

Abstract
Background
Peripheral blood leukocytes are the most commonly used surrogates to study epigenome-induced risk and epigenomic response to disease-related stress. We considered the hypothesis that the various classes of peripheral leukocytes differentially regulate the synthesis of 5-methylcytosine (5mCG) and its removal via Ten-Eleven Translocation (TET) dioxygenase catalyzed hydroxymethylation to 5-hydroxymethylcytosine (5hmCG), reflecting their responsiveness to environment. Although it is known that reductions in TET1 and/or TET2 activity lead to the over-proliferation of various leukocyte precursors in bone marrow and in development of chronic myelomonocytic leukemia and myeloproliferative neoplasms, the role of 5mCG hydroxymethylation in peripheral blood is less well studied.
Results
We developed simplified protocols to rapidly and reiteratively isolate non-overlapping leukocyte populations from a single small sample of fresh or frozen whole blood. Among peripheral leukocyte types we found extreme variation in the levels of transcripts encoding proteins involved in cytosine methylation (DNMT1, 3A, 3B), the turnover of 5mC by demethylation (TET1, 2, 3), and DNA repair (GADD45A, B, G) and in the global and gene-region-specific levels of DNA 5hmCG (CD4+ T cells ≫ CD14+ monocytes > CD16+ neutrophils > CD19+ B cells > CD56+ NK cells > Siglec8+ eosinophils > CD8+ T cells).
Conclusions
Our data taken together suggest a potential hierarchy of responsiveness among classes of leukocytes with CD4+, CD8+ T cells and CD14+ monocytes being the most distinctly poised for a rapid methylome response to physiological stress and disease.
Keywords: 5-Hydroxymethylcytosine, Surrogate cells, Disease, Epigenetic control, Epigenome-induced risk, Cellular memory
1. Background
Peripheral blood leukocytes are the most commonly used cell types to assess human disease states (Javierre et al., 2010; Huang et al., 2014; Di Francesco et al., 2015; Ellinger et al., 2015). Because of their accessibility, leukocytes are used in preference to other tissues such as the brain, muscle, adipose tissue, bone or various non-blood-borne cancer cells, even when these latter cell types are the focus of disease. As a result, the methylome and transcriptome of peripheral blood leukocytes often act as proxies for disease states centered in other tissues and cell types. One implicit assumption is that genetic and epigenetic reprogramming caused by disease states in other tissues are systemically reflected in blood-borne leukocytes. There are numerous published studies examining the response of genome-methylation in whole blood leukocytes to diseases and disease progression (Javierre et al., 2010; Wang et al., 2010; Weiss et al., 2011; Saied et al., 2012; Schroeder et al., 2012; Smith et al., 2012, 2014; Sun et al., 2013; Xu et al., 2013; Almen et al., 2014). However, the genome-wide methylation data from these studies comprises the epigenomic information of several major leukocyte cell types combined as a weighted average of their fractional representation in blood.
Epigenetics, from its modern inception, predicts that different cell types within a tissue or organ are epigenetically distinct (Nanney, 1958). Reinius et al. (2012) made pairwise comparisons of the methylomes of seven major leukocyte types to reveal that even the two most closely related peripheral blood mononuclear cells (PBMCs), CD4+/CD8± and CD8+/CD4± T cells, differ significantly in DNA methylation levels. Over 45,000 of the 485,000 cysteine-guanine dinucleotide (CG) sites measured (~9%), which are concentrated in gene regions, are distinct. Equally remarkable, the PBMC CD8+ T cells and granulocyte Siglec8+ eosinophils differ in approximately 193,000 of the CG sites measured (~40%) (Reinius et al., 2012). Different leukocyte types all have quantitatively different global methylation profiles, with the relatively hypomethylated granulocytes (Siglec8+ eosinophils, CD16+ neutrophils) showing 5 to 10 times less methylation across different gene regions than the hypermethylated PBMC lymphoid cell types (CD4+ and CD8+ T cells, CD19+ B cells, CD56+ NK cells, CD14+ monocytes) (Reinius et al., 2012). When these cell types are analyzed together in DNA methylation studies using total peripheral blood DNA, the methylation profiles from the seven leukocyte types with high and low levels of methylation, as well as their sequence specific differences in methylation, are obscured as a weighted averaged depending upon cell type frequency. Many statistically significant differences in the methylation profiles of individual cell types are lost, making the results from whole blood less intelligible and less meaningful than they would be from individual cell types.
In the Reinius et al. (2012) study they started with large whole blood samples, separated PBMCs and granulocytes by density centrifugation on Ficoll-Paque gradients and lysed the remaining red blood cells with NH4Cl. Seven leukocyte subtypes were then isolated from separate aliquots of the PBMC and granulocyte fractions by immuno-paramagnetic bead capture. Fluorescence activated multichannel cell sorting (FACS) can also be used to isolate various classes of leukocytes after Ficoll gradient separation (Roederer et al., 1997; Melzer et al., 2015). Herein, as a potentially less expensive and rapid alternative to these two methods we develop the approach of first lysing red blood cells by NH4Cl treatment or by freezing and thawing and then reiteratively isolating six or seven cell types on immuno-paramagnetic beads starting from a single small starting sample. A reiterative approach (Lyons et al., 2007) has the potential to eliminate or include specific cell types expressing overlapping markers in each isolated class, depending upon the order of isolation, and providing relatively pure distinct leukocyte populations for subsequent epigenetic analysis. The utility of examining non-overlapping populations of leukocytes is demonstrated herein by our own analysis of 5hmCG levels and supports the previous benefits of reiterative methods presented in Lyons et al. (2007).
Recent evidence suggests the gene sequence-specific differences in 5mCG levels at 20% of CG sites are tissue specific and may be important to changes in gene regulation, whereas 80% of global 5mCGs appear to have little impact on gene expression (Lister et al., 2013; Meagher, 2014; Wu and Zhang, 2014; Gu et al., 2016). By contrast, 5hmCG levels are much lower than 5mCG levels, but highly correlated with differential gene regulation (Mellen et al., 2012; Lister et al., 2013; Tsagaratou et al., 2014). 5hmCG is concentrated in euchromatin (Ficz, 2015), which has an open-chromatin conformation facilitating transcriptional regulation. Genes and gene regions enriched for 5hmCG are said to be “poised” to be differentially regulated (Pastor et al., 2011). For example, during chondrogenesis there is an increase in 5hmCG associated with important chondrogenic genes, while minimal changes in 5hmCG were observed in housekeeping genes (Taylor et al., 2016). Finally, 5hmCG enriched gene sets appear to be relatively distinct for each tissue (Nestor et al., 2012). Therefore, it is reasonable to propose that differences in gene region specific 5hmCG levels among peripheral blood might reflect their potential to respond to their environment with changes in gene expression.
We recently presented strong evidence that the majority of functional 5mCG sites, turn over rapidly (Fig. 1), with half-lives of less than an hour (Meagher, 2014). Many of the proteins essential to the establishment and maintenance of 5mCG (DNA methyltransferases, DNMT1, DNMT3A, DNMT3B), removal of 5mCG by oxidation to 5hmCG (Ten eleven translocation dioxygenase, TET1, 2, 3), and repair back to cytosine (Growth arrest and DNA damage induced, GADD45A, B, G) are expressed in leukocytes (Calabrese et al., 2014). TET catalyzed oxidation may be a rate-limiting step in 5mCG removal (Sabag et al., 2014; Wu and Zhang, 2014), and hence, the model presented in Fig. 1 emphasizes the central importance of TET activity to the turnover of modified cytosine and its potential impact on gene regulation.
Fig. 1.
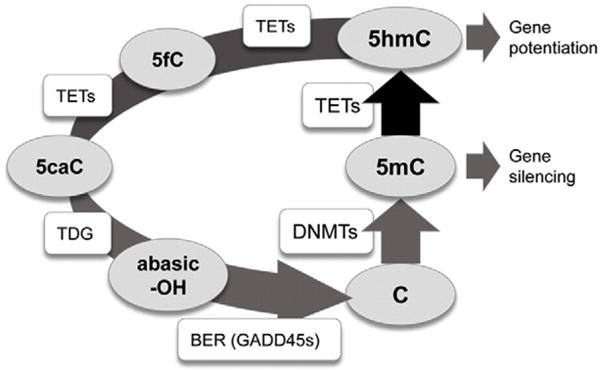
The dynamic modification cycle of DNA cytosine and its impact on gene activity. This model of the turnover of modified cytosine (C) residues emphasizes the central role of DNMTs in the methylation of C to 5-methylcytosine (5mC) and TETs in the rate limiting removal of 5mC by oxidation to 5-hydroxymethylcytosine (5hmC). The dynamic turnover of 5mC appears critical to regulating rapid changes in linked gene expression (Meagher, 2014; Wu and Zhang, 2014). TETs may further oxidize 5hmC to 5-formalcytosine (5fC) and 5-carboxycytosine (5caC). Thymine DNA glycosidase TDG removes the modified 5fC or 5caC bases leaving an abasic nucleotide (–OH). Base excision repair (BER) repairs the single nucleotide gap in double stranded DNA back to a C residue. Enzymes are in square boxes and nucleotide bases are in ovals. The diagram was modified from (Kohli and Zhang, 2013), based on the data in (Chen et al., 2012; Ramon et al., 2012; Dubois-Chevalier et al., 2014; Haseeb et al., 2014; Oger et al., 2014).
There are limited data showing the importance of the DNA methylation cycle (Fig. 1) to peripheral blood leukocytes. TET enzyme oxidation of 5mCG to 5hmCG is critical in T cell development as well as in the expression of T cell lineage specific genes (Tsagaratou et al., 2014; Ichiyama et al., 2015). RNAi mediated silencing of TET2 in cord blood progenitor CD34+ cells lowers 5hmCG levels and skews differentiation toward granulocytes and away from lymphoid and erythroid lineages (Pronier et al., 2011). When both TET2 and TET3 are mutated in zebrafish, there are reductions in the number of hematopoietic stem cells emerging during embryonic development (Li et al., 2015). Furthermore, TET2 mutations are implicated in the development of T-cell lymphomas, myeloproliferation, and myeloid malignancies, where bone marrow precursor cells are affected (Moran-Crusio et al., 2011; Pronier et al., 2011; Muto et al., 2014). Although the relationship of the cytosine modification cycle to health is limited, it is reasonable to consider that each of the divergent leukocyte lineages may regulate the cycle differently.
The roles for TETs and 5hmCG in leukocyte development led us to hypothesize that various classes of peripheral leukocytes differentially regulate the establishment of 5mCG and its removal via oxidation to 5hmCG (Fig. 1). These and other data also point to potential cause-and-effect relationships, that these differences in sequence specific 5mCG and 5hmCG impart to each cell type more or less potential to respond to physiological stresses and disease and in a cell type specific manner. We looked for initial evidence that non-overlapping leukocyte classes, isolated by our reiterative isolation protocol, might vary in the expression of machinery controlling the rates of 5mCG turnover, through changes in their DNA cytosine hydroxymethylome. Our results identify CD4+ T cells and CD14+ monocytes as having the highest levels of 5hmCG, but identified CD8+ T cells as having the highest levels of TET gene expression that might reflect turnover rates.
2. Results
2.1. Isolation of cell populations
After a number of preliminary studies, we developed three different isolation methods to successively and rapidly isolate a few to seven leukocyte types (helper T cells, inflammatory T cells, monocytes, neutrophils, B cells, natural killer cells, and eosinophils) from single 5 ml samples of fresh or frozen whole blood as summarized in Fig. 2. The three methods included: (1) the isolation of CD4+ T cells, CD8+ T cells, and CD14+ monocytes directly from whole fresh blood, (2) the isolation of six or seven leukocyte types from whole blood using prior red blood cell lysis, and (3) the isolation of six or seven leukocyte types from frozen whole blood.
Fig. 2.
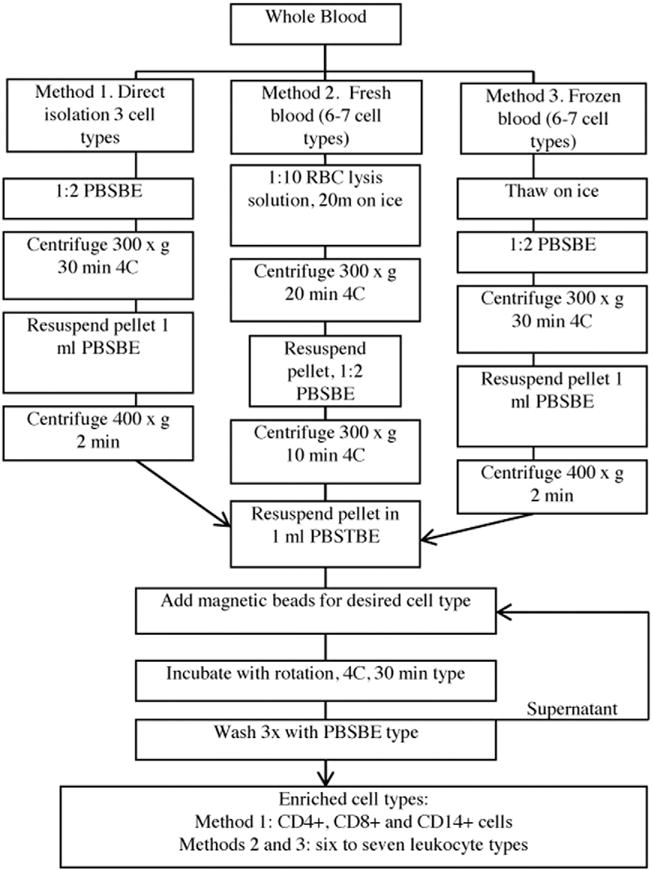
Description of isolation protocols. Graphical outline of the three isolation methods (1, 2, 3) each starting with 5 ml of peripheral blood.
In determining the order of isolation that would yield the purest samples of the seven leukocyte types, we had to consider that each of the seven leukocyte populations are complex and often express more than one of the common plasma membrane antigens (PMAs) used to isolate each population (Supplemental Table 1). Our results represent an attempt to optimize isolation of defined leukocyte populations free of unwanted cell types without seriously compromising the recovery of cell types. Three different orders of isolation were identified, where isolation order A was used for isolation method 1 and isolation order B was found to yield the purest cell populations for the isolated cell types (methods 2 and 3) with the exception of one cell type, NK cells. Isolation order C resulted in relatively pure populations of some of the leukocyte types (e.g., CD16+ neutrophils), but not others, and is shown to highlight the importance of the order of isolation in recovering pure cell populations.
The efficiencies of recovery of leukocyte types from each isolation method are estimated in Table 1. Method 1 produced the highest recovery of CD4+ T cells and CD14+ monocytes while Method 2 generated the highest recovery of CD8+ T cells, CD16+ neutrophils, CD19+ B cells, CD56+ NK cells, and Siglec8+ eosinophils. In general there was a 30 to 80% reduction in recovery depending upon leukocyte types for Method 3, resulting from cell lysis during the freeze-thaw process.
Table 1.
Efficiency of recovery of each isolation method.
| CD4+ T cells | CD8+ T cells | CD14+ monocytes | CD16+ neutrophils | CD19+B cells | CD56+ NK cells | Siglec8+ eosinophils | |
|---|---|---|---|---|---|---|---|
| Method | |||||||
| 1 | 72,300 ± 6500a | 34,500 ± 7400a,b | 11,100 ± 1600a | NR | NR | NR | N |
| 2 | 44,900 ± 4700b | 39,300 ± 10,600a | 3500 ± 1600b | 211,000 ± 53,500a | 3900 ± 800a | 54,300* ± 11,100a | 6400 ± 2900a |
| 3 | 13,800 ± 2100c | 7100 ± 2300b | 4800 ± 1500a,b | 43,200 ± 6700b | 2800 ± 900a | 14,100* ± 4500b | 3600 ± 200a |
Numbers of cells recovered from each isolation method starting with 5 ml of blood reported as Mean ± SEM (N = 3). Fresh blood with RBC lysis and frozen blood protocols were isolated following isolation order A, except *CD56+ cells which were isolated directly from white blood cells isolated with the respective method, as they are unable to be recovered in isolation order A. NR: Not Recovered. A one-way ANOVA followed by Tukey HSD post hoc was performed between methods 1, 2 and 3 for the three cell types where cells were recovered in all three methods. A two tailed t test was performed between methods 2 and 3 for the four cell types where cells were only recovered in these methods. Significant differences between the different methods for each of the seven cell types are designated by having different letters.
2.1.1. Confirmation of purity of isolated cell types
Initial analysis of the purity of the seven isolated cell types was performed using the four distinct nuclear morphologies (round for CD4+, CD8+, CD19+, CD56+ cells; kidney shaped for CD14+ and CD56+ cells; multilobular for CD16+ cells; bilobed for Siglec8+ cells) of peripheral blood leukocytes (Alberts et al., 1994). Purity was assessed based on the absence of three uncharacteristic nuclear phenotypes for six cell types with relatively unique morphologies. For CD56+ NK cells the estimate is based on the absence of two nuclear morphologies, multilobed and bilobed. The fluorescent nuclear morphology we observed for the seven isolated leukocyte types after staining with DAPI and propidium iodide (PI) are shown in Fig. 3. After examining several fields of cells by fluorescent microscopy, each cell type isolated by the methods and isolation orders described in Fig. 2 and Table 1, were found to be at least 95% pure, thus meeting our criteria to be initially considered as a highly enriched cell population pending qRT-PCR transcript analysis. Some of the isolated cell types were bound to numerous dynabeads during the isolation process, making the nuclear morphology of these cells difficult to assess. Cells with obscured nuclear morphologies are not included in our estimates of purity. Purity was further confirmed through qRT-PCR analysis of leukocyte-specific transcripts.
Fig. 3.
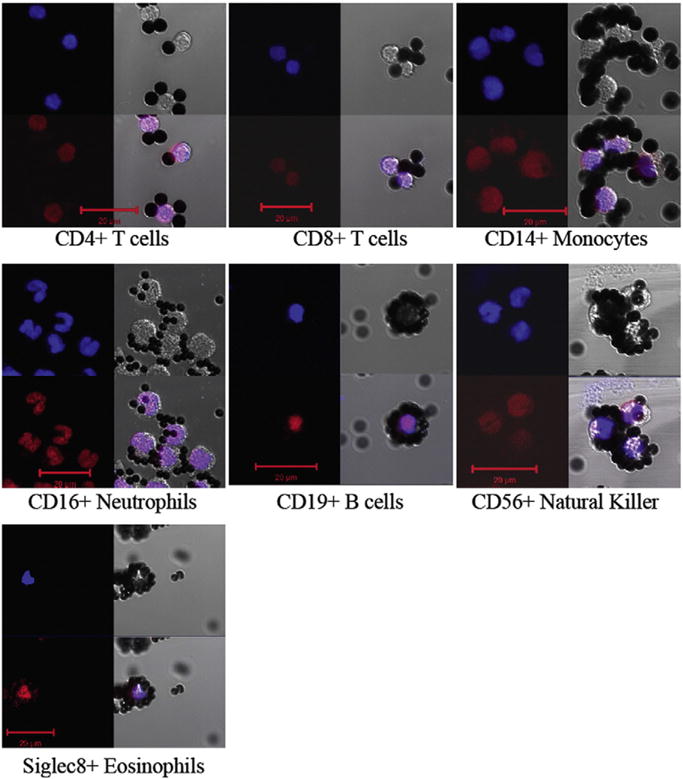
Nuclear morphologies of isolated cell types. Isolated leukocytes, bound to Dynabeads were stained with: DAPI (upper left blue) and PI (lower left red) and photographed by fluorescence (left) and DIC microscopy (upper right) of each of the seven panels. The three images were merged to yield the image in the lower right. Scale bar = 20 μm.
Most studies of isolated leukocytes use cytometry to demonstrate that the cell types express common PMAs consistent with expectations (i.e., CD4+ T cells are CD4+/CD3+/CD14−) (Reinius et al., 2012). However, this approach does not quantitatively address the level of contamination from other unwanted cell types, without the use of many other marker antibodies during cytometry. The binding of multiple large Dynabeads to cells complicates cytometry and bead removal may damage cells. Therefore, we developed qRT-PCR assays using a panel of eight leukocyte-specific marker transcripts to assess the purity of the seven isolated cell types. The relative quantity (RQ) of expression of each transcript in each cell type is shown in Supplemental Fig. 1A (isolation order B, yielding the most pure leukocyte populations) and isolation order C (shown to demonstrate the importance of isolation order) in Supplemental Fig. 1B. Unexpectedly high levels of CD4 mRNA were observed in CD14+ monocytes and CD16+ neutrophils (Supplemental Fig. 1A). Therefore, IFM was used to assess CD4 protein localization in these cell types and in CD4+ T cells (Supplemental Fig. 2). We found CD4 protein in the membrane of isolated CD4 cells, but only in the cytoplasm of CD14+ monocytes and CD16+ neutrophils. The results of this analysis provided evidence that the CD4 protein is only trafficked to the membrane in the CD4+ T cells. Considering the IFM analysis of nuclear morphology showed no round nuclei in either the isolated monocytes or neutrophils, we can conclude that there is no contamination of CD4+ T cells in these isolated cell populations.
As shown in Supplemental Fig. 1, NK cells captured using the CD56+ antigen express transcripts for many of the common PMAs used for cell isolation (Supplemental Table 1), and thus represent a heterogeneous population of cells (Kelly-Rogers et al., 2006; Poli et al., 2009). If the order of the isolation is manipulated to isolate CD56+ NK cells last, very few cells remain, as the majority of CD56+ cells have been isolated in the other cell populations as sub-types of these cell populations. This is due to the co-expression of CD16 (Kelly-Rogers et al., 2006; Poli et al., 2009; Accomando et al., 2014), CD4 (Kelly-Rogers et al., 2006; Zloza and Al-Harthi, 2006; Gruenbacher et al., 2009), CD8 (Kelly-Rogers et al., 2006; Zloza and Al-Harthi, 2006; Gruenbacher et al., 2009), and CD14 (Gruenbacher et al., 2009) surface markers in some CD56+ NK cells. For the cells assayed in Supplemental Fig. 1A, CD56+ cells were isolated first, hence, CD56+ cells expressing all these other markers were included.
2.2. Utilization of isolated leukocytes for epigenetic analysis
Based on the purity of the isolated cell populations as defined by nuclear morphology and the expression of cell type specific transcripts, isolation order B was selected and used for the remaining experimental analysis in this manuscript, with the exception of only the first six cell types were isolated following this order, while CD56+ NK cells were isolated in parallel from an additional blood sample.
2.2.1. 5hmCG levels among leukocytes
To explore the differences in 5hmCG levels and gene-region specific distribution of 5hmCG among leukocytes, we performed a TET assisted bisulfite sequencing (TAB-Seq) genome wide analysis on DNA isolated from each of the seven isolated cell types. The majority of the 5hmCG modification is found in the CG dinucleotide context, resulting from the specificity of TET enzymes and associated DNA binding machinery to CG dinucleotides (Lister et al., 2013; Wu and Zhang, 2014). And thus, we first quantified the overall level of 5hmCG in each of the cell types (Table 2) and found that CD4+ T cells had by far the highest level of 5hmCG at CG dinucleotides: 3.67% of CG sites assayed contained a 5hmCG. Monocytes, neutrophils, and B cells had intermediate levels of 5hmCG: 2.69%, 2.62% and 2.38%, respectively. NK cells, eosinophils and CD8+ T cells had the lowest levels with 2.12%, 1.99%, and 1.91%, respectively. These values were based on the analysis of ~10 × 106 different CG dinucleotides from each cell type.
Table 2.
5hmC levels are distinct among the seven classes of peripheral leukocytes.
| Leukocyte type | Total 5hmCG sites | Total CG sites | % 5hmCG | Scaled % 5hmCG |
|---|---|---|---|---|
| CD4+ T cell | 268,707 | 12,379,005 | 2.17% | 3.67% |
| CD14+ monocyte | 155,335 | 10,332,947 | 1.50% | 2.69% |
| CD16+ neutrophil | 176,386 | 12,621,471 | 1.40% | 2.62% |
| CD19+ B cell | 144,885 | 10,154,660 | 1.43% | 2.38% |
| CD56+ NK cell | 174,264 | 12,397,889 | 1.41% | 2.12% |
| Siglec8+ eosinophil | 117,257 | 8,357,101 | 1.40% | 1.99% |
| CD8+ T cell | 158,512 | 13,045,103 | 1.22% | 1.91% |
Quantification of 5hmC levels from TAB-Seq data demonstrated a modestly wide range of 5hmC levels among the peripheral leukocytes. See the Materials and methods section.
As an independent confirmation of this range of 5hmCG levels among classes of leukocytes we performed immuno-fluorescent microscopy (IFM) (Fig. 4) of 5hmC in preparations of total leukocytes costained with DAPI for DNA. Preparations of leukocytes where prepared from both fresh (method 2, Fig. 4A) and frozen (method 3, Fig. 4B) blood to determine the impact of using freezing on recovery. Observation of the lowest levels of 5hmC in some cells required longer photographic exposures and a further assessment of antibody reagent controls (Supplemental Fig. 3), however all leukocyte nuclei, as identified by DAPI staining (Supplemental Fig. 3A), expressed 5hmC (Supplemental Fig. 3B–C) in varying degrees after this assessment, agreeing with our TAB-Seq data. After quantification of the 5hmC signal of at least 100 cells of each of the four distinct nuclear morphologies (round: T cells, B cells and NK cells; kidney: monocytes and NK cells; multilobed: neutrophils; bilobed: eosinophils) the 5hmC signal for each morphology was classified as minimal, low, medium or high (Fig. 4C). A two-way MANOVA revealed that freezing had no significant effect on the distribution of 5hmC signal among nuclear types. However, as expected, the various nuclear morphologies were a significant determinant of 5hmC levels (p = 0.001). In particular, among nuclei with the high 5hmC signal, 92.7% of the variance in 5hmC levels was explained by the different nuclear morphologies (p = 2.07 × 10−7) with the kidney and round morphologies much more commonly represented than the others. Furthermore, nuclear morphology significantly impacted 5hmC signal in the medium, low and minimal groups (p < 0.05, explaining 61.6%, 76.3%, and 45.5% of the variance in 5hmC signal respectively). However, all the different nuclear morphologies were represented in these groups. The differences identified were similar to the TAB-seq analysis, where the CD4+ T cells with round nuclei and monocytes with kidney shaped nuclei have the highest level of 5hmC, and lower levels of 5hmC are found among all nuclear morphologies (i.e. all cell types).
Fig. 4.
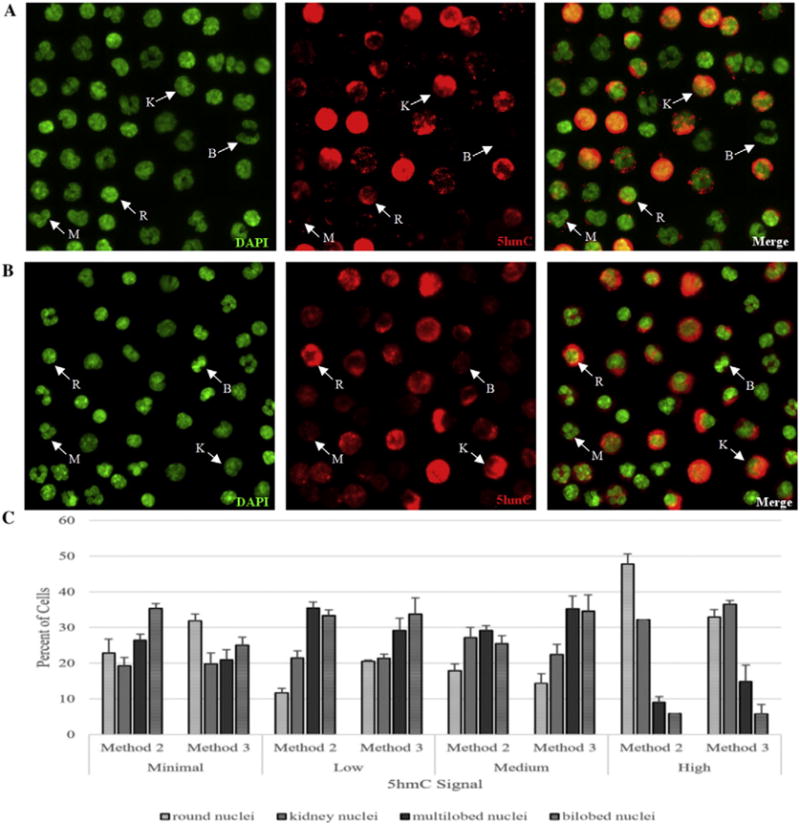
Immuno-fluorescent analysis showed a wide distribution of 5hmC levels among various classes of leukocytes. A. Total human leukocyte fraction from fresh peripheral blood (method 2) were labeled with DAPI for DNA (fluorescent green), primary antibody to 5hmC and secondary R-PE (red fluorescence), and then the merged image of DAPI and 5hmC is also presented. The same field of cells is shown in all images. B. Total human leukocyte fraction from frozen peripheral blood (method 3) were labeled with DAPI for DNA (fluorescent green), primary antibody to 5hmC and secondary R-PE (red fluorescence), and then the merged image of DAPI and 5hmC is also presented. The same field of cells is shown in all images. Example cells are labeled based on nuclear morphologies. K: kidney shaped (monocytes or natural killer cells), R: Round (T cells and B cells), M: Multilobed (neutrophils), B: Bilobed (eosinophils). C. 5hmC signal was quantified, and categorized as minimal, low, medium or high for each of the nuclear morphologies in each isolation method (fresh blood: method 2, frozen blood: method 3) and the percent of cells for each nuclear morphology was plotted for each 5hmC signal. Error bars represent standard error of the mean.
2.2.2. Gene-region specific 5hmCG distribution
We next assessed the gene-region specific 5hmCG profile of the seven different leukocyte types (Fig. 5). Three regions were defined for all genes, 100 kb upstream of the transcription start site (TSS), the gene body (i.e., start to stop of transcript) and 100 kb downstream of the transcription stop site (TTS). The gene regions examined are the same as those used to examine gene region distribution of 5hmCG in brain (Lister et al., 2013). 18,000 transcripts detected in leukocytes (Palmer et al., 2006) were broken into five expression quintiles (5 of 5 representing transcripts with the highest steady state level and 1 of 5 representing transcripts with the lowest levels). All of the leukocyte types had roughly the same pattern of gene-region 5hmCG distribution per transcript expression quintile with the most highly expressed transcripts having the highest levels of 5hmCG, with decreasing 5hmCG correlating with decreasing quintile of expression (Fig. 5B). Although the highest expression quintile for each cell type had the highest level of 5hmCG, the absolute level was different for each cell type. CD4+ T cells had by far the highest levels of 5hmCG with a peak at 4.53% and CD8+ T cells had the lowest levels of 5hmCG with a peak at 2.57% (Fig. 5B). The relative differences among cell types held true across gene location within each quintile of expression, where for the highest expression transcript quintile, the level of 5hmCG was highest in the CD4+ T cells and lowest in the CD8+ T cells following the same pattern as the global levels presented in Table 2. The highest quintile data are shown in Fig. 5C and lower quintile data in Supplemental Fig. 4. The highest 5hmCG peak for each cell type and transcript expression quintiles occurred immediately after the TSS, at the beginning of the gene body, following a valley of 5hmCG just prior to the TSS site. A dip in the level of 5hmCG across all cell types and expression quintiles can be seen at the end of the gene body with a second smaller peak of 5hmCG at or near the TTS (Fig. 5B).
Fig. 5.
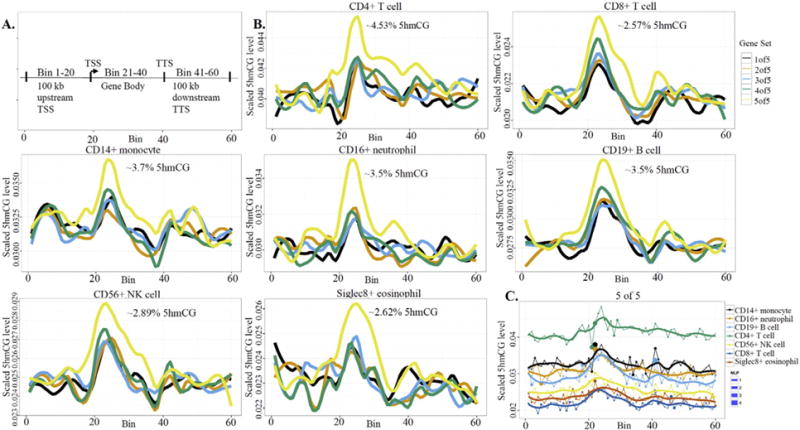
Gene-region-specific 5hmCG levels are distinct among the peripheral leukocyte types and vary by transcript level. A. Map defining the three gene regions assayed (Lister et al., 2013). B. 5hmCG levels were plotted for each of the seven leukocyte types by quintile of transcript expression level. The peak percentage of 5hmCG relative to total CG content for each cell type is estimated at the top of each graph. C. The 5hmCG levels for the quintile of the highest quintile of transcript for each cell type was plotted together. The dots plotted with each line on the graph represent the degree of change from the previous regions level of 5hmCG to the current level. The figure legend to the right of panel C shows the varying levels of significance of this change as determined by NLP. The relative position of each cell type is the same when these data are plotted for the other 4 quintiles of transcript expression (Supplemental Fig. 4).
We then examined the differences in the levels of 5hmCG for several different leukocyte-relevant gene-ontology (GO) term categories (Fig. 6). For GO category analysis each gene was divided into seven smaller gene sequence locations (Fig. 6) than used in Fig. 5. This analysis allowed us to examine both the total variance in 5hmCG levels in all cell types assayed for each GO term, and also assess how different each cell type is from all the others in each gene sequence location. The total leukocyte data for all but two of the GO term based gene lists (Adaptive Immune Response and Leukocyte Migration) showed a dip in 5hmCG levels around the TTS, in agreement with this analysis of larger gene regions in Fig. 5. By pooling the exons into one single bin in the box plot we no longer resolve the spike in 5hmCG at the start of the gene body (Fig. 5). With a few exceptions, the two regions immediately flanking the TTS had some of the highest levels of 5hmCG for all leukocyte types, which was obscured when we looked at the larger downstream region in Fig. 5.
Fig. 6.
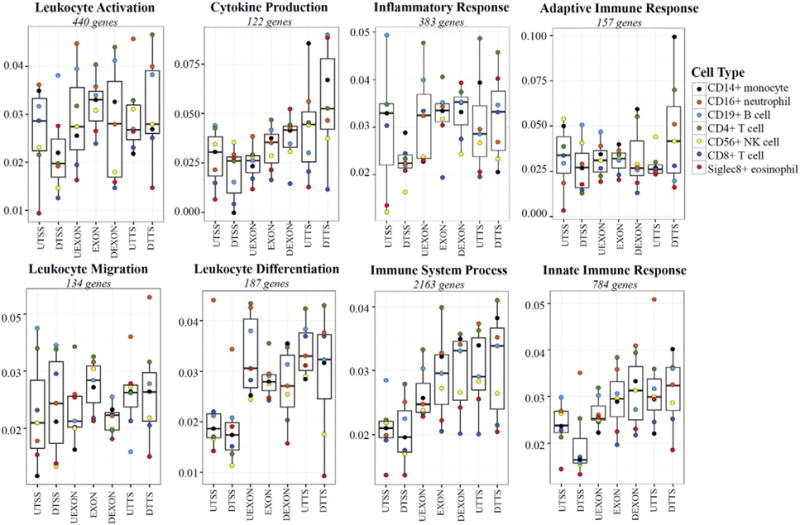
Gene-sequence-specific distribution of 5hmCG levels in the peripheral leukocytes for relevant GO term gene lists. The fraction of 5hmCG relative to all CG dinucleotides for seven gene sequence locations in all leukocytes (100 bp upstream of the TSS (UTSS), 100 bp downstream TSS (DTSS), 100 bp upstream of all exons (UEXON), within exons (EXON), 100 bp downstream of exons (DEXON), 100 bp upstream TTS (UTTS), and 100 bp downstream TTS (DTTS)) were plotted for different GO terms related to leukocyte function as box plots with the bar representing the median, and the box extending from the 25th to 75th percentiles. The whiskers represent ±1.5 times the interquartile range. Within each box plot the weighted average of 5hmCG for each leukocyte type was plotted as a colored dot, while the box is the weighted average for all 7 cell types. Similarly plotted data for several other genes grouped by GO terms are presented in Supplemental Fig. 5.
When the differences in the weighted average of 5hmCG for all genes in the GO term list for each cell type are examined in relation to the 5hmCG levels observed in all leukocytes, striking differences from the total leukocyte population emerge. As an example, for the Leukocyte Differentiation genes, CD16+ neutrophils had much higher weighted average of 5hmCG levels in the two regions around the TSS, far exceeding 1.5 times the interquartile range (IQR) for this region of the 5hmCG levels observed in the total leukocytes (Fig. 6). As an additional example, CD4+ and CD8+ T cells also distinguish themselves from the other leukocyte types with much higher and much lower levels of 5hmCG, respectively, within exons of Inflammatory Response genes (Fig. 6).
Not only are there differences among cell types for the different GO terms, but there are also differences in the level of 5hmCG for total leukocytes across GO terms. Most of the GO terms related to leukocyte function had variable levels of 5hmCG across the different gene regions assayed, with the exception of the Adaptive Immune Response gene list, where the level of 5hmCG is relatively constant across gene regions with the exception of the 100 nt downstream of the TTS, where levels of 5hmCG are three times higher (Fig. 6). The peak level of 5hmCG across these smaller gene regions among the gene lists are variable with some reaching more than 7.5% (Cytokine Production and Adaptive Immune Response), while others only reach ~4% (Immune System Process, Leukocyte Differentiation, and Leukocyte Activation). Additional GO terms related to general cell function were also examined and are presented in Supplemental Fig. 5.
It seemed possible that any one of three factors (1) the genomic bin (the gene-region specific location), (2) the cell type, and/or (3) the GO term-based gene set might account disproportionately for the variance in our data, weighting its biological relevance. Therefore, we examined the variance in 5hmCG levels separately for these three factors (Supplemental Fig. 6). For all leukocyte types, the gene-region specific location and the cell type explained approximately 31% of the variance in 5hmCG levels, ~16% and ~15%, respectively. The different gene ontology gene lists explained less (~7%) of the variance in 5hmCG levels. Hence, there are many factors controlling 5hmCG levels and its gene-region distribution in leukocytes.
2.2.3. Expression of factors involved in regulation of DNA 5hmCG levels in peripheral leukocyte types
We used qRT-PCR to determine the relative quantity (RQ) of each transcript for nine factors involved in the DNA cytosine methylation cycle (DNMT1, 3A and 3B, TET1, 2, and 3, GADD45A, B, and G) as shown in Fig. 7. DNMT1 was 8- to 16-fold more highly expressed in the CD4+ and CD8+ T cells (p < 0.0005), while DNMT3A and 3B were 8- and 4-fold more highly expressed in the CD4+ T cells (p < 0.0005) and CD8+ T cells (p < 0.05), respectively, than in the 5 other cells types (Fig. 7A). TET1 was 50-fold more highly expressed in the two T cell types than other leukocytes (p < 0.0005). TET2 was 10- to 50-fold higher in the CD8+ T cells and eosinophils than in 4 other cell types (p < 0.005). TET3 was most highly expressed in eosinophils, monocytes, and CD4+ T cells than the other leukocyte types (p < 0.05) (Fig. 7B). GADD45A was 50-fold higher in CD8+ T cells than in all other cell types (p < 0.0005). GADD45B was 8- to 16-fold higher in both T cell populations relative to all other cell types (p < 0.0005), except for eosinophils, where it was modestly expressed. GADD45G was 15-fold higher in the CD4+ T cells than in the other cell types (p < 0.0005), again with exception of modest expression in eosinophils (Fig. 7C). In summary, there are dramatically different expression profiles for many of the enzymes central to the turnover of modified cytosine residues (Fig. 1) among classes of leukocytes.
Fig. 7.
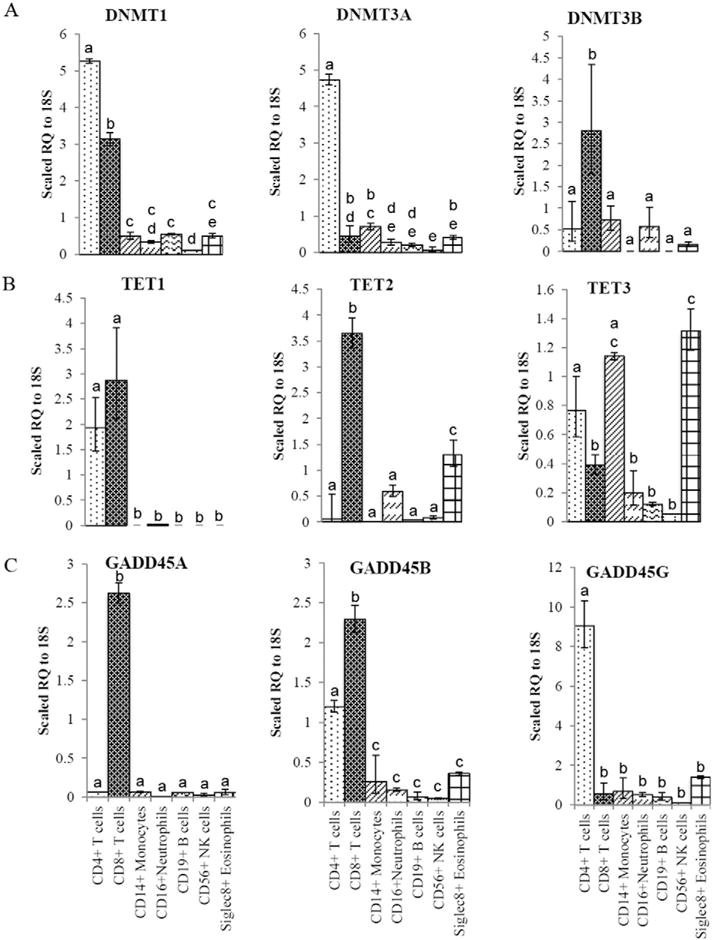
Expression of transcripts encoding enzymes involved in the establishment and removal of modified DNA cytosine. A–C. qRT-PCR analysis of the relative transcript expression was performed on cDNA prepared from seven leukocyte types. Values are expressed as a scaled Relative Quantity (RQ) of transcript in each cell type using the dCT method. Letters designate significant differences of at least p < 0.05. A. Analysis of transcripts for DNMTs (Fig. 1). The RQ value for each cell type is presented as a scaled value of 104, 105, and 106 times their RQ value for DNMT1, DNMT3A, and DNMT3B, respectively. B. Analysis of transcripts for TETs (Fig. 1). The RQ value for each cell type is presented as a scaled value of 106, 105, and 105 times their RQ value for TET1, TET2, and TET3, respectively. C. Analysis of transcripts for GADD45s (Fig. 1). The RQ value for each cell type is presented as a scaled value of 105, 105, and 106 times their RQ value for GADD45A, GADD45B, and GADD45G, respectively.
3. Discussion
3.1. Isolating sub-populations of leukocytes
Before testing our hypothesis that the various classes of peripheral leukocytes differentially regulate the establishment of 5mCG and its removal via oxidation to 5hmCG, we first optimized methods to quickly and reiteratively isolate a few to seven of the peripheral blood leukocyte types from small samples of fresh or frozen stored blood. Our approach represents an extension of the partially reiterative methods of Lyons et al. (2007), who started with two aliquots of PBMCs and then successively isolated CD14+ monocytes and then CD4+ T cells from one aliquot and the CD19+ B cells followed by CD8+ T cells from the other. Most other reports of blood cell methylome analysis start with large amounts of blood (e.g., 100 to 450 ml) and isolate single cell types from separate aliquots (Reinius et al., 2012; Zilbauer et al., 2013). Such large blood samples are not easily adapted to the analysis of large patient populations particularly if multiple cell types are to be isolated. Furthermore, typical protocols for leukocyte isolation rely on Ficoll density centrifugation to first purify the leukocyte population from the red blood cells prior to individual cell type isolation, which takes time and effort and results in a significant loss of cells. This is of concern with epigenetic analysis, as the half-life of promoter region specific DNA methylation for many genes is measured in fractions of an hour (Meagher, 2014). Hence, any additional time of manipulation may result in the loss of the original methylation profile. Our protocol(s) allows for rapid isolation of several relatively pure leukocyte populations with minimal time and labor compared to the current methods.
If multiple cell types are to be isolated from one blood sample, six to ten color flow cytometry and sorting has the potential to identify and eliminate contamination of subpopulations expressing multiple markers (Roederer et al., 1997; Granja et al., 2015; Melzer et al., 2015) and provide more information about the cell populations than magnetic bead isolation. However, multichannel sorting is more technically challenging and more costly. In addition, during cytometry “contamination” of one cell type with another depends not only upon co-expressed markers, but also on the choice of fluorophores and their overlapping emission spectra. While the reiterative isolation of cell types employed herein is less complex, less expensive, and more rapid, it may not achieve the level of purity possible using multichannel FACS.
The current common criterion for assessing purity of isolated leukocyte types relies on fluorescently labeling isolated cells with a panel of antibodies and subsequent analysis by flow cytometry (Reinius et al., 2012; Accomando et al., 2014). However, we were confronted with the problem that the large 2.8 or 4.5 micron diameter Dynabeads used to capture cells would have to be dissociated from cells before cytometric analyses. Therefore, to confirm purity by a technique independent of cytometry we employed qRT-PCR analysis of transcripts encoding eight defining PMA marker proteins.
3.2. Evidence for extreme variability in the regulation of 5mCG and 5hmCG levels among classes of leukocytes
We examined the levels of 5hmCG and related proteins regulating synthesis and decay of 5mCG levels among leukocytes. We were exploring the idea that the leukocyte types with higher levels of 5hmCG and enzymes leading to 5hmCG synthesis and removal would be more readily poised to respond to different environmental stimuli via changes in DNA demethylation and rapid changes in transcription. We hoped these data would begin to define those classes of blood-borne leukocytes with the greatest potential to respond rapidly to changes in the cell, tissue, and blood environment. However, we have not yet directly tested the response of the methylome of different cell types to environmental stress.
Our goal was a broad survey of 5hmCG across all of the peripheral leukocyte types. When we examined 5hmC by IFM in a total leukocyte preparation, we found that all cells examined expressed 5hmC at some level (Supplemental Fig. 3), and that the level of 5hmC varied dramatically among leukocyte types (Fig. 4). However, there was no difference in the 5hmC levels as determined by IFM for total leukocytes isolated with either method 2 (fresh blood) or 3 (frozen blood) (Fig. 4) and thus we only examined one isolation method in our further analysis. The analysis of 5hmC levels was expanded and quantified using TAB-Seq and showed a wide variation among leukocytes (Table 2). The modestly high levels of 5hmCG in all peripheral leukocyte types suggests that all peripheral leukocytes are able to alter their 5mCG levels via hydroxymethylation, presumably in response to different physiological signals.
However, it should be mentioned that a direct relationship between the loss of 5mCG and the gain of 5hmCG should not be expected, as there are many regulated steps in the cycle, and the half-lives of all the intermediates may vary in a site specific manner. In other words, these two modifications may change independently in different gene sequences and in different cell types in a tissue (Ruzov et al., 2011; Salvaing et al., 2012; Hahn et al., 2013). With a few exceptions, we did not observe a clear relationship between the relative levels of 5hmCG among the 7 leukocyte types and the levels of 5mCG reported previously (Reinius et al., 2012). This comparison supports the view that the cytosine modification cycle is quite distinctly regulated in each cell type.
Further, we did not find a simple correlation between the expression of transcripts encoding enzymes involved in the turnover of modified cytosine residues (e.g., TETs, GADD45s, Fig. 1) and the levels of 5hmCG among the seven leukocyte types, although some speculation about these relationships seems warranted. CD4+ T cells have by far the highest levels of 5hmCG (Table 2, Fig. 5) and they have the highest levels of 5mCG of any leukocyte type assayed (Reinius et al., 2012). It is tempting to speculate then, that they are the most highly potentiated to respond to stress by 5hmCG mediated changes in 5mCG levels followed by changes in gene expression. Yet, CD4+ T cells did not distinguish themselves with high levels of expression of those factors controlling the synthesis of 5hmCG. They did highly express TET1, DNMT1, DNMT3A, and GADD45G. These data suggest there is another level of regulation controlling the balance among the factors in the turnover cycle of cytosine modification (Fig. 1).
Overall, CD8+ T cells expressed relatively high steady state levels of transcripts encoding most of the factors involved in this cycle (DNMT1, DNMT3B, TET1, TET2, GADD45A, and GADD45B). Despite this, CD8+ T cells had the lowest levels of 5hmCG for all quintiles of transcript expression. CD8+ T cells have moderately high levels of 5mCG relative to other PBMCs (Reinius et al., 2012). Hence, the balance among these modifying activities (examine Figs. 1 and 7) for CD8+ T cells must be weighted toward accumulation of 5mCG and rapid removal of 5hmCG. The high levels of TET and GADD45 suggests these cells may be highly responsive in changing 5mCG levels, despite having low levels of 5hmCG. It has been shown there are dynamic changes in DNA cytosine methylation in CD8+ T cells in response to acute infection (Scharer et al., 2013), supporting the view that the cycle turns over rapidly in these cells. This emphasizes the importance of both DNA demethylation and methylation in the ability of CD8+ T cells to respond to changes in their environment (Scharer et al., 2013).
CD14+ monocytes have relatively low levels of all the factors assayed with the exception of TET3, and yet they have the second highest levels of 5hmCG. By contrast they have 2 to 3 times lower levels of 5mCG relative to other PBMCs (Reinius et al., 2012). Hence, these modifying activities must be weighted toward removal of 5mCG and accumulation of 5hmCG. Granulocytes, CD16+ neutrophils and Siglec8+ eosinophils have the lowest levels of 5mCG (Reinius et al., 2012), but have moderate and low levels of 5hmCG, respectively (Table 2, Fig. 5).
Interestingly, the two cell types with the highest levels of 5hmCG, CD4+ T cells and CD14+ monocytes, both expressed high levels of transcripts involved in the removal of 5hmCG (TETs and GADDs), which may seem counterintuitive. Perhaps the TET activity in these two cell types efficiently carry out the first oxidation step from 5mC to 5hmC, but are less efficient at further oxidation. Those sites that are oxidized further to 5fC and 5caC may be repaired by the base excision repair (BER) pathway back to C and then further modified to 5mC and then back to 5hmC. This proposed mechanism emphasizes the importance of the modification cycle for DNA cytosine (Fig. 1). Both of these cell types also express high to modest levels of the DNMTs, further strengthening this explanation.
When we examined the 5hmCG distribution across gene regions among the quintiles of gene expression (Fig. 5) for the seven leukocyte types, we observed a strong depletion of 5hmCG at the TSS and an enrichment over the gene body. These data agree with previous work assessing 5hmCG distribution in T cells (Tsagaratou et al., 2014; Ichiyama et al., 2015), as well as other non-leukocyte cell types and tissues (Song et al., 2011; Mellen et al., 2012; Yu et al., 2012b; Chapman et al., 2015; Taylor et al., 2016). In genes with the highest levels of expression (Fig. 5), we observed an enrichment of 5hmCG in the gene body, suggesting that higher gene body 5hmCG correlates with the activation of these genes. Gene body enrichment of 5hmCG and its association with highly expressed cell type specific genes have been observed in studies of the brain, neurons, colonocytes, and chondrocytes (Mellen et al., 2012; Hahn et al., 2013; Chapman et al., 2015; Taylor et al., 2016).
Higher and lower levels of 5hmCG in both the total leukocytes and the individual cell types were observed in our analysis of smaller gene regions (Fig. 6) than what we had observed in larger gene regions (Fig. 5), because the extreme levels are averaged out by adjacent regions when larger gene regions are examined. The highest 5hmCG level in the different gene regions and GO terms were not always in the CD4+ T cells as we have observed for large gene regions Fig. 5. For example, in the Cytokine Production GO gene list in the region upstream of the TTS, CD14+ monocytes have high 5hmCG levels (~8%) and in the region downstream of the TTS, Siglec8+ eosinophils and CD19+ B cells have levels reaching ~8.5% (Fig. 6). The Siglec8+ eosinophils have the second lowest global 5hmCG level as well as the second lowest peak 5hmCG level when examined by transcript expression quintile and larger gene regions. This may suggest that although globally there are generally low levels of 5hmCG in the eosinophils, there are gene-region and gene-type differences in 5hmCG in this cell type (and others) that may be important to gene regulation of specific genes and would be missed by both global analysis and transcript expression quintile analysis of larger gene regions.
Cell type specific changes in 5hmCG in relation to different GO classes of genes and gene region may be relevant to the role of 5hmCG in potentiating gene expression. For example, during chondrogenesis those genes for which increases in 5hmCG correlate with increases in gene expression there was a enrichment of 5hmCG both prior to the TSS and in the gene body and a sharp drop in between (Taylor et al., 2016). This distribution of 5hmCG was not observed in the genes with either no change in expression or a decrease in expression associated with large changes in 5hmCG (Taylor et al., 2016). Our data (Fig. 6) also shows this relationship. For example, when neutrophils were examined for the leukocyte activation GO gene list we saw enrichment of 5hmCG in the region prior to the TSS and in the gene body with a valley in between. By contrast, when the adaptive immune response GO gene list is applied to neutrophils, an innate immune cell type, this enrichment pattern of 5hmCG is not observed. These data support the idea that gene region specific enrichment of 5hmCG is cell type specific in leukocytes, potentiating the expression of cell type specific genes for regulated gene expression.
3.3. The role of hydroxymethylcytosine in potentiating gene regulation
A current view is that high levels of gene region specific 5hmCG potentiate genes for “on demand gene regulation”(Irier et al., 2014). Further, there is direct evidence that this cytosine modification is essential for normal regulation of gene expression. For example, during erythropoiesis in zebra fish, there is both increased expression and demethylation of scl, gata-1, and cmyb. However, if TET2 is knocked down there is increased methylation and decreased expression of these genes that results in anemia (Ge et al., 2014) suggesting a direct role for 5mCG oxidation in essential gene regulation. In embryonic stem cells, when TET1 and TET2 are knocked down, there is an increase in methylation of the pluripotency related genes with a corresponding decrease in expression, altering their differentiation potential (Ficz et al., 2011).
5hmCG rich regions are associated with a potentiated, open chromatin state allowing access to various transcription and chromatin remodeling factors. For example, in embryonic stem cells enhancer regions enriched for 5hmCG are also enriched for nucleosomal histone modifications H3K4Me1 and H3K27Ac, which are associated with active transcription (Szulwach et al., 2011). In addition, in CD4+/CD8+ double positive thymocytes, 5hmCG is enriched in active thymus-specific enhancers, which have high levels of H3K4Me1 and H3K27Ac (Tsagaratou et al., 2014). There is also a negative correlation between 5hmC and the repressive histone modification H3K27me3 (Tsagaratou et al., 2014). It has been suggested that the combination of these chromatin structures indicates genes that are poised for transcriptional activation or silencing in response to environmental cues (Lister et al., 2013; Tsagaratou and Rao, 2013; Tsagaratou et al., 2014).
Considering that oxidation of 5mC to 5hmC must impact 5mC levels it is worth considering the contribution of 5mC binding protein to the concept of 5hmC function. 5mCG dinucleotides attract a wide variety of chromatin remodeling machinery such as methyl-CG binding proteins MBD4 and MeCP2 (Du et al., 2015). MBD4 recognizes 5hmCG and has a glycosylase domain (Otani et al., 2013) (see Fig. 1, TDG) and therefore, may contribute to the cyclic modification and removal of cytosine (Fig. 1) in various classes of leukocytes. The binding and activity of MeCP2 appears enhanced at promoter 5hmCG sites in the brain (Zhubi et al., 2014) and its activity is considered particularly important, because in the brain MeCP2 activity responds to external stimuli (e.g., cocaine, ethanol) (Pol Bodetto et al., 2013; Liyanage et al., 2015). MeCP2 binding in the brain to modified C residues can work to enhance transcription if bound to 5hmCG, or repress transcription if bound to 5mCG (Mellen et al., 2012). Appropriate levels of MeCP2 activity are essential for the differentiation of naive CD4+ T cells into a variety of T cell types (Jiang et al. 2014; Yang et al. 2012). Thus it appears that the balance between 5mCG and 5hmCG is allowing the DNA sequence to be poised for activation and expression or repression via a combination of interactions with methyl-binding proteins.
In summary, Pastor et al. (2011) presented an initial simple view of the role of 5hmCG that may still be valid, stating that “5hmC contributes to the ‘poised’ chromatin signature found at developmentally-regulated genes”. On the other hand, Nestor et al. (Nestor et al., 2012) presented solid evidence that “tissue type” was “a major modifier of the 5-hydroxymethylcytosine content” of genes. It is reasonable for us to interpret tissue type as leukocyte type and expect gene specific differences among cell types when a genome-wide analysis is performed. Our data herein and that from mouse brain (Lister et al., 2013) agree with both views, suggesting that gene expression levels and cell type are both significant determinants of overall 5hmCG levels. In short, gene region-specific 5hmCG level differences between active and inactive chromatin regions are preserved within a background of higher or lower total 5hmCG, which is determined by the cell type. Perhaps it is important to recall that the seven major classes of leukocytes examined are indeed quite divergent.
3.4. Conclusions
In summary, we optimized protocols for the reiterative isolation of the major leukocyte types from single small whole blood samples that are rapid and efficient. The isolated populations of the various leukocyte types appear sufficiently pure for epigenome and transcriptome studies. This was confirmed with the analysis of both nuclear morphology and of leukocyte cell-type specific transcripts.
We showed that these seven isolated leukocyte types, (1) differentially express factors involved in the cycle of DNA cytosine methylation and demethylation, (2) have rank order differences in 5hmCG levels and gene region dispersal, and (3) collectively our data suggest that the CD4+ and CD8+ T cells and CD14+ monocytes may be potentiated to turnover their 5mCG more rapidly via oxidation to 5hmCG than other leukocytes. However, there is not a simple relationship between TET expression levels and 5hmCG levels as suggested by our hypothesis, implying that a much better understanding of every step in the cytosine modification cycle is needed. Recall that TETs catalyze the further oxidation of 5hmCG to 5fCG and 5caCG (Fig. 1). Taken together our data suggest that each leukocyte type uniquely regulates their cycle of DNA cytosine modification, which imparts to each cell type a distinct ability to regulate gene expression in response to different physiological cues. Definitive identification of the optimal surrogate leukocyte cell types in peripheral blood to report the methylome’s response to health status awaits further experimental analysis. It still needs to be established that 5hmCG levels and components of the turnover cycle for cytosine modification enable some cell types to respond more rapidly or more definitively than others to distinct physiological stresses and diseases.
4. Materials and methods
4.1. Methods of reiteratively isolating leukocytes
Building on previous efforts to reiteratively isolate some classes of peripheral blood leukocytes (Lyons et al., 2007) we developed simplified protocols that could be executed in 5 hours on six or seven leukocyte classes (Figs. 2–3, Supplemental Figs. 1–2, Table 1). Venous blood samples were collected in EDTA tubes in the morning between 8 and 9 AM from a nonfasted healthy 65-year-old male volunteer free of cardiovascular and other diseases with a BMI of 29. These studies were approved by the Institutional Review Board at the University of Georgia. Analyzing the methylome of seven cell types from one individual in this initial study eliminated genetic variation so as to strengthen the interpretation of complex data, as was done with the early studies of bisulfite conversion for analyzing 5mCG residues, TET-assisted bisulfite sequencing for analyzing 5hmCG residues, and six color sorting for the analysis of multiple leukocyte types (Frommer et al., 1992; Roederer et al., 1997; Yu et al., 2012a). However, we have not yet addressed the natural variability in the leukocyte populations or in chromatin structures that may exist among healthy individuals differing in age and/or sex.
Leukocytes were isolated following the methods described in detail below and are depicted in Fig. 2. For each method the cells were isolated in the following order: (A) CD4+ T cells, CD8+ T cells, and CD14+ monocytes, respectively, (B) CD16+ neutrophils, Siglec8+ eosinophils, CD4+ T cells, CD8+ T cells, CD19+ B cells, CD14+ monocytes, and CD56+ NK cells, respectively, and (C) CD14+ monocytes, CD4+ T cells, CD16+ neutrophils, CD56+ NK cells, CD8+ T cells, CD19+ B cell, and Siglec8+ eosinophils, respectively. The majority of leukocyte data presented in the text utilize Method 2 in which six of the cell types were reiteratively isolated from one 5 ml fresh blood sample in isolation order B, while the seventh type, CD56+ NK cells were isolated in parallel from a second 5 ml blood sample, because NK cells are a highly mixed cell type in terms of the affinity markers being utilized, their recovery is poor when isolated at the end of isolation order B.
4.1.1. Dynabead preparation
Anti-CD4, anti-CD8, and anti-CD14 antibodies bound to 4.5 μm paramagnetic Dynabeads were obtained from Life Technologies (Grand Island, NY, USA Cat# 11145D, 11147D, 11149D). 25 μl of suspended anti-CD4+ and anti-CD8+ Dynabeads and 20 μl of suspended anti-CD14+ Dynabeads were used per 5 ml of blood. The Dynabeads we prepared per manufacturer’s instructions and stored in 50 μl of PBSBE (phosphate buffered saline, 1% BSA, 2 mM EDTA, pH 7.4) on ice until needed. Protein G Dynabeads (2.8 μm, Life Technologies, Grand Island, NY, USA Cat# 10009D) were coupled to anti-CD16 (Santa Cruz Biotech Dallas, TX, USA Cat# sc-19620), anti-CD19 (Bio legend San Diego, CA, USA Cat# 302202), anti-CD56 (Bio Legend, San Diego, CA, USA Cat# 31824), or anti-Siglec8 (Bio Legend, San Diego, CA, USA Cat# 347102) antibodies for the isolation of 4 other leukocyte classes. Protein G Dynabeads were first washed with 1 ml of PBSBE, resuspended in 200 μl of PBSBE where the antibody was then added (40 μl Dynabeads and 4 μl anti-CD16 or 10 μl Dynabeads and 0.5 μl of anti-CD19, anti-CD56, and anti-Siglec8). Beads and antibodies were incubated at room temperature with rotation for 15 min, washed twice with 1 ml PBSBE, resuspended in 50 μl of PBSBE and stored on ice for no more than 4 h until use. Washing was implemented with the use of strong neodymium magnets that pulled the Dynabead-bound cells to the side of a 1.5 ml microfuge tube (MagnaRack, Invitrogen, Grand Island, NY, USA Cat # CS15000) in a minute.
4.1.2. Method 1: isolation of leukocytes from fresh whole blood without red blood cell lysis (Fig. 2)
Five ml of venous blood samples were collected in EDTA tubes in the morning between 8 and 9 AM in a nonfasted male subject (kept on ice immediately after collection until leukocyte isolation) and were diluted 1:2 with PBSBE and centrifuged at 300 ×g for 30 min at 4 °C. Supernatant was discarded, and the cell pellet was resuspended in 1 ml of PBSBE. Cells were centrifuged at 400 ×g for 2 min, supernatant was discarded, and cells resuspended in 1 ml of PBSTBE (phosphate buffered saline, 2% Tween 20, 1% BSA, 2 mM EDTA, pH 7.4). Three leukocyte types (CD4+, CD8+, and CD14+) were then successively isolated following the general isolation protocol (see below).
4.1.3. Method 2: isolation from fresh whole blood with controlled red blood cell lysis (Fig. 2)
Five ml of venous blood samples were collected in EDTA tubes in the morning between 8 and 9 AM in a nonfasted male subject (kept on ice immediately after collection until leukocyte isolation) and were diluted 1:10 with freshly prepared 1× cold red blood cell lysis solution form a 10× stock (10× red blood cell lysis solution: 1.5 M NH4Cl 100 mM NaHCO3, 10 mM EDTA, pH 7.4) (Bossuyt et al., 1997) and placed on ice for 20 min. Samples were then centrifuged at 300 ×g for 20 min at 4 °C. Supernatant was discarded, and cells were resuspended in 10 ml of PBSBE and centrifuged at 300 ×g for 10 min at 4 °C. Supernatant was again discarded, and cells were resuspended in 1 ml of PBSTBE. Six to seven leukocyte types were then successively isolated following the general isolation protocol.
4.1.4. Method 3: isolation from frozen whole blood (Fig. 2)
Venus blood samples were collected in EDTA tubes in the morning between 8 and 9 AM in nonfasted a male subject and frozen at −80 °C. Five millilitres of frozen blood (at −80 °C) was thawed on ice. Approximately 90% of red blood cells lyse during the freeze-thaw process, but most white cells do not (Fiebig et al., 1997). Thawed blood was diluted 1:2 with PBSBE and centrifuged at 300 ×g for 30 min at 4 °C. The supernatant was discarded, and cells were resuspended in 1 ml of PBSBE. Cells were centrifuged at 400 ×g for 2 min, supernatant was discarded, and cells were resuspended in 1 ml of PBSTBE. Again, six to seven leukocyte types were then successively isolated following the general isolation protocol.
4.1.5. General isolation protocol
Dynabeads prepared for the first cell type to be isolated were added to the cells. Cells were incubated with Dynabeads with rotation at 4 °C for 30 min for CD4+ T cells, CD8+ T cells, CD14+ monocytes, CD19+ B cells, CD56+ natural killer (NK) cells, and Siglec8+ eosinophils or 1 h for CD16+ neutrophils. Samples were then placed on a magnetic rack (Invitrogen, Grand Island, NY, USA Cat # CS15000) for 2 min, and the supernatant was carefully removed, placed in a fresh microcentrifuge tube for the next leukocyte type to be isolated, and stored on ice until processing. The cells bound to paramagnetic beads were resuspended in 1 ml of PBSBE and washed three times with PBSBE using the magnetic rack. Cells were finally resuspended in 200 μl of PBS (phosphate buffered saline, pH 7.4) and stored according to future use (e.g., the cell pellet was frozen with liquid nitrogen and stored at −80 °C for RNA, frozen at −80 °C for DNA extraction or fixed with 3.7% formaldehyde for Immuno-Fluorescent Microscopy (IFM)). The Dynabeads prepared for the next cell type to be isolated were added to the microcentrifuge tube containing the uncaptured cells, and the next cell type was isolated following the same protocol. This process was repeated until all desired leukocyte types had been isolated. When we tried to use this capture method with the white blood cells suspended in 10 ml the recovery was not quantitative, even with longer incubation times, using twice as much antibody and beads, and larger neodymium magnets.
4.1.6. Determination of cell recovery for each isolation method
Ten μl of the formalin fixed cells of each cell type were incubated for 20 min in the dark at 4 °C with 0.25 μl of Propidium Iodine (PI, 1 mg/ml). Stained cells were examined on a hemocytometer under combined fluorescence and DIC, and cells were counted following standard protocols. Florescence microscopy was performed on a Leica TR600 epifluorescence microscope using Hamamatsu SimplePCI Image Analysis software or on a confocal microscope (Zeiss SM710) using ZEN 2011 software. A one-way ANOVA followed by Tukey’s HSD post hoc was performed using SPSS software (IBM) to determine differences in cell recovery between the three methods of isolation for the three cell types where cells were recovered for all three methods. Significance was set at p < 0.05. A two tailed t test was performed on the cell recoveries between methods 2 and 3 to determine any significant differences in cell recovery between the two methods for the four cell types in which cells were only recovered in these methods. Significance was set at p < 0.05. Data are presented in Table 1.
4.1.7. Assessment of nuclear morphology by fluorescent microscopy
Initial cell purity was assessed by microscope analysis of the four distinct nuclear morphologies characteristic of the various leukocyte types (Alberts et al., 1994). Samples for analysis were prepared the same as used to determine cell recovery. Analysis of at least 100 cells showed that all cell preparations assayed were greater than 95% pure, based on the presence of the correct nuclear morphology and the absence of the alternate morphologies. Red blood cells were seldom observed. Data are presented in Fig. 3.
To confirm the purity of CD16+ neutrophils and CD14+ monocytes, we fluorescently labeled the total leukocyte fraction with DAPI and anti-CD4-Texas RPE (Invitrogen, Grand Island, NY, USA Cat# MHCD0417). First the cells were blocked for 45 min with PBSBE, washed 3×, and stained with DAPI and the fluorescent antibody for 1 h with rotation in the dark. Cells were then washed 3× with PBSBE and a 20 μl aliquot was set aside for immediate microscope analysis while the remaining cells were split into three equal aliquots. CD4+, CD14+, and CD16+ were then isolated from one of the three samples of labeled leukocytes and then immediately analyzed by fluorescent microscopy. Cells positive for the CD4 antibody were classified by their nuclear morphology and all cells were counted into their respective classifications. The percentage of each cell type was then calculated. A one-way ANOVA followed by Tukey HSD post hoc test was used to determine significant differences between each of cell populations for each cell type using SPSS software (IBM).
4.1.8. qRT-PCR analysis of transcript levels
To further confirm the purity of the cell types, we performed qRT-PCR assays for eight transcripts specific to the seven isolated cell types (CD3, CD4, CD8, CD14, CD16, CD19, CD56 and Siglec8. Leukocytes bound to Dynabeads (Method 3, Order B) were washed in PBS and frozen in liquid nitrogen and stored at −80 °C. RNA was extracted using RNeasy Mini Kit (Qiagen, Frederick, MD, USA Cat# 74104) following manufacture instructions. RNA concentrations were quantified using Qubit RNA assay kit (Life technologies, Grand Island, NY, USA Cat# Q32855) and 400 ng of RNA was used for cDNA synthesis using qScript cDNA synthesis supermix (Quanta Biosciences, Gaithersburg, MD, USA Cat# 95148-100). Relative quantities (RQ) of cell type specific transcripts were normalized to an endogenous control, Beta Actin (ACTB) (Vandesompele et al., 2002) using the dCT method (Livak and Schmittgen, 2001). ACTB was used when we were examining the expression profile of the transcripts of cell type markers within a single cell type, and not making comparisons among cell types. ACTB mRNA abundance, and hence, its CT values were closer to those of the target transcripts being examined, however expression was variable across cell types (Supplemental Fig. 7). Oligonucleotide primer sequences (Supplemental Table 2) were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Two to six primer sets were tested for each of the target genes, ensuring the specific gene target was being amplified, and those having a single sharp dissociation peak and the lowest CT values were selected for subsequent use. A 25 μl reaction using SYBR green master mix (Life Technologies, Grand Island, NY, USA Cat# 43677659) and 4 ng of cDNA was used for analysis of the eight gene panel in all seven isolated leukocyte types. To determine statistical relevance of differences in transcript levels, a one-way ANOVA was used to examine the effects of cell type on expression with Tukey’s HSD test as a post hoc using Statistica software 7.1 (StatSoft; Tulsa, OK, USA).
4.2. TAB-Seq library preparation and sequencing
DNA (0.5 to 1 μg) was prepared from all seven isolated cell types (1-to 2- × 105 cells) while still attached to Dynabeads using DNeasy kit (Qiagen, Frederick, MD, USA #69506) according to the manufacturer’s recommendations. DNA was quantified using a Qubit 2.0 Instrument and dsDNA HS Reagent (Life Technologies, Grand Island, NY, USA #Q32866 and #Q32851 respectively). TET-enzyme assisted bisulfite sequencing (TAB-Seq) was performed as described previously (Yu et al., 2012a). Briefly, 0.5 ng of M. SssI methylated Lambda DNA and 0.25 ng of hydroxymethylated pUC19 DNA was added per 1 μg of DNA prior to treatment as C/5mCG and 5hmCG control respectively and then sequencing libraries were prepared following the MethylC-seq protocol (Urich et al., 2015) (Supplemental Table 3). Deep sequencing was performed using an Illumina NextSeq500 Instrument at the University of Georgia Genomics Facility. The limitation of our TAB-Seq analysis is that there is only moderate to low coverage of the genome and thus it is only statistically valid to look at groupings of genes and not individual genes. Our coverage for the different cell types ranged from 34% to 55% of the human genome (Supplemental Table 3), which is considerably above accepted levels for a meta-analysis (Popp et al., 2010) and hence the analysis of gene groups with more than 100 genes gave statistically sound results.
4.2.1. TAB-Seq data analysis
The raw sequence reads were trimmed for adapters, preprocessed to remove low quality reads and aligned as previously described in (Yu et al., 2012b) to the Homo sapiens GRCh38 reference genome. Fully unmethylated lambda DNA was treated by M. SssI to methylate all cytosines in the CG context to 5mCG. These 5mCG sites in CG contexts were used to calculate the 5mCG non-conversion rate upon TET and bisulfite treatment. Non-CG sites were used to compute the non-conversion rate of unmodified cytosines upon bisulfite treatment. For the 5hmCG control, a ~1.64 kb region of the pUC19 vector was constructed by PCR amplification with all cytosines being synthesized as 5hmCGs. These 5hmCG sites were used to evaluate the protection rate of 5hmCGs. For this analysis, only cytosines in the CG context were considered (Supplemental Table 3). The TAB-Seq data set supporting the results of this article is available in NCBI GEO repository, accession number GSE70519.
For the data plotted in Fig. 5 and Supplemental Fig. 4, we grouped genes into five quintile groups based on the ranking of expression levels for ~18,000 transcripts in peripheral blood mononuclear cells in a highly cited previous study (Palmer et al., 2006). For each quintile (~3600 transcripts), the level of 5hmCG was determined using weighted methylation level calculations (Schultz et al., 2012) for each of 20 bins upstream, 20 bins within genes (between annotated TSS and TTS), and 20 bins downstream of the gene region. Each of the upstream and downstream bins spanned 5 kb of the total of 100 kb. The within-gene regions, which varied in length, were evenly divided between the 20 bins. Figures were prepared using ggplot2 (Wickham, 2009).
A related analysis was used to investigate the level of the 5hmCG marks in 100 bp windows in the context of transcriptionally- and translationally-important locations: around the TSS, exon boundaries, and around the TTS and in a larger window containing the collated exons (i.e., seven genomic bins in all). For this analysis, numerous immune cell-relevant GO terms were used. The genes in each GO term set were extracted, and all CG sites within the bins were examined to determine the level of 5hmCG. The total levels of 5hmCG in all leukocytes were presented as box plots in order to show the variance in the level of 5hmCG in each region. The weighted average of 5hmCG for each cell type was also plotted as a colored dot within the diagram. A table describing the GO terms used and the lists of genes is provided in Supplemental File 1. In order to determine the amount of variance in 5hmCG levels explained by genomic bin, cell type, and GO term set, an ANOVA was performed between the three factors using R and the amount of variance explained by each factor was determined by the proportion of their total sum of square (Supplemental Fig. 6).
4.3. Fluorescent labeling of 5hmCG
Total white blood cells were isolated from both fresh (method 2) and frozen blood (method 3) (Fig. 4), fixed with 3.7% formaldehyde for 15 min, and then centrifuged at 400 × g for 2 min, after which the supernatant was discarded. Cells were resuspended in 500 μl of 50% methanol in PBS (phosphate buffered saline, pH 7.4), incubated for 1 min at room temperature, and centrifuged at 400 ×g for 3 min, after which the supernatant was discarded. Cells were heated to 95 °C for 5 min, snap cooled on ice, washed two times with PBSBE (phosphate buffered saline, 1% BSA, 2 mM EDTA, pH 7.4), and finally resuspended in 200 μl PBSBE and incubated for 45 min to block non-specific binding of antibodies. Rabbit anti-5hmC polyclonal antibody (Active Motif, Carlsbad, CA, USA Cat# 39769) was added to cells at a 1:200 dilution in PBSBE and incubated at room temperature with rotation for 1 h. Cells were washed three times with PBSBE and resuspended in 1:500 dilution of goat anti-rabbit conjugated with phycoerythrin (PE) (Abcam, Cambridge, MA, USA Cat# ab97070) in PBSBE and incubated at room temperature with rotation in the dark for 1 h. Cells were washed three times with PBSBE and finally resuspended in 30 μl of PBSBE. 10 μl of labeled cells were stained with 1 μl of DAPI (1 mg/ml) for ten minutes and examined with fluorescence microscopy, all under the same exposure conditions. Cells were classified by nuclear morphology into four groups, round nuclei, kidney shaped nuclei, multilobed nuclei, or bilobed nuclei, and Hamamatsu SimplePCI Image Analysis software was used to assess the 5hmCG signal for at least 100 cells for each morphology in each isolation method. Background fluorescent was subtracted from each raw 5hmCG signal. The 5hmGC signal for each of the morphologies and each isolation method was divided into 4 quintiles of data, labeled as minimal, low, medium or high 5hmCG signal. The cells for each morphology were randomly separated into three groups and the percent of cells falling in each 5hmCG signal range were identified for each morphology in both isolation methods. To determine if there was an interaction between the isolation method and the nuclear morphology on the number of cells in each 5hmCG signal group a two-way MANOVA was performed using Pillai trace test using SPSS software (IBM). A significant effect of only nuclear morphology was identified and Bonferroni post-hoc analysis was used to assess differences between nuclear morphology and 5hmCG signal for this analysis with significance set at p < 0.05.
4.4. qRT-PCR analysis of transcript levels
To assess the potential for turnover of 5mCG, three transcripts related to DNA methylation (DNMT1, 3A, and 3B) and six related to DNA demethylation (TET1, 2, 3 and GADD45A, B, G) were also assayed. Isolated leukocytes bound to Dynabeads (Method 3, Order B) were washed in PBS and frozen in liquid nitrogen and stored at −80 °C. RNA was extracted using RNeasy Mini Kit (Qiagen, Frederick, MD, USA Cat# 74104) following the manufacturer’s instructions. RNA concentrations were quantified using the Qubit RNA assay kit (Life technologies, Grand Island, NY, USA Cat# Q32855) and 400 ng of RNA was used for cDNA synthesis using qScript cDNA synthesis supermix (Quanta Biosciences, Gaithersburg, MD, USA Cat# 95148-100). Relative quantities (RQ) of cell type specific transcripts were normalized to endogenous control ribosomal 18S rRNA (Vandesompele et al., 2002) using the dCT method (Livak and Schmittgen, 2001). We found beta-actin was far more variably expressed across the seven cell types than 18S rRNA, relative to a constant cDNA input (Supplemental Fig. 7). Therefore, 18S rRNA was used as an endogenous control when comparing transcripts related to establishment and removal of 5mCG across cell types. 18S rRNA has the disadvantage as an endogenous control that it is 1000-fold more abundant than any mRNAs being assayed. Therefore, the low number of PCR cycles needed to reach cycle threshold levels are far removed from those of the target, which could be another source of error. Oligonucleotide primer sequences (Supplemental Table 2) were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Two to six primer sets were tested for each of the target genes and those having a single sharp dissociation peak, ensuring the specific gene target was being amplified, and the lowest CT values were selected for subsequent use. A 25 μl reaction using SYBR green master mix (Life Technologies, Grand Island, NY, USA Cat# 43677659) and 4 ng of cDNA was used for analysis of the gene panel. To determine statistical relevance of differences in transcript levels, a one-way ANOVA was used to examine the effects of cell type on expression with Tukey’s HSD test as a post hoc using Statistica software 7.1 (StatSoft; Tulsa, OK, USA).
Supplementary Material
Acknowledgments
Several colleagues at the University of Georgia and Emory University made helpful contributions to the work including Alicia Smith, Emily Rose England, Ping Yu, and Anthony Kim. The work was funded by grants from the National Institutes of Health NIDDK Grants DK096300 and DK100392 and UGA’s Research Foundation (10-21RX093900) to RBM, NHGRI HG006827 to CH, National Science Foundation grant IOS-1339194 and the National Institutes of Health grant R00GM100000 to BJS and from Georgia Research Alliance Eminent Scholar Fund (1025AR154280) to the late Dr. Clifton A. Baile. CH is an investigator of the Howard Hughes Medical Institute. MY is an international predoctoral fellow of the Howard Hughes Medical Institute.
Abbreviations
- ACTB
cytoplasmic beta actin
- DNMT1
3A, 3B, DNA methyltransferase 1, 3A, 3B
- CG
cytosine-guanine dinucleotide
- CNS
conserved noncoding sequence
- CT
cycle threshold
- GADD45A
B, G, Growth arrest DNA damage inducible proteins 45a, 45b, 45g
- 5hmC
5′-hydroxymethylcytosine
- GO
gene ontology
- 5hmCG
5-hydroxymethylcytosine-guanine dinucleotide
- IFM
Immuno-Fluorescence Microscopy
- 5mC
5′-methylcytosine
- 5mCG
5′-methyl CG dinucleotide
- MBD2
MBD4, MeCP2, Methyl-CpG Binding Domain Proteins
- NLP
natural log p-value
- nt
nucleotide
- PE
phycoerythrin
- RQ
relative quantity
- SLE
systemic lupus erythematous
- TAB-Seq
TET assisted bisulfite sequencing
- TSS
transcription start site
- TTS
(transition termination site)
- TET1, 2, 3
Ten-eleven Translocation Enzymes 1, 2, 3, methylcytosine dioxygenases 1, 2, 3
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jim.2016.05.003.
Footnotes
Competing interests
The authors declare no competing interests related to this manuscript.
Author contributions
NMH participated in conceptualization of study and experiments, performed most of the laboratory experiments, developed most of cell isolation methods, prepared RNA and DNA, and drafted the manuscript. MMK performed the confocal microscopy. BGP helped with planning the study and collected the blood samples. CAB was essential to planning the study and experiments. RBM was essential in the conceptualization, troubleshooting and experimental design of the study, directed the project, and assisted in revising the manuscript. CH and MY processed the DNA samples for TAB-Seq, while LX, KL, and RJS performed the sequencing and bioinformatic analysis of the sequencing data.
Contributor Information
Natalie M. Hohos, Email: nhohos@uga.edu.
Kevin Lee, Email: klee84@uga.edu.
Lexiang Ji, Email: lxji@uga.edu.
Miao Yu, Email: miaoyu1988@uchicago.edu.
Muthugapatti M. Kandasamy, Email: kandu@uga.edu.
Bradley G. Phillips, Email: bgp@uga.edu.
Chuan He, Email: chuanhe@uchicago.edu.
Robert J. Schmitz, Email: schmitz@uga.edu.
References
- Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol. 2014;15:R50. doi: 10.1186/gb-2014-15-3-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Garland Science. Taylor and Francis Group; New York: 1994. Molecular biology of the cell. [Google Scholar]
- Almen MS, Nilsson EK, Jacobsson JA, Kalnina I, Klovins J, Fredriksson R, Schioth HB. Genome-wide analysis reveals DNA methylation markers that vary with both age and obesity. Gene. 2014;548:61–67. doi: 10.1016/j.gene.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Bossuyt X, Marti GE, Fleisher TA. Comparative analysis of whole blood lysis methods for flow cytometry. Cytometry. 1997;30:124–133. [PubMed] [Google Scholar]
- Calabrese R, Valentini E, Ciccarone F, Guastafierro T, Bacalini MG, Ricigliano VA, Zampieri M, Annibali V, Mechelli R, Franceschi C, Salvetti M, Caiafa P. TET2 gene expression and 5-hydroxymethylcytosine level in multiple sclerosis peripheral blood cells. Biochim Biophys Acta. 2014;1842:1130–1136. doi: 10.1016/j.bbadis.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Chapman CG, Mariani CJ, Wu F, Meckel K, Butun F, Chuang A, Madzo J, Bissonette MB, Kwon JH, Godley LA. TET-catalyzed 5-hydroxymethylcytosine regulates gene expression in differentiating colonocytes and colon cancer. Sci Rep. 2015;5:17568. doi: 10.1038/srep17568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Tian Y, Shu W, Bo X, Wang S. Comprehensive identification and annotation of cell type-specific and ubiquitous CTCF-binding sites in the human genome. PLoS One. 2012;7:e41374. doi: 10.1371/journal.pone.0041374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Francesco A, Arosio B, Falconi A, Micioni Di Bonaventura MV, Karimi M, Mari D, Casati M, Maccarrone M, D’Addario C. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain Behav Immun. 2015;45:139–144. doi: 10.1016/j.bbi.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Dubois-Chevalier J, Oger F, Dehondt H, Firmin FF, Gheeraert C, Staels B, Lefebvre P, Eeckhoute J. A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res. 2014;42:10943–10959. doi: 10.1093/nar/gku780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Q, Luu PL, Stirzaker C, Clark SJ. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics. 2015:1–23. doi: 10.2217/epi.15.39. [DOI] [PubMed] [Google Scholar]
- Ellinger J, Muller SC, Dietrich D. Epigenetic biomarkers in the blood of patients with urological malignancies. Expert Rev Mol Diagn. 2015:1–12. doi: 10.1586/14737159.2015.1019477. [DOI] [PubMed] [Google Scholar]
- Ficz G. New insights into mechanisms that regulate DNA methylation patterning. J Exp Biol. 2015;218:14–20. doi: 10.1242/jeb.107961. [DOI] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- Fiebig EW, Johnson DK, Hirschkorn DF, Knape CC, Webster HK, Lowder J, Busch MP. Lymphocyte subset analysis on frozen whole blood. Cytometry. 1997;29:340–350. doi: 10.1002/(sici)1097-0320(19971201)29:4<340::aid-cyto11>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Zhang RP, Wan F, Guo DY, Wang P, Xiang LX, Shao JZ. TET2 plays an essential role in erythropoiesis by regulating lineage-specific genes via DNA oxidative demethylation in a zebrafish model. Mol Cell Biol. 2014;34:989–1002. doi: 10.1128/MCB.01061-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja T, Schad J, Schussel P, Fischer C, Haberle H, Rosenberger P, Straub A. Using six-colour flow cytometry to analyse the activation and interaction of platelets and leukocytes — a new assay suitable for bench and bedside conditions. Thromb Res. 2015;136:786–796. doi: 10.1016/j.thromres.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Gruenbacher G, Gander H, Rahm A, Nussbaumer W, Romani N, Thurnher M. CD56+ human blood dendritic cells effectively promote TH1-type gammadelta T-cell responses. Blood. 2009;114:4422–4431. doi: 10.1182/blood-2009-06-227256. [DOI] [PubMed] [Google Scholar]
- Gu J, Stevens M, Xing X, Li D, Zhang B, Payton JE, Oltz EM, Jarvis JN, Jiang K, Cicero T, Costello JF, Wang T. Mapping of Variable DNA Methylation across Multiple Cell Types Defines a Dynamic Regulatory Landscape of the Human Genome (G3 Bethesda) 2016 doi: 10.1534/g3.115.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP, Lu Q. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 2013;3:291–300. doi: 10.1016/j.celrep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseeb A, Makki MS, Haqqi TM. Modulation of ten-eleven translocation 1 (TET1), Isocitrate Dehydrogenase (IDH) expression, alpha-Ketoglutarate (alpha-KG), and DNA hydroxymethylation levels by interleukin-1beta in primary human chondrocytes. J Biol Chem. 2014;289:6877–6885. doi: 10.1074/jbc.M113.512269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YT, Maccani JZ, Hawley NL, Wing RR, Kelsey KT, McCaffery JM. Epigenetic patterns in successful weight loss maintainers: a pilot study. Int J Obes. 2014 doi: 10.1038/ijo.2014.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, Tian Q, Aifantis I, Wei L, Dong C. The methylcytosine dioxygenase tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. 2015;42:613–626. doi: 10.1016/j.immuni.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irier H, Street RC, Dave R, Lin L, Cai C, Davis TH, Yao B, Cheng Y, Jin P. Environmental enrichment modulates 5-hydroxymethylcytosine dynamics in hippocampus. Genomics. 2014;104:376–382. doi: 10.1016/j.ygeno.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O’Hanlon TP, Rider LG, Jacinto FV, Lopez-Longo FJ, Dopazo J, Forn M, Peinado MA, Carreno L, Sawalha AH, Harley JB, Siebert R, Esteller M, Miller FW, Ballestar E. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li C, McRae G, Lykken E, Sevilla J, Liu SQ, Wan Y, Li QJ. MeCP2 reinforces STAT3 signaling and the generation of effector CD4+ T cells by promoting miR-124-mediated suppression of SOCS5. Sci Signal. 2014;7:ra25. doi: 10.1126/scisignal.2004824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Rogers J, Madrigal-Estebas L, O’Connor T, Doherty DG. Activation-induced expression of CD56 by T cells is associated with a reprogramming of cytolytic activity and cytokine secretion profile in vitro. Hum Immunol. 2006;67:863–873. doi: 10.1016/j.humimm.2006.08.292. [DOI] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Lan Y, Schwartz-Orbach L, Korol E, Tahiliani M, Evans T, Goll MG. Overlapping requirements for Tet2 and Tet3 in normal development and hematopoietic stem cell emergence. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Liyanage VR, Zachariah RM, Davie JR, Rastegar M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp Neurol. 2015;265:102–117. doi: 10.1016/j.expneurol.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons PA, Koukoulaki M, Hatton A, Doggett K, Woffendin HB, Chaudhry AN, Smith KG. Microarray analysis of human leucocyte subsets: the advantages of positive selection and rapid purification. BMC Genomics. 2007;8:64. doi: 10.1186/1471-2164-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher RB. ‘Memory and molecular turnover,30 years after inception. Epigenetics Chromatin. 2014;7:37. doi: 10.1186/1756-8935-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer S, Zachariae S, Bocsi J, Engel C, Loffler M, Tarnok A. Reference intervals for leukocyte subsets in adults: results from a population-based study using 10-color flow cytometry. Cytometry B Clin Cytom. 2015;88:270–281. doi: 10.1002/cyto.b.21234. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, Perna F, Pandey S, Madzo J, Song C, Dai Q, He C, Ibrahim S, Beran M, Zavadil J, Nimer SD, Melnick A, Godley LA, Aifantis I, Levine RL. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto H, Sakata-Yanagimoto M, Nagae G, Shiozawa Y, Miyake Y, Yoshida K, Enami T, Kamada Y, Kato T, Uchida K, Nanmoku T, Obara N, Suzukawa K, Sanada M, Nakamura N, Aburatani H, Ogawa S, Chiba S. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014;4:e264. doi: 10.1038/bcj.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanney DL. Epigenetic Control Systems. Proc Natl Acad Sci U S A. 1958;44:712–717. doi: 10.1073/pnas.44.7.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–477. doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oger F, Dubois-Chevalier J, Gheeraert C, Avner S, Durand E, Froguel P, Salbert G, Staels B, Lefebvre P, Eeckhoute J. Peroxisome proliferator-activated receptor gamma regulates genes involved in insulin/insulin-like growth factor signaling and lipid metabolism during adipogenesis through functionally distinct enhancer classes. J Biol Chem. 2014;289:708–722. doi: 10.1074/jbc.M113.526996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani J, Arita K, Kato T, Kinoshita M, Kimura H, Suetake I, Tajima S, Ariyoshi M, Shirakawa M. Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J Biol Chem. 2013;288:6351–6362. doi: 10.1074/jbc.M112.431098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, Tahiliani M, Daley GQ, Liu XS, Ecker JR, Milos PM, Agarwal S, Rao A. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol Bodetto S, Carouge D, Fonteneau M, Dietrich JB, Zwiller J, Anglard P. Cocaine represses protein phosphatase-1Cbeta through DNA methylation and methyl-CpG binding protein-2 recruitment in adult rat brain. Neuropharmacology. 2013;73:31–40. doi: 10.1016/j.neuropharm.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009;126:458–465. doi: 10.1111/j.1365-2567.2008.03027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, Masse A, Le Couedic JP, Pendino F, Carbonne B, Larghero J, Ravanat JL, Casadevall N, Bernard OA, Droin N, Solary E, Godley LA, Vainchenker W, Plo I, Delhommeau F. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–2555. doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon S, Bancos S, Thatcher TH, Murant TI, Moshkani S, Sahler JM, Bottaro A, Sime PJ, Phipps RP. Peroxisome proliferator-activated receptor gamma B cell-specific-deficient mice have an impaired antibody response. J Immunol. 2012;189:4740–4747. doi: 10.4049/jimmunol.1200956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M, De Rosa S, Gerstein R, Anderson M, Bigos M, Stovel R, Nozaki T, Parks D, Herzenberg L, Herzenberg L. 8 color, 10-parameter flow cytometry to elucidate complex leukocyte heterogeneity. Cytometry. 1997;29:328–339. doi: 10.1002/(sici)1097-0320(19971201)29:4<328::aid-cyto10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Ruzov A, Tsenkina Y, Serio A, Dudnakova T, Fletcher J, Bai Y, Chebotareva T, Pells S, Hannoun Z, Sullivan G, Chandran S, Hay DC, Bradley M, Wilmut I, De Sousa P. Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res. 2011;21:1332–1342. doi: 10.1038/cr.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabag O, Zamir A, Keshet I, Hecht M, Ludwig G, Tabib A, Moss J, Cedar H. Establishment of methylation patterns in ES cells. Nat Struct Mol Biol. 2014;21:110–112. doi: 10.1038/nsmb.2734. [DOI] [PubMed] [Google Scholar]
- Saied MH, Marzec J, Khalid S, Smith P, Down TA, Rakyan VK, Molloy G, Raghavan M, Debernardi S, Young BD. Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS One. 2012;7:e33213. doi: 10.1371/journal.pone.0033213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvaing J, Aguirre-Lavin T, Boulesteix C, Lehmann G, Debey P, Beaujean N. 5-Methylcytosine and 5-hydroxymethylcytosine spatiotemporal profiles in the mouse zygote. PLoS One. 2012;7:e38156. doi: 10.1371/journal.pone.0038156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. 2013;191:3419–3429. doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JW, Smith AK, Brennan PA, Conneely KN, Kilaru V, Knight BT, Newport DJ, Cubells JF, Stowe ZN. DNA methylation in neonates born to women receiving psychiatric care. Epigenetics. 2012;7:409–414. doi: 10.4161/epi.19551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, Schmitz RJ, Ecker JR. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012;28:583–585. doi: 10.1016/j.tig.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AK, Conneely KN, Newport DJ, Kilaru V, Schroeder JW, Pennell PB, Knight BT, Cubells JC, Stowe ZN, Brennan PA. Prenatal antiepileptic exposure associates with neonatal DNA methylation differences. Epigenetics. 2012;7:458–463. doi: 10.4161/epi.19617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AK, Conneely KN, Pace TW, Mister D, Felger JC, Kilaru V, Akel MJ, Vertino PM, Miller AH, Torres MA. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun. 2014;38:227–236. doi: 10.1016/j.bbi.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, Smith JA, Almli LM, Binder EB, Klengel T, Cross D, Turner ST, Ressler KJ, Kardia SL. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet. 2013;132:1027–1037. doi: 10.1007/s00439-013-1311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Han JW, Kim S, Namburi S, Hermetz K, Kim JJ, Rudd MK, Yoon YS, Ren B, He C, Jin P. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SE, Li YH, Smeriglio P, Rath M, Wong WH, Bhutani N. Stable 5-hydroxymethylcytosine (5hmC) acquisition marks gene activation during chondrogenic differentiation. J Bone Miner Res. 2016;31:524–534. doi: 10.1002/jbmr.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagaratou A, Rao A. TET proteins and 5-methylcytosine oxidation in the immune system. Cold Spring Harb Symp Quant Biol. 2013;78:1–10. doi: 10.1101/sqb.2013.78.020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE, Lahdesmaki H, Rao A. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A. 2014;111:E3306–E3315. doi: 10.1073/pnas.1412327111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich MA, Nery JR, Lister R, Schmitz RJ, Ecker JR. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat Protoc. 2015;10:475–483. doi: 10.1038/nprot.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhu H, Snieder H, Su S, Munn D, Harshfield G, Maria BL, Dong Y, Treiber F, Gutin B, Shi H. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010;8:87. doi: 10.1186/1741-7015-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss T, Skelton K, Phifer J, Jovanovic T, Gillespie CF, Smith A, Umpierrez G, Bradley B, Ressler KJ. Posttraumatic stress disorder is a risk factor for metabolic syndrome in an impoverished urban population. Gen Hosp Psychiatry. 2011;33:135–142. doi: 10.1016/j.genhosppsych.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. ggplot2: Elegant Graphics for Data Analysis. Use R. 2009:1–212. [Google Scholar]
- Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Su S, Barnes VA, De Miguel C, Pollock J, Ownby D, Shi H, Zhu H, Snieder H, Wang X. A genome-wide methylation study on obesity: differential variability and differential methylation. Epigenetics. 2013;8 doi: 10.4161/epi.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Ramocki MB, Neul JL, Lu W, Roberts L, Knight J, Ward CS, Zoghbi HY, Kheradmand F, Corry DB. Overexpression of methyl-CpG binding protein 2 impairs T(H)1 responses. Sci Transl Med. 2012;4:163ra158. doi: 10.1126/scitranslmed.3004430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Jin P, Ren B, He C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 2012a;7:2159–2170. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, Min JH, Jin P, Ren B, He C. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012b;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A, Grayson DR. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl Psychiatry. 2014;4:e349. doi: 10.1038/tp.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilbauer M, Rayner TF, Clark C, Coffey AJ, Joyce CJ, Palta P, Palotie A, Lyons PA, Smith KG. Genome-wide methylation analyses of primary human leukocyte subsets identifies functionally important cell-type-specific hypomethylated regions. Blood. 2013;122:e52–e60. doi: 10.1182/blood-2013-05-503201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zloza A, Al-Harthi L. Multiple populations of T lymphocytes are distinguished by the level of CD4 and CD8 coexpression and require individual consideration. J Leukoc Biol. 2006;79:4–6. doi: 10.1189/jlb.0805455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.