 0.0084* (n = 11) | 0.64 ± 0.14 (n = 10) | 3.7 ± 0.82 (n = 11) | 4.2 ± 0.62 (n = 10) |
| ISOP/IBMX + CChd | 2.03 ± 0.20 (n = 6) | 0.16 ± 0.019 (n = 9) | 1.1 ± 0.10 (n = 6) | 4.11 ± 0.51 (n = 9) | 5.3 ± 0.67 (n = 6) |
| ISOP/IBMX 1 h + CChd | 1.57 ± 0.20 (n = 8) | 0.031 ± 0.0062† (n = 10) | 0.85 ± 0.21 (n = 8) | 1.2 ± 0.53* (n = 10) | 4.6 ± 0.65 (n = 8) |
| ISOP/IBMX + CChe | 1.8 ± 0.24 (n = 7) | 0.16 ± 0.022 (n = 9) | 1.2 ± 0.22 (n = 7) | 3.7 ± 0.60 (n = 9) | 4.7 ± 0.92 (n = 7) |
| ISOP/IBMX + GPN + CChe | 1.5 ± 0.17 (n = 12) | 0.21 ± 0.038 (n = 12) | 0.83 ± 0.14 (n = 12) | 3.8 ± 0.71 (n = 12) | 4.4 ± 0.57 (n = 12) |
Note: threshold and maximal reference the concentration of CCh used to evoke a Ca2+ response.
BAF-treated acini did not show any differences between all parameters listed.
FORS + CCh vs. FORS + CCh + BAF Ca2+ responses were not significantly altered for the different parameters except for a significant decrease in the rate of initial Ca2+ rise (*P = 0.011) in BAF-treated acini.
ISOP/IBMX + CCh vs. ISOP/IBMX + CCh + BAF Ca2+ responses were not altered at the maximal peak amplitude, maximal rate, or at either stimulation level in terms of oscillation number. However, the rate of initial Ca2+ rise in ISOP/IBMX + CCh + BAF-treated acini was significantly decreased (*P = 0.039).
ISOP/IBMX + CCh vs. ISOP/IBMX 1HR + CCh responses were unaffected at maximal peak amplitude, rate, and oscillations. ISOP/IBMX 1 HR + CCh-treated acini had a significantly decreased rate of initial Ca2+ rise (†P < 0.0001) and significantly fewer oscillations (*P = 0.021) under threshold signaling conditions.
GPN-treated acini did not show any changes at all parameters listed.
Cylic AMP-dependent recruitment of acidic store Ca2+ release.
Although initial experiments demonstrated acidic organelles had no observable effect on CCh-evoked Ca2+ signals, the aforementioned experiments assessed only activation of the parasympathetic pathway. Given that autonomic inputs are known to synergistically regulate secretory output in the salivary gland (2, 25, 52), we hypothesized recruitment of acidic organelle Ca2+ release depended on coincident activation of sympathetic input. Sympathetic signaling via norepinephrine activation of β-ARs increases intracellular cAMP, a messenger shown to enhance IP3-evoked Ca2+ release through cAMP-activated protein kinase A (PKA)-mediated regulation (11, 60–61). Although this enhancement largely resulted from IP3 receptor (IP3R) phosphorylation, it is also possible the enhanced Ca2+ response is in part due to amplification of local acidic organelle Ca2+ release. Thus we designed experiments to elevate cAMP to assess its potential to recruit acidic organelle Ca2+ release and modulate CCh-evoked Ca2+ dynamics in parotid acini. First, parotid acinar clusters were preincubated for 10 min in 10 μM forskolin (FORS), an adenylate cyclase agonist, to induce a rise in cytosolic cAMP and then stimulated with CCh at either a threshold or maximal concentrations. Following BAF treatment, the average FORS-induced enhancement of the peak Ca2+ amplitude evoked by threshold CCh stimulation was reduced 65% compared with control responses (Fig. 3, C and D). Furthermore, the FORS-induced enhancement was diminished by 33% at a maximal CCh stimulation (Fig. 3, C and D). Additionally, there was a significant decrease in the average rate of evoked Ca2+ rise following BAF treatment while the number of Ca2+ oscillations were not altered (Table 1).
FORS robustly and globally elevates cAMP by direct activation of adenylate cyclase, but it likely does not recapitulate physiological agonist-induced changes in cAMP. Thus isoproterenol (ISOP), a β-AR agonist, was used to elevate cAMP in a more physiologically relevant manner. In these set of experiments, parotid acinar clusters were treated with 1 μM BAF for 1 h before imaging. The acini were then treated with ISOP for 5 min to elevate cytosolic cAMP and enhance Ca2+ release. In addition to ISOP, 3-isobutyl-1-methylxanthine (IBMX), a phosphodiesterase inhibitor was used to block cAMP degradation. As shown in Fig. 4, A and B, the average enhancement of peak Ca2+ release by ISOP at threshold cholinergic stimulation was reduced by ∼55% in cells that received BAF. In addition, the average rate of Ca2+ rise was significantly decreased in parotid acini treated with BAF (Table 1). The peak amplitude of Ca2+ rise was unaffected at maximal stimulation while the number of Ca2+ oscillations remained unaffected at both threshold and maximal CCh concentrations (Table 1). These data are consistent with the idea that acidic organelle recruitment and Ca2+ release required agonist-evoked elevation of cAMP.
Fig. 4.
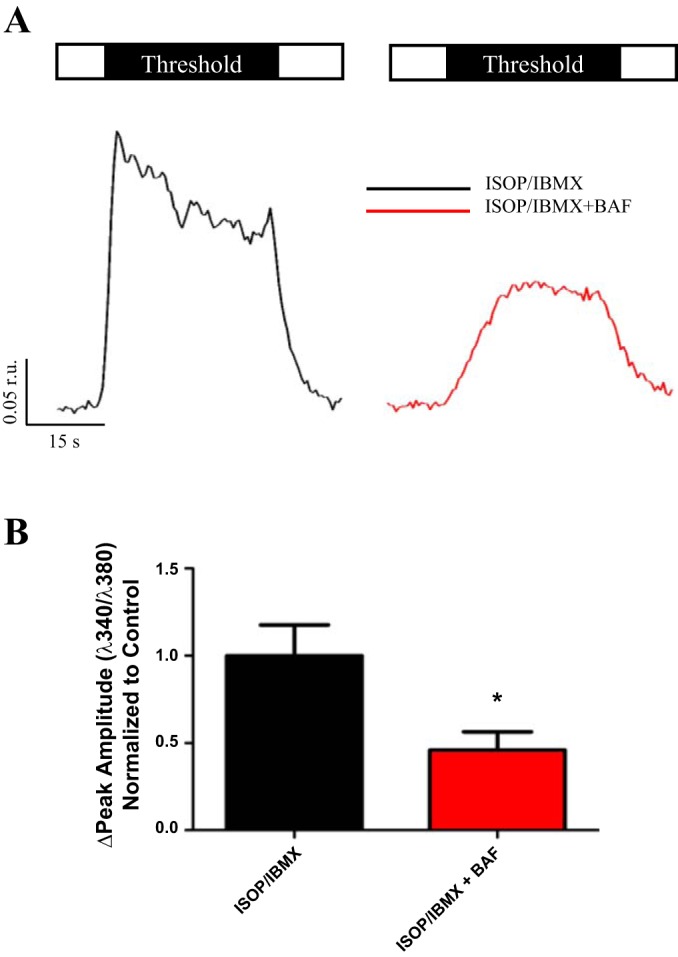
ISOP evoked elevations in cAMP. Open box represents saline, and black box indicates duration and concentration of CCh stimulus. Acini were stimulated at a threshold concentration of CCh following elevation of cAMP via ISOP/IBMX in the absence (A, black trace) or presence (A, red trace) of BAF pretreatment. Compared with ISOP/IBMX alone responses (0.27 ± 0.048 r.u., n = 13), there was a significant reduction in the peak amplitude of Ca2+ release following BAF treatment (0.13 ± 0.029 r.u., n = 11, *P = 0.020) at a threshold level of stimulation (B). Responses to maximal stimulation were not significantly different following BAF treatment.
Secretory granules are the relevant acidic organelle Ca2+ release site.
While use of BAF revealed a role for acidic Ca2+ stores in Ca2+ signaling, BAF treatment was relatively unselective and ubiquitously dissipated all the acidic stores. Thus additional functional studies were performed using more selective disruptors of acidic organelles to further probe the contribution of individual acidic compartments to the observable Ca2+ signal. To address whether secretory granules served as a predominant site for acidic store Ca2+ release, we treated acini with prolonged ISOP stimulation (1 h) to deplete granular stores. This degranulation was confirmed using live-cell calcein/LysoTracker staining (Fig. 5A). Ca2+ signals were assessed once again at threshold and maximal levels of cholinergic stimulation following extended ISOP treatment and compared with control acini that underwent the standard 5–10 min ISOP treatment. ISOP treatment for 1 h followed by stimulation at a threshold concentration of CCh produced a 66% diminishment in the peak amplitude of Ca2+ release (Fig. 5, B and C). Additionally, the rate of Ca2+ rise was significantly decreased following secretory granule depletion and the number of Ca2+ oscillations were also diminished at threshold cholinergic stimulation (Table 1). Secretory granule depletion had no significant effects on Ca2+ signaling dynamics when acini were stimulated at a maximal CCh concentration (Table 1).
Fig. 5.

Secretory granules appeared to be the dominant acidic organelle Ca2+ release site. Confocal images show acinar cells (A) labeled with LysoTracker (red, acidic organelles) and calcein (green, cytoplasm). Compared with a control treated cell, prolonged stimulation with ISOP for 1 h largely depleted the secretory granule population indicated by a nearly complete abolishment of the LysoTracker signal. Scale bar is 7.5 μm for control and 10 μm for ISOP 1 h. Following depletion of secretory granules by ISOP treatment for 1 h, acini were stimulated with a threshold concentration of CCh (B, red trace) and compared with acini treated with ISOP/IBMX for 10 min (B, black trace). Open box represents saline, and black box indicates duration and concentration of CCh stimulus. Granule depleted acini had diminished peak amplitudes (0.15 ± 0.027 r.u., n = 10) compared with controls (0.44 ± 0.034 r.u., n = 9, ***P = <0.0001) at threshold stimulation (C). Responses to maximal stimulation were unaffected. GPN-treated acini were stimulated with a threshold concentration of CCh (D, green trace) and compared with acini treated with ISOP/IBMX for 10 min (D, black trace). Lysosome-depleted acini at threshold stimulation (E) had peak amplitudes (0.44 ± 0.048 r.u., n = 12) that were not significantly different from control acini (0.45 ± 0.038 r.u., n = 9, P = 0.72). Responses to maximal stimulation were unaffected.
Imaging experiments indicated that lysosomes did not contribute significantly to the overall LysoTracker (or acidic organelle) signal. However, it was still important to assess the functional consequences of depleting lysosomal stores. In these experiments we used GPN in the presence of cathepsin inhibitor 1 to osmotically rupture lysosomes. Fura-2 AM loaded acini were then treated with ISOP/IBMX to elevate cAMP and stimulated with threshold and maximal concentrations of CCh. Compared with controls, GPN-treated acini did not exhibit significant changes in the peak amplitude of Ca2+ rise under threshold CCh stimulation (Fig. 5, D and E). This remained true for all examined analytical parameters at either CCh concentration (Table 1). Interestingly, there was a subtle but statistically significant increase in the resting 340/380 ratio following GPN treatment (1.4 to 1.6, P = 0.0019). However, whether this increase affected CCh-evoked Ca2+ responses was not determined. Therefore, the significant reduction in the Ca2+ signal following secretory granule depletion, coupled with minimal changes in Ca2+ signals observed in GPN-treated cells, identified secretory granules as a dominant site for acidic organelle Ca2+ release within parotid acini.
NAADP as a potential mediator of acidic store Ca2+ release.
Having demonstrated that acidic organelles participated in the enhancement of CCh-evoked Ca2+ signals at threshold levels of stimulation following cAMP elevation, we tested the role of a previously identified mediator of acidic store Ca2+ release-the potent small molecule NAADP. NAADP was first discovered in sea urchin eggs to target an IP3, cyclic ADP ribose insensitive Ca2+ store (38), eventually shown to be an acidic lysosomal-related organelle (19). Moreover, the putative enzyme that converts NADP to NAADP has been shown to be regulated by cAMP in other cell types (12, 54, 65). To test the hypothesis that NAADP contributed to Ca2+ release from acidic stores, parotid acini were incubated for 30 min with 10 μM NED19, a NAADP receptor antagonist, and stimulated with either a threshold or maximal concentration of CCh. Treatment with NED19 had no effect on Ca2+ dynamics in acini stimulated with CCh alone at either concentrations of cholinergic agonist compared with controls (Fig. 6, A and C, and Table 2). However, when ISOP was used to elevate cAMP, NED19 reduced the CCh-evoked peak Ca2+ release by 40% at threshold stimulation compared with cells treated with ISOP alone (Fig. 6, B and C). Similar to BAF pretreatment, the rate in which Ca2+ reached its peak amplitude was significantly slower in NED19/ISOP-treated acini (Table 2) at threshold CCh-evoked signals but without affect at maximal stimulation. The number of oscillations was unaffected at threshold and maximal CCh concentrations (Table 2). The significant NED19-induced decrease in the threshold Ca2+ response mimicked BAF treatment and was consistent with other studies that identified NAADP as a second messenger releasing Ca2+ from acidic organelles.
Fig. 6.

NED19-dependent inhibition of Ca2+ release. Open box represents saline, and black box indicates duration and concentration of CCh stimulus. Acini were stimulated at a threshold concentration of CCh with (A, red trace) or without (A, black trace) NED19 pretreatment. There was no change in the peak amplitude of Ca2+ release following NED19 treatment (0.13 ± 0.038 r.u., n = 10) when compared with control (0.17 ± 0.032 r.u., n = 14, P = 0.44) at a threshold level of stimulation (C). In contrast, following cAMP elevation in the absence (B, black trace) or presence (B, red trace) of NED19 pretreatment, there was a significant reduction in the peak amplitude (ISOP/IBMX, 0.14 ± 0.017 r.u., n = 13 vs. ISOP/IBMX + NED19, 0.088 ± 0.016, n = 13, *P = 0.026) of threshold responses (C).
Table 2.
NED19-dependent inhibition of Ca2+ release
| Treatment | Maximal Peak Amplitude, r.u. | Threshold Rate, r.u./s | Maximal Rate, r.u./s | Threshold Oscillations | Maximal Oscillations |
|---|---|---|---|---|---|
| CCh* | 1.1 ± 0.081 (n = 14) | 0.043 ± 0.011 (n = 14) | 0.52 ± 0.058 (n = 14) | 3.6 ± 0.68 (n = 14) | 4.9 ± 0.73 (n = 14) |
| CCh + NED19* | 0.95 ± 0.12 (n = 10) | 0.026 ± 0.010 (n = 10) | 0.49 ± 0.079 (n = 10) | 3.2 ± 0.63 (n = 10) | 4.7 ± 0.79 (n = 10) |
| ISOP/IBMX + CCh† | 0.97 ± 0.087 (n = 13) | 0.044 ± 0.012 (n = 13) | 0.62 ± 0.099 (n = 13) | 3.2 ± 0.78 (n = 13) | 4.9 ± 1.0 (n = 13) |
| ISOP/IBMX + NED19 + CCh† | 0.90 ± 0.069 (n = 13) | 0.016 ± 0.0049‡ (n = 13) | 0.41 ± 0.056 (n = 12 | 2.1 ± 0.60 (n = 13) | 3.1 ± 0.66 (n = 13) |
Note: threshold and maximal reference the concentration of CCh used to evoke a Ca2+ response.
NED19-treated acini had no significant differences between the listed parameters.
The rate of initial Ca2+ rise at threshold stimulation was significantly decreased in ISOP/IBMX + CCh + NED19-treated acini (‡P = 0.044) with no other observable differences.
Presence of acidic store Ca2+ release machinery.
Two-pore channels (TPCs) have been implicated as the lysosomal Ca2+ channel responsible for acidic store Ca2+ release (9, 13, 73). There are three isoforms of TPCs (TPC1-3) and RNA message for both TPC1 and TPC2 has been found in salivary tissue, although only the TPC2 protein has shown to be expressed. Using immunoflourescent methods and confocal microscopy we determined the subcellular distribution of TPC2. The acini clusters displayed punctate immunoreactivity for TPC2 (Fig. 7A) prominently along the lateral and apical membranes with very minimal staining basally. We next investigated redistribution of TPC2 following inducement of secretory granule exocytosis. To achieve this, cells were treated with 4 μM ISOP for 30 or 60 min and then stained for TPC2. Thirty minutes of ISOP treatment resulted in a gradual increase of TPC2 immunopositive staining (Fig. 7B); however, treatment for 60 min produced a robust increase in the signal of TPC2 along the apical and lateral membranes (Fig. 7C). Our results identify that TPC2 redistributes following prolonged ISOP treatment, suggesting TPC2 might be localized on secretory granules that are known to redistribute following exocytosis.
Fig. 7.

The presence of TPC2. TPC2 (green, A) was subcellularly distributed along the apical and lateral boarders of acinar cells, a pattern reminiscent of the LysoTracker staining and VAMP8 distribution. Attempts were made to determine whether TPC2 was redistributed following exocytosis. Cells were pretreated with ISOP for 30 min (B) or 1 h (C) before labeling. There was some accumulation of TPC2 along the apical and lateral boarders after ISOP treatment for 30 min (B, *) and robust accumulation following ISOP treatment for 1 h (C, *). All scale bars are 5 μm.
Downstream effects of disrupted acidic organelle Ca2+ release.
Our results show acidic organelle Ca2+ release contributes to an agonist-evoked global Ca2+ signal in parotid acinar cells. These Ca2+ signals are known to drive a variety of downstream processes such as control of the exocytotic release of salivary proteins and activation of Ca2+-regulated ion channels coordinating fluid transport. To explore whether acidic organelle Ca2+ release was important for regulating salivary protein release, experiments were performed to assess whether CCh + ISOP-induced exocytosis from acini was altered following pretreatment with 1 μM BAF for 1 h. To assess exocytotic function, we used a variety of techniques that included 1) time-differentiated imaging, 2) Texas Red dextran labeling, and 3) Western blotting for secreted amylase. To quantify data from time-differentiated imaging, the number of exocytotic events were normalized to the number of acini present during recording. During a threshold level of CCh stimulation (30 s) following pretreatment with ISOP/IBMX, acini displayed 1.52 events/cell, which was slightly albeit unsignificantly reduced in the presence of BAF (1.10 events/cell, Fig. 8A). Due to potential difficulties discerning changes in optical density, additional experiments measured exocytosis by uptake of a dextran conjugated to Texas Red. This dextran was used as a fluid phase probe that was likely incorporated via fusion pores during exocytosis (and endocytosis). To identify sites of exocytosis, control or treated cells were fixed and the exocytotic sites revealed by Texas Red fluorescence using confocal microscopy (Fig. 8B). Images were quantified and the percentage of Texas Red signal to total area of the cell was determined. At thresholds level of CCh stimulation in the presence of ISOP, acini had ∼8.5% of total area stained with Texas Red, which was not significantly different following BAF treatment (11.1%, Fig. 8C). A final set of experiments quantified amylase release with or without BAF pretreatment using Western blotting (Fig. 8D). Acini were stimulated with a high concentration of CCh in the presence of ISOP to promote maximal amylase release. CCh+ISOP stimulated acini secreted ∼51.5% of the total amylase content. Consistent with our other assessments of exocytotic activity, CCh + ISOP stimulated acini pretreated with BAF did not appear to have a significantly altered secretion of the total amylase content at 45.2%. Interestingly, although there was a significant effect on a global Ca2+ signal following disruption of acidic organelles it appeared this change did not alter the exocytotic profile. It is possible that acidic organelle Ca2+ signaling may selectively modulate salt secretion in parotid acini.
Fig. 8.

Acidic organelle Ca2+ release does not affect exocytosis. A: using time-differentiated imaging, disruption of acidic organelle Ca2+ signaling with BAF did not significantly diminish exocytosis at a threshold level of CCh stimulation following cAMP elevation with ISOP/IBMX (n = 9 for ISOP/IBMX + CCh, n = 8 for ISOP/IBMX + CCh + BAF, P = 0.3905). B and C: whereas there was an increase in the percent of Texas Red dextran labeling of acini when the cells were stimulated with ISOP and CCh indicating exocytotic activity (B, scale bars are 10 μm), BAF treatment did not diminish the agonist-evoked signal (C, n = 5 for all groups, P = 0.3762). D: consistent with imaging experiments, Western blotting for secreted amylase revealed that the percentage of total amylase released increased significantly following ISOP and CCh costimulation for both control and BAF-treated acini (n = 4 for both groups, P = 0.6943). Inset (D) is a representative Western blot for amylase. Control refers to ISOP + CCh-treated cells, and BAF-treated represents acini stimulated with ISOP + CCh following preincubation with BAF. S, secreted amylase; L, lysed amylase fraction.
DISCUSSION
Generation of Ca2+ signals is a complex and highly regulated process in terms of time, space, and converging messengers. To date, considerable effort has focused on intracellular Ca2+ signals that control the release of Ca2+ from the ER and ER resident Ca2+ channels. However, recent studies have begun to define the role of acidic organelles as a releasable Ca2+ store, thus adding an additional layer of regulation to an already inherently complex system.
In the current study, the identification, subcellular distribution, and contribution of acidic organelles to agonist-evoked Ca2+ dynamics was investigated in parotid salivary gland acinar cells. Acidic organelles were prominently localized to the apical membrane and subsequent immunofluorescent labeling showed these stores included secretory granules, lysosomes, and endosomes. A surprising and important finding of this study was a coincident signal in the form of cAMP was necessary to mobilize release of Ca2+ from acidic organelles and modulate Ca2+ signals evoked by threshold cholinergic stimulation. Threshold signaling represents the modest stimuli likely predominating under most physiological situations and may indicate acidic stores act as an important sensitizing factor for the initiation of saliva secretion. In contrast, we hypothesize that the contribution by acidic organelles during maximal CCh stimulation is overwhelmed by large-scale ER release and store operated Ca2+ entry important for sustained fluid production (1, 44–46, 53).
The observation of cAMP-dependent recruitment of acidic organelles in the parotid salivary gland aligns with the idea that specific agonists may produce specific signatures of Ca2+ signals (6, 14–16, 27, 29, 66–67). Previous work in pancreatic acini demonstrated that Ca2+ signaling patterns derived from the circulating hormone cholecystokinin were induced by NAADP (15–16, 67). On the other hand, acetylcholine-evoked Ca2+ release from a nonacidic pool (27, 66) and did not significantly elevate intracellular NAADP (67). We propose in parotid acinar cells that β-AR activation elevated NAADP and induced Ca2+ release from an acidic pool whereas CCh alone elicited signals were unaffected by BAF or NED19 treatment as IP3 production primarily released Ca2+ from a nonacidic, thapsigargin-sensitive store. Thus parotid acini, like pancreatic acini, can use agonist specific intracellular messengers to coordinate signal specificity or intensity.
Although the current study suggested acidic organelle Ca2+ release is initialized by a rise in NAADP following elevation of cAMP, the intermediaries of this pathway are unknown. A likely candidate for activation of the putative enzyme for NAADP production is PKA, shown previously to regulate ADP-ribosyl cyclase activity, a NAADP synthesizing enzyme (35, 54, 56). However, experiments using inhibitors of PKA, such as PKI, would presumably not only block acidic store Ca2+ release but also block PKA-dependent phosphorylation of IP3Rs. The type II IP3R subtype is the most abundant IP3R in parotid acinar cells (71) and in acini have been shown to be phosphorylated by PKA (11), which results in enhanced Ca2+ release. Thus we were unable using current approaches to distinguish the effects of PKI on acidic store Ca2+ release vs. PKI's dampening effect on IP3R phosphorylation. Another possibility is that cAMP can also activate proteins known as exchange proteins directly activated by cAMP (EPAC) that are known to regulate Rap1 (a guanine nucleotide exchange factor). Indeed, EPAC has been shown to sensitize r yanodine receptors to accelerate Ca2+ responses in pancreatic acini (55) and recent work by our lab showed EPAC activation can enhance Ca2+ signals evoked by purinergic receptor activation via a CICR mechanism (6).
Although localization of TPC2 in parotid acini to a region similar to acidic organelle distribution would suggest these proteins are candidates for mediating acidic organelle Ca2+ release, the literature is equivocal when it comes to whether TPCs are the primary site of action by NAADP. Work by Xu and colleagues (63) also argues that TPCs are in fact sodium, not Ca2+, conducting channels. Moreover, several studies demonstrated NAADP can mobilize Ca2+ through ryanodine receptors (26, 32, 47) or through closely related mucolipin channels of the acidic endolysosomal system (4, 69–70). Interestingly, demonstrating NAADP's binding site on TPCs remains elusive. An alternative hypothesis is that NAADP, through a binding partner (40), can regulate the variety of ion channels shown to be important for NAADP-induced Ca2+ signaling (30). Nevertheless, there is a general consensus that TPCs are important for NAADP-mediated Ca2+ release from an acidic pool.
Functionally, secretory granules appeared to be the predominant site of acidic organelle Ca2+ release as acini depleted of secretory granules produced diminished Ca2+ signals whereas osmotic rupturing of lysosomes had no discernable effect. However, the contribution of endosomes could not be directly assessed using current pharmacological approaches. Other groups have addressed the contribution of zymogen granules to a global Ca2+ signal by inducing depletion via intraperitoneal injection of ISOP (48) or parotid ductal ligation (41). Their results indicated there were no observable changes in a CCh-evoked Ca2+ response and suggested secretory granules likely were not key players in generating Ca2+ signals. However, the former study used CCh concentrations that produced robust rather than threshold Ca2+ signals. In contrast, our results support a role for acidic organelle Ca2+ release only at threshold stimulation and this signal is less important or can be bypassed at stronger stimulation. In the work by Liu et al. (41) experiments probed threshold CCh signaling, however, this group performed experiments in the absence of ISOP or other cAMP elevators. Taken together, we hypothesize secretory granules function as acidic Ca2+ stores that are mobilized for Ca2+ release following agonist-evoked rises in cAMP. We believe localized Ca2+ release can sensitize proximally located canonical channels, IP3Rs and ryanodine, allowing for enhanced release from the ER upon activation by IP3, consistent with the “trigger” model for acidic organelle Ca2+ release (15, 27).
Experiments revealed that although release of Ca2+ from acidic secretory granules enhanced the release from ER Ca2+ stores, the abolishment of this “trigger” using BAF did not result in any downstream changes to exocytosis. This finding was unexpected as studies in salivary gland acini demonstrated changes in pH to granules using weak bases (our experiments used BAF) affected the retention of proteins in mature secretory granules (59) and induced more basolateral secretion (34). Moreover, studies in T cells have shown that release of Ca2+ from acidic cytolytic granules through NAADP acts in an autocrine fashion to regulate release of the granules themselves (21–22). One caveat is that the contributions of acidic organelle Ca2+ release may have been overwhelmed by cAMP-dependent exocytosis which promotes the bulk of exocytosis in parotid acini.
The findings presented here may have clinical relevance for those suffering from Sjogren's Sydrome, an autoimmune disease characterized by dry mouth and dry eye. Early stage effects on the parotid include decreased cholinergic sensitivity (23, 36), a subsequent decrease of cholinergic-induced IP3 signaling, reduced Ca2+ release, and ultimately diminished saliva production i.e., chronic dry mouth. Interestingly, a recent publication by Teos et al. (57) also demonstrated IP3R decreases in Sjogren's patients that reduced CCh-evoked Ca2+ mobilization. Although the acidic organelle population in the minor salivary glands used in the Teos et al. study was unknown, our work suggests that Ca2+ release from acidic organelles could be selectively used as a pharmacological target to sensitize any residual IP3Rs to augment Ca2+ release to in turn increase fluid secretion, and reduce symptoms of dry mouth. The parotid is not the only exocrine gland targeted by Sjogren's. Initial results to test the acidic organelle population in submandibular and lacrimal glands showed differential staining patterns. Abundant and polarized acidic organelles were seen in lacrimal tissues while submandibular acini had a more diffuse pattern of labeling. This diffuse pattern of labeling may explain results from Harmer et al. (31), who demonstrated NAADP did not evoke a K+ or Cl− current response indicative of Ca2+ release, presumably because of the sparse acidic organelle population in submandibular acini. Future work is ongoing to characterize the functional recruitment of the abundant acidic organelles in lacrimal acini. Acidic organelles may have a key role in regulating fluid secretion and offer therapeutic targets to treat dry eye in addition to dry mouth.
GRANTS
This work was funded by National Institutes of Health National Institute of Dental and Craniofacial Research Grant DE-023418.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.F.I. and D.R.G. conception and design of research; J.F.I., S.B., S.K., A.W., P.G., and A.K.I. performed experiments; J.F.I., S.B., S.K., A.W., P.G., and A.K.I. analyzed data; J.F.I., S.B., S.K., A.W., P.G., and D.R.G. interpreted results of experiments; J.F.I. prepared figures; J.F.I. drafted manuscript; J.F.I., S.B., and D.R.G. edited and revised manuscript; J.F.I. and D.R.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Andrea Kalinoski in the Advanced Microscopy and Imaging Core at the University of Toledo Health Science Campus for assistance with the confocal microscope; Brooke Saepoo, Branden Stansley, and Carmen Mitchell for helpful scientific discussions; the University of Toledo Summer Under Graduate Research Fellowship for sponsoring A. Weiss; and a Medical Student Summer Research Fellowship to S. Khuder.
REFERENCES
- 1.Ambudkar IS. Regulation of calcium in salivary gland secretion. Crit Rev Oral Biol Med 11: 4–25, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Asking B. Sympathetic stimulation of amylase secretion during a parasympathetic background activity in the rat parotid gland. Acta Physiol Scand 124: 535–542, 1985. [DOI] [PubMed] [Google Scholar]
- 3.Baum BJ. Principles of saliva secretion. Ann NY Acad Sci 694: 17–23, 1993. [DOI] [PubMed] [Google Scholar]
- 4.Beck A, Kolisek M, Bagley LA, Fleig A, Penner R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J 20: 962–964, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 361: 315–325, 1993. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya S, Imbery JF, Ampem PT, Giovannucci DR. Crosstalk between purinergic receptors and canonical signaling pathways in the mouse salivary gland. Cell Calcium 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhattacharya S, Verrill DS, Carbone KM, Brown S, Yule DI, Giovannucci DR. Distinct contributions by ionotropic purinoceptor subtypes to ATP-evoked calcium signals in mouse parotid acinar cells. J Physiol 590: 2721–2737, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol 3: 122–131, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol 186: 201–209, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brailoiu E, Hoard JL, Filipeanu CM, Brailoiu GC, Dun SL, Patel S, Dun NJ. Nicotinic acid adenine dinucleotide phosphate potentiates neurite outgrowth. J Biol Chem 280: 5646–5650, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Bruce JI, Shuttleworth TJ, Giovannucci DR, Yule DI. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J Biol Chem 277: 1340–1348, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Bruzzone S, Moreschi I, Usai C, Guida L, Damonte G, Salis A, Scarfi S, Millo E, De Flora A, Zocchi E. Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc Natl Acad Sci USA 104: 5759–5764, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459: 596–600, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancela JM. Specific Ca2+ signaling evoked by cholecystokinin and acetylcholine: the roles of NAADP, cADPR, and IP3. Annu Rev Physiol 63: 99–117, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Cancela JM, Churchill GC, Galione A. Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature 398: 74–76, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV, Petersen OH. Two different but converging messenger pathways to intracellular Ca(2+) release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. EMBO J 19: 2549–2557, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 50: 279–290, 1998. [PubMed] [Google Scholar]
- 18.Chini EN, Beers KW, Dousa TP. Nicotinate adenine dinucleotide phosphate (NAADP) triggers a specific calcium release system in sea urchin eggs. J Biol Chem 270: 3216–3223, 1995. [DOI] [PubMed] [Google Scholar]
- 19.Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 111: 703–708, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Dai YS, Ambudkar IS, Horn VJ, Yeh CK, Kousvelari EE, Wall SJ, Li M, Yasuda RP, Wolfe BB, Baum BJ. Evidence that M3 muscarinic receptors in rat parotid gland couple to two second messenger systems. Am J Physiol Cell Physiol 261: C1063–C1073, 1991. [DOI] [PubMed] [Google Scholar]
- 21.Davis LC, Galione A. Cytolytic granules supply Ca(2+) for their own exocytosis via NAADP and resident two-pore channels. Commun Integr Biol 6: e24175, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis LC, Morgan AJ, Chen JL, Snead CM, Bloor-Young D, Shenderov E, Stanton-Humphreys MN, Conway SJ, Churchill GC, Parrington J, Cerundolo V, Galione A. NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr Biol 22: 2331–2337, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson LJ, Field EA, Harmer AR, Smith PM. Acetylcholine-evoked calcium mobilization and ion channel activation in human labial gland acinar cells from patients with primary Sjogren's syndrome. Clin Exp Immunol 124: 480–485, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekstrom J. Autonomic control of salivary secretion. Proc Finn Dent Soc 85: 323–331; discussion 361–323, 1989. [PubMed] [Google Scholar]
- 25.Emmelin N. Nerve interactions in salivary glands. J Dent Res 66: 509–517, 1987. [DOI] [PubMed] [Google Scholar]
- 26.Gerasimenko JV, Maruyama Y, Yano K, Dolman NJ, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol 163: 271–282, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerasimenko JV, Sherwood M, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J Cell Sci 119: 226–238, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Gill DL, Ghosh TK, Mullaney JM. Calcium signalling mechanisms in endoplasmic reticulum activated by inositol 1,4,5-trisphosphate and GTP. Cell Calcium 10: 363–374, 1989. [DOI] [PubMed] [Google Scholar]
- 29.Giovannucci DR, Groblewski GE, Sneyd J, Yule DI. Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. J Biol Chem 275: 33704–33711, 2000. [DOI] [PubMed] [Google Scholar]
- 30.Guse AH. Linking NAADP to ion channel activity: a unifying hypothesis. Sci Signal 5: pe18, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Harmer AR, Gallacher DV, Smith PM. Role of Ins(1,4,5)P3, cADP-ribose and nicotinic acid-adenine dinucleotide phosphate in Ca2+ signalling in mouse submandibular acinar cells. Biochem J 353: 555–560, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hohenegger M, Suko J, Gscheidlinger R, Drobny H, Zidar A. Nicotinic acid-adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem J 367: 423–431, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes AR, Takemura H, Putney JW Jr. Kinetics of inositol 1,4,5-trisphosphate and inositol cyclic 1:2,4,5-trisphosphate metabolism in intact rat parotid acinar cells. Relationship to calcium signalling. J Biol Chem 263: 10314–10319, 1988. [PubMed] [Google Scholar]
- 34.Hoque AT, Baccaglini L, Baum BJ. Hydroxychloroquine enhances the endocrine secretion of adenovirus-directed growth hormone from rat submandibular glands in vivo. Hum Gene Ther 12: 1333–1341, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Kim BJ, Park KH, Yim CY, Takasawa S, Okamoto H, Im MJ, Kim UH. Generation of nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 57: 868–878, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Kovacs L, Torok T, Bari F, Keri Z, Kovacs A, Makula E, Pokorny G. Impaired microvascular response to cholinergic stimuli in primary Sjogren's syndrome. Ann Rheum Dis 59: 48–53, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laniyonu A, Sliwinski-Lis E, Fleming N. Muscarinic M3 receptors are coupled to two signal transduction pathways in rat submandibular cells. Eur J Pharmacol 188: 171–174, 1990. [DOI] [PubMed] [Google Scholar]
- 38.Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem 270: 2152–2157, 1995. [DOI] [PubMed] [Google Scholar]
- 39.Lim D, Kyozuka K, Gragnaniello G, Carafoli E, Santella L. NAADP+ initiates the Ca2+ response during fertilization of starfish oocytes. FASEB J 15: 2257–2267, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, Marchant JS. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem 287: 2296–2307, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu P, Scott J, Smith PM. Intracellular calcium signalling in rat parotid acinar cells that lack secretory vesicles. Biochem J 330: 847–852, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masgrau R, Churchill GC, Morgan AJ, Ashcroft SJ, Galione A. NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic beta cells. Curr Biol 13: 247–251, 2003. [DOI] [PubMed] [Google Scholar]
- 43.McMillian MK, Soltoff SP, Lechleiter JD, Cantley LC, Talamo BR. Extracellular ATP increases free cytosolic calcium in rat parotid acinar cells. Differences from phospholipase C-linked receptor agonists. Biochem J 255: 291–300, 1988. [PMC free article] [PubMed] [Google Scholar]
- 44.Melvin JE, Koek L, Zhang GH. A capacitative Ca2+ influx is required for sustained fluid secretion in sublingual mucous acini. Am J Physiol Gastrointest Liver Physiol 261: G1043–G1050, 1991. [DOI] [PubMed] [Google Scholar]
- 45.Melvin JE, Yule D, Shuttleworth T, Begenisich T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol 67: 445–469, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Mertz LM, Horn VJ, Baum BJ, Ambudkar IS. Calcium entry in rat parotid acini: activation by carbachol and aluminum fluoride. Am J Physiol Cell Physiol 258: C654–C661, 1990. [DOI] [PubMed] [Google Scholar]
- 47.Mojzisova A, Krizanova O, Zacikova L, Kominkova V, Ondrias K. Effect of nicotinic acid adenine dinucleotide phosphate on ryanodine calcium release channel in heart. Pflügers Arch 441: 674–677, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Nezu A, Tanimura A, Morita T, Irie K, Yajima T, Tojyo Y. Evidence that zymogen granules do not function as an intracellular Ca2+ store for the generation of the Ca2+ signal in rat parotid acinar cells. Biochem J 363: 59–66, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel S, Docampo R. Acidic calcium stores open for business: expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol 20: 277–286, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen OH, Gerasimenko OV, Tepikin AV, Gerasimenko JV. Aberrant Ca(2+) signalling through acidic calcium stores in pancreatic acinar cells. Cell Calcium 50: 193–199, 2011. [DOI] [PubMed] [Google Scholar]
- 51.Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev 74: 595–636, 1994. [DOI] [PubMed] [Google Scholar]
- 52.Proctor GB, Carpenter GH. Regulation of salivary gland function by autonomic nerves. Auton Neurosci 133: 3–18, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Putney JW., Jr Identification of cellular activation mechanisms associated with salivary secretion. Annu Rev Physiol 48: 75–88, 1986. [DOI] [PubMed] [Google Scholar]
- 54.Rah SY, Mushtaq M, Nam TS, Kim SH, Kim UH. Generation of cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate by CD38 for Ca2+ signaling in interleukin-8-treated lymphokine-activated killer cells. J Biol Chem 285: 21877–21887, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah AU, Grant WM, Latif SU, Mannan ZM, Park AJ, Husain SZ. Cyclic AMP accelerates calcium waves in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 294: G1328–G1334, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song EK, Rah SY, Lee YR, Yoo CH, Kim YR, Yeom JH, Park KH, Kim JS, Kim UH, Han MK. Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport. J Biol Chem 286: 44480–44490, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teos LY, Zhang Y, Cotrim AP, Swaim W, Won JH, Ambrus J, Shen L, Bebris L, Grisius M, Jang SI, Yule DI, Ambudkar IS, Alevizos I. IP3R deficit underlies loss of salivary fluid secretion in Sjogren's Syndrome. Sci Rep 5: 13953, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner RJ, Sugiya H. Understanding salivary fluid and protein secretion. Oral Dis 8: 3–11, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Von Zastrow M, Castle AM, Castle JD. Ammonium chloride alters secretory protein sorting within the maturing exocrine storage compartment. J Biol Chem 264: 6566–6571, 1989. [PubMed] [Google Scholar]
- 60.Wagner LE 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol 586: 3577–3596, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagner LE 2nd, Li WH, Yule DI. Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2- splice variants. J Biol Chem 278: 45811–45817, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Wang CC, Shi H, Guo K, Ng CP, Li J, Gan BQ, Chien Liew H, Leinonen J, Rajaniemi H, Zhou ZH, Zeng Q, Hong W. VAMP8/endobrevin as a general vesicular SNARE for regulated exocytosis of the exocrine system. Mol Biol Cell 18: 1056–1063, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 151: 372–383, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watson EL, Abel PW, DiJulio D, Zeng W, Makoid M, Jacobson KL, Potter LT, Dowd FJ. Identification of muscarinic receptor subtypes in mouse parotid gland. Am J Physiol Cell Physiol 271: C905–C913, 1996. [DOI] [PubMed] [Google Scholar]
- 65.Wilson HL, Galione A. Differential regulation of nicotinic acid-adenine dinucleotide phosphate and cADP-ribose production by cAMP and cGMP. Biochem J 331: 837–843, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamasaki M, Masgrau R, Morgan AJ, Churchill GC, Patel S, Ashcroft SJ, Galione A. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J Biol Chem 279: 7234–7240, 2004. [DOI] [PubMed] [Google Scholar]
- 67.Yamasaki M, Thomas JM, Churchill GC, Garnham C, Lewis AM, Cancela JM, Patel S, Galione A. Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr Biol 15: 874–878, 2005. [DOI] [PubMed] [Google Scholar]
- 68.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev 57: 387–395, 2005. [DOI] [PubMed] [Google Scholar]
- 69.Zhang F, Li PL. Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J Biol Chem 282: 25259–25269, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Zhang F, Xu M, Han WQ, Li PL. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(−/−) cells. Am J Physiol Cell Physiol 301: C421–C430, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang X, Wen J, Bidasee KR, Besch HR Jr, Wojcikiewicz RJ, Lee B, and Rubin RP. Ryanodine and inositol trisphosphate receptors are differentially distributed and expressed in rat parotid gland. Biochem J 340: 519–527, 1999. [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu MX, Ma J, Parrington J, Calcraft PJ, Galione A, Evans AM. Calcium signaling via two-pore channels: local or global, that is the question. Am J Physiol Cell Physiol 298: C430–C441, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rotzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C. The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflügers Arch 458: 891–899, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]