

Abstract
The inward rectifier potassium (Kir) channel Kir7.1 (KCNJ13) has recently emerged as a key regulator of melanocortin signaling in the brain, electrolyte homeostasis in the eye, and uterine muscle contractility during pregnancy. The pharmacological tools available for exploring the physiology and therapeutic potential of Kir7.1 have been limited to relatively weak and non-selective small-molecule inhibitors. Here, we report the discovery in a fluorescence-based high-throughput screen of a novel Kir7.1 channel inhibitor, VU714. Site-directed mutagenesis of pore-lining amino acid residues identified Glutamate 149 and Alanine 150 as essential determinants of VU714 activity. Lead optimization with medicinal chemistry generated ML418, which exhibits sub-micromolar activity (IC50 = 310 nM) and superior selectivity over other Kir channels (at least 17-fold selective over Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, and Kir4.1) except for Kir6.2/SUR1 (equally potent). Evaluation in the EuroFins Lead Profiling panel of 64 GPCRs, ion-channels and transporters for off-target activity of ML418 revealed a relatively clean ancillary pharmacology. While ML418 exhibited low CLHEP in human microsomes which could be modulated with lipophilicity adjustments, it showed high CLHEP in rat microsomes regardless of lipophilicity. A subsequent in vivo PK study of ML418 by intraperitoneal (IP) administration (30 mg/kg dosage) revealed a suitable PK profile (Cmax = 0.20 µM and Tmax = 3 hours) and favorable CNS distribution (mouse brain:plasma Kp of 10.9 to support in vivo studies for in vivo studies. ML418, which represents the current state-of-the-art in Kir7.1 inhibitors, should be useful for exploring the physiology of Kir7.1 in vitro and in vivo.
Keywords: KCNJ13, thallium flux, electrophysiology, comparative modeling, melanocortin signaling, myometrium
INTRODUCTION
Inward rectifier potassium (Kir) channels play fundamental roles in diverse organ systems, and could in some cases represent unexploited drug targets for neurological, cardiovascular, endocrine, and muscle disorders 1, 2. Kir7.1, which is encoded by KCNJ13, one of sixteen genes comprising the Kir channel family, is expressed in the eye, brain, uterus, kidney, gut, and thyroid gland3–9. The genetic loss of Kir7.1 function in retinal pigmented epithelial cells of the eye leads to derangements in subretinal electrolyte homeostasis and cell degeneration underlying leber congenital amaurosis and snowflake vitreoretinopathy10–13. Using a panel of mostly non-specific inhibitors with differential activities against Kir channels, Ghamari-Langroudi and colleagues recently identified Kir7.1-like currents in neurons of the paraventricular nucleus (PVN) that are functionally coupled to the melanocortin-4 receptor (MC4R)14. Agonist binding to MC4R inhibits Kir7.1 activity, depolarizes the membrane potential, and increases neuronal firing, whereas competitive antagonist binding increases Kir7.1 activity and dampens neuronal excitability. This model suggests that Kir7.1 coupling to MC4R plays a key role in the central regulation of food intake and energy homeostasis by the PVN14. Kir7.1 expression in uterine muscles increases dramatically during pregnancy, thereby hyperpolarizing the membrane potential, inhibiting calcium signaling, and inducing uterine quiescence during fetal development15. Inhibiting Kir7.1 expression with microRNAs or inhibiting channel function with small-molecule inhibitors (i.e. VU59016, MRT0020076917) induces long-lasting uterine muscle contractions, lending support to the idea that Kir7.1 represents a novel drug target for augmentation of labor and treating post-partum hemorrhage17.
The current pharmacological ‘toolkit’ for exploring the physiology and therapeutic potential of Kir7.1 is inadequate, prompting us to perform a high-throughput screen (HTS) and lead optimization campaign in order to identify more potent and selective inhibitors. Here, we report the development and in vitro characterization of ML418, a new state-of-the-art inhibitor of Kir7.1.
RESULTS AND DISCUSSION
Kir7.1-M125R Assay Development
HTS was performed using a fluorescence-based assay that reports the inward movement of the potassium (K+) congener thallium (Tl+) through Kir7.1 channels expressed in the plasma membrane of T-REx-HEK293 cells. The higher conductance Kir7.1-M125R mutant was used as a surrogate to circumvent the weak Tl+ flux observed for wild type (WT) Kir7.1 (see ref. 18). As shown in Fig. 1, the assay reports robust Kir7.1-M125R–dependent Tl+ flux after induction with tetracycline (Fig. 1A), is DMSO tolerant up to 0.6% v/v (Fig. 1B; screening DMSO concentration = 0.1% DMSO v/v), and is sufficiently reproducible for HTS (Fig. 1C; mean ± SEM Z’= 0.67 ± 0.03; n = 3 plates on 3 separate days).
Figure 1. Kir7.1 Tl+ flux assay used for HTS.

(A) Representative Thallos fluorescence traces recorded from T-REx-HEK-293-Kir7.1-M125R cells cultured overnight with (grey line) or without (black line) tetracycline. Thallium stimulus buffer was added to each well simultaneously as indicated with the arrow. (B) DMSO tolerance test indicating that DMSO has no effect on Kir7.1-M125R–mediated Tl+ flux as concentrations up to 1.3% (v/v). (C) Determination of assay reproducibility. Alternate wells of a 384-well plate were treated with DMSO (vehicle) or Kir7.1 inhibitor VU573 (30 µM) before initiating Tl+ flux. Mean fluorescence and 3 S.D. from the mean for each well population are indicated with a blue dashed line and solid black line, respectively. The mean ± SEM. Z’ for 3 plates assayed on 3 separate days was Z’ = 0.67 ± 0.03.
Discovery and Characterization of VU714
From a pilot screen of 5,230 compounds in the Vanderbilt Institute of Chemical Biology (VICB) library, 11 putative Kir7.1-M125R inhibitors, comprising 5 distinct scaffolds, and with differing levels of selectivity over other Kir channels, were identified (data not shown). VU714 (Fig. 2A) was the most potent and selective inhibitor from the screen, and was therefore re-synthesized and confirmed from powder to be an authentic Kir7.1-M125R inhibitor. VU714 inhibited Kir7.1-M125R-mediated Tl+ flux in a dose-dependent manner with an IC50 of 5.6 µM (95% Confidence Interval [CI]: 1.9 µM - 17.5 µM) (Fig 2B–C). In “gold-standard” whole-cell voltage clamp experiments, the rate of Kir7.1-M125R inhibition by VU714 was concentration dependent (Fig. 2D), 10 µM VU714 fully inhibited outward and inward Kir7.1-M125R–mediated current (Fig. 2E), and the IC50 was 1.5 µM (CI: 1.3 µM - 1.7µM) (Fig. 2F). The 3.7-fold shift in IC50 determined with patch clamp electrophysiology, as compared with Tl+ flux, is consistent with previous observations of other Kir channel inhibitors 18–20. Quantitative Tl+ flux assays were utilized to evaluate the selectivity of VU714 for Kir7.1 over Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, Kir4.1, and Kir6.2/SUR1, as reported previously 16, 21, 22. The concentration-response curves (CRCs) shown in Fig. 3A revealed that VU714 is only moderately selective, and inhibits other Kir channels with a rank order potency of Kir7.1 (IC50 = 5.6 µM) > Kir4.1 (IC50 = 13 µM) > Kir1.1 (IC50 = 16 µM) > Kir6.2/SUR1 (IC50 = 30 µM) > Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2 (IC50 > 30 µM). Kir2.2,Kir2.3, and Kir3.1/3.2 CRCs have been excluded from Fig. 3A for clarity.
Figure 2. Discovery and characterization of VU714.

(A) Chemical structure of VU714. (B) Dose-dependent inhibition of Kir7.1-M125R–dependent Tl+ flux by VU714. Cells were pre-treated with the indicated concentrations of VU714 for 10 min before adding Tl+ stimulus buffer (arrow). (C) Mean ± SEM % control fluorescence recorded in the indicated concentrations of VU714 (n = 4). (D) Representative whole-cell patch clamp experiment showing timecourse of VU714-dependent inhibition of Kir7.1 current recorded at −120 mV. VU714 concentrations (in µM) are indicated at the top. Experiments were terminated by bath application of 2 mM barium (Ba). (E) Current-voltage plot showing inhibition of Kir7.1 by 10 µM VU714 or 2 mM Ba. (F) Mean ± SEM % Kir7.1 inhibition at −120 mV. IC50 values were derived by fitting CRC data with a 4-parameter logistical function.
Figure 3. Analysis of VU714 and ML418 selectivity for Kir7.1 over other Kir channels.

(A) VU714 CRCs constructed for Kir7.1-M125R over Kir6.2/SUR1 (open diamonds), Kir1.1 (closed circles), Kir2.1 (closed squares), Kir4.1 (open squares) in Tl+ flux assays. Kir2.2,Kir2.3, Kir3.1/3.2 (IC50s >30 µM) have been excluded for clarity. Data are means ± SEM % control fluorescence (n = 4–10 per concentration). (B) ML418 CRCs constructed for the same channels in Tl+ flux assays. (C) Representative whole-cell patch clamp experiment showing dose-dependent inhibition of Kir7.1 current at −120 mV by the indicated concentration of ML418. The experiment was terminated by bath application of 2 mM Ba. (D) Comparison of CRCs for VU714 (grey line, data from Fig. 2) and ML418 determined in patch clamp electrophysiology experiments.
VU714 Requires Pore-lining E149 and A150 Residues for Activity
Kir channels are tetrameric proteins consisting of 8 membrane-spanning domains, an ion-conduction pore, and cytoplasmic domain. Most small-molecule inhibitors studied to date appear to block the conduction pathway by interacting with amino acid residues lining the transmembrane pore (reviewed in 1). We therefore performed scanning mutagenesis and voltage-clamp electrophysiology to test if VU714 interacts with pore-lining amino acids in Kir7.1. Residues that are predicted from homology modeling (Fig. 4A) to face the pore were mutated to the corresponding residues in Kir2.1 and Kir1.1, which are only weakly inhibited by (Kir1.1) or insensitive (Kir2.1) to VU714 (Fig. 3A). Mutations were first introduced into WT Kir7.1, and if they impaired channel activity, were retested in the M125R background due to its more robust functional expression and ability to rescue the activity of some mutants. For clarity, only non-functional mutants in the M125R background are indicated in Fig. 4B; however, these mutations were first tested in the WT background and found to be non-functional. The M125R mutation does not alter VU714 sensitivity (Fig. 4C).
Figure 4. Identification of pore-lining residues in Kir7.1 required for VU714 activity.

(A) Alignment of pore-lining M2 helices from human Kir7.1, Kir2.1, and Kir1.1, with predicted pore-facing residues indicated with arrowheads. (B) Effects of pore mutations on Kir7.1-WT or Kir7.1-M125R sensitivity to 3 µM VU714. Data are mean ± SEM % inhibition at −120 mV. * P <0.05 compared to respective control. N.F., not functional. (C) VU714 CRC for Kir7.1-WT (closed squares; IC50=1.4 µM ), Kir7.1-M125R (open squares; IC50=1.6 µM), Kir7.1-M125R–E148Q (open circles; IC50=18.1 µM), Kir7.1-WT-A150S (closed circles; IC50=6.9 µM). (D) Kir7.1 homology model showing low-energy pose of VU714 near residues E149 and A150. (E) Higher-magnification view (from white box in D) of VU714 near E149 and A150.
Mutations L146V, I160M, and A161S had no effect on Kir7.1 inhibition by 3 µM VU714, whereas the T153C/WT mutation significantly (P <0.05) increased channel inhibition. Mutation of E149 to the corresponding Asparagine residue in Kir1.1 (E149N) abolished Kir7.1 activity in both WT and M125R backgrounds. However, the more conservative mutation, E149Q/M125R, led to a partial, albeit significant (P<0.05), loss of sensitivity to VU714. The negative charge at position 149 appears to be important for VU714 activity since mutating E149 to Aspartate (E149D) had no effect on block. Mutation of the adjacent residue A150 to Serine (A150S) led to a partial loss of inhibition. As shown in Fig. 4C, the E149Q and A150S mutations significantly (P <0.0001) shifted the IC50 for VU714 from ~2 µM in the WT or M125R mutants to ~18 µM and ~7 µM, respectively. Mutation of both residues simultaneously led to an additive loss of VU714 sensitivity (Fig. 4B).
Molecular modeling was used to visualize how VU714 might interact with Kir7.1 to induce channel block. Interactive ligand placement near E149 and A150, combined with energy minimization, revealed the best theoretical configuration of VU714 in the Kir7.1 channel pore (Fig. 4D–E). The limited volume of the pore in the putative binding region results in obstruction of the channel by VU714, just below the selectivity filter. This model suggests a straightforward steric mechanism for the observed conduction block by VU714. It is notable that this general region of pore is also involved in block of Kir1.1 by VU59123, Kir2.1 by ML13324, and Kir4.1 by fluoxetine25 (reviewed in ref. 1). Pentamidine26 and chloroquine27 require residues in the cytoplasmic pore for block; however, it is unlike that these residues participate in VU714 inhibition of Kir7.1 since the double mutation E149Q and A150S virtually eliminated block of the channel (Fig. 4B).
VU714 Structure-Activity Relationships
Medicinal chemistry was employed in an effort to identify VU714 analogs with improved potency and selectivity for Kir7.1. Our first priority was to reduce the high lipophilicity of VU714 (clogP = 5.83), which may be a liability for off-target selectivity, metabolism and toxicity. The initial library to explore SAR (Structure-Activity Relationship) in the right-hand portion of VU714 identified several equipotent moieties, including a simple methyl compound (1, IC50 = 4.8 µM, clogP = 4.41) without the benzyl moiety (Table 1). Subsequent libraries with a hydrophilic handle indicated that changing the piperidine ring system to a piperazine was not productive (amide, sulfonamide, urea and carbamate, 2 – 6), including the direct benzyl analog which resulted in a 4.5-fold loss of potency compared to VU714 (5, IC50 = 22.0 µM). However, attaching a hydrophilic handle (−NHR’) at the 4-position of the piperidine ring system endowed less lipophilic compounds with equal potency to the HTS hit compound, namely the amide compound (7, IC50 = 8.3 µM, clogP = 3.43) and the carbamate (11, IC50 = 1.7 µM, clogP = 3.65) which were generally more potent than the corresponding N-alkylated derivatives (12 – 16) (Table 1). Based on these results, we next evaluated a number of carbamate analogs in order to explore the SAR around this moiety (Table 2). While the simple methyl carbamate (17, inactive) and ethyl carbamate (18, IC50 = 9.8 µM) analogs were less active, all of the branched alkyl carbamates (19 – 22) were equipotent with the HTS hit compound. Among them, the isopropyl (19, IC50 = 1.3 µM, clogP = 3.25) and the tert-butyl carbamate (11, IC50 = 1.7 µM, clogP = 3.65) were the most active compounds. The benzyl compound (23), ring contracted compounds (24 and 25) and the spirocyclic compounds (26 and 27) were also tolerated, but less active than the HTS hit VU714. The next library was focused on right-hand 4-aminopiperidine amide analogs. The most active amides were aryl analogs in nature (28 – 40, 43) with the 3-chlorophenyl (32, IC50 = 3.1 µM, clogP = 4.34) being the most potent in this class. Branched alkyl amides (41 – 42) were also evaluated, but they were less active than the parent compound (Table 2).
Table 1.
SAR on attaching hydrophilic handles in a right-hand portion
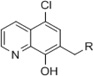 | |||
|---|---|---|---|
| Entry | R | R’ | Kir7.1 IC50 (µM)a |
| 1 | - | 4.8 | |
| 2 | COPh | Inactive | |
| 3 | SO2Ph | Inactive | |
| 4 | CONHPh | Inactive | |
| 5 | CH2Ph | 22.6 | |
| 6 | CO2But | Inactive | |
| 7 | 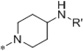 |
COPh | 8.3 |
| 8 | SO2Ph | 14.7 | |
| 9 | CONHPh | 15.8 | |
| 10 | CH2Ph | 12.4 | |
| 11 | CO2But | 1.7 | |
| 12 | 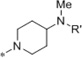 |
COPh | Inactive |
| 13 | SO2Ph | 12.2 | |
| 14 | CONHPh | 23.6 | |
| 15 | CH2Ph | 7.4 | |
| 16 | CO2But | 4.1 | |
hKir7.1 IC50 reported as average from our Thallium flux assay, n = 3
Table 2.
SAR on the carbamate and amide analogs
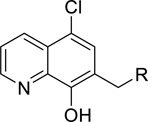 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | R’ | Kir7.1 IC50a (µM) |
Entry | R | R’ | Kir7.1 IC50a (µM) |
| 17 | Me | Inactive | 7 | Ph | 8.3 | ||
| 18 | Et | 9.8 | 28 | 2-F-Ph | 9.6 | ||
| 19 (ML418) | i-Pr | 1.3 | 29 | 3-F-Ph | 5.7 | ||
| 11 | 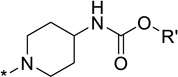 |
t-Bu | 1.7 | 30 | 4-F-Ph | 12.1 | |
| 20 | i-Bu | 4.1 | 31 | 2-Cl-Ph | 15.8 | ||
| 21 | Cyclopentyl | 2.8 | 32 | 3-Cl-Ph | 3.1 | ||
| 22 | Cyclohexyl | 4.2 | 33 | 4-Cl-Ph | 3.9 | ||
| 23 | CH2Ph | 6.3 | 34 | 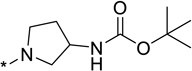 |
2-OMe-Ph | 7.1 | |
| 24 | 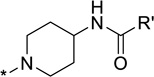 |
6.4 | 35 | 3-OMe-Ph | 9.3 | ||
| 36 | 4-OMe-Ph | 10.1 | |||||
| 25 | 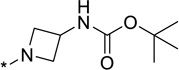 |
10.6 | 37 | 3-Me-Ph | 4.2 | ||
| 38 | 3-CF3-Ph | 6.1 | |||||
| 26 | 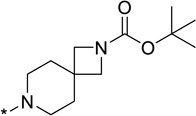 |
7.7 | 39 | 3-CN-Ph | 12.4 | ||
| 40 | 3-Br-Ph | 4.0 | |||||
| 41 | t-Bu | Inactive | |||||
| 27 | 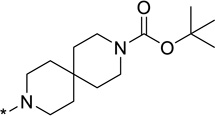 |
6.1 | 42 | Cyclohexyl | 9.6 | ||
| 43 | 2-Naphthyl | 12.3 | |||||
hKir7.1 IC50 reported as average from our Thallium flux assay; n = 3.
Next, we explored SAR around a left-hand quinoline moiety keeping the 4-methyl piperidine in the right-hand portion constant. Unfortunately, structural modifications of this portion of the molecule were not well tolerated, even minor changes such as changing the chlorine to fluorine and bromine atoms were less active. The only modification tolerated in our SAR exploration was an introduction of a methyl group at the 4-position of the quinoline ring (Fig. S1).
The SAR data (Fig. S1) is in qualitative agreement with the docking model that illustrates a plausible binding mode (Fig. 4E). Specifically, position 2 of the quinoline ring faces towards the protein backbone. There is no space for additional bulk. This is in contrast to positions 3 and 4 of the quinoline ring where methyl substitution is tolerated. The Nitrogen atom of the quinoline points to a polar pocket that could provide space for an additional, hydrogen-bound water molecule. A Carbon atom could not engage in these favorable interactions. The chloride and hydroxyl substituents are in tight, polar pockets. No larger substituents will fit into these pockets. A quantitative analysis of the agreement is beyond the predictive power of the homology model used for the docking simulation.
Discovery and Characterization of ML418
Having identified several VU714 analogs with lower lipophilicity and similar potencies in Tl+ flux assays, we next evaluated their activity toward Kir7.1 using voltage clamp electrophysiology and selectivity among Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, Kir4.1 and Kir6.2/SUR1 in Tl+ flux assays. The methyl piperidine analog (1) was similar in activity and selectivity to VU714 (Table 3). However, the carbamate (11 and 19) and amide (32) analogs exhibited superior inhibitory activity and selectivity. The isopropyl carbamate analog is the most potent in Kir7.1 activity (19 = ML418) in our library and is at least 23-fold selective against the Kir channels tested, with the exception of Kir6.2/SUR1 (Fig. 3B; Table 3). ML418 inhibits Kir7.1 dose-dependently (Fig. 3C) with an IC50 in patch clamp electrophysiology experiments of 310 nM (Fig. 3D; Table 3). While the tert-butyl carbamate (11) showed similar potency and selectivity to ML418, the amide analog (32) exhibited modest activity and greater than 10-fold selectivity over the Kir channels tested (Table 3). Although compounds 44 and 45 also show selectivity preference over Kir6.2/SUR1, and should be considered for use as an in vitro tool compound, the totality of the properties favors ML418 as the first selective Kir7.1 inhibitor.
Table 3.
Potency in patch clamp assay and selectivity over related Kir channels in Tl+ assay for selected analogs. ND, not determined.
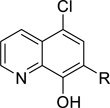 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R | Patch clamp |
Thallium flux | |||||||
| Kir7.1 IC50 (µM) |
Kir7.1 IC50 (µM) |
Kir Channel Selectivity (Fold) | ||||||||
| 1.1 | 2.1 | 2.2 | 2.3 | 4.1 | 6.2 | 3.1/3.2 | ||||
| VU714 | 1.5 | 5.6 | 3 | >5 | 2 | 5 | >5 | |||
| 1 | 1.4 | 4.8 | 6 | >7 | 3 | ND | ||||
| 44 | 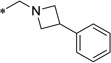 |
0.93 | 3.0 | 9 | 6 | 9 | 5 | 10 | 6 | ND |
| 45 | 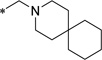 |
0.59 | 2.3 | 8 | >13 | ND | ||||
|
19 (ML418) |
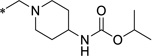 |
0.31 | 1.3 | >23 | 1 | >23 | ||||
| 11 | 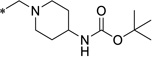 |
0.47 | 1.7 | >18 | 1 | ND | ||||
| 32 | 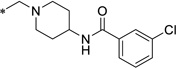 |
0.86 | 3.1 | >10 | ND | |||||
In a Lead Profiling Screen (Eurofins) of 64 potential off-targets, ML418 showed a relatively clean ancillary pharmacology having significant interactions (i.e. >50% radioligand displacement) with the L-type calcium channel, voltage-gated sodium channel, Dopamine D2S and D4.2 receptors, Sigma σ1 receptor, and Norepinephrine receptor (Table S1).
DMPK Evaluation for Selected Analogs
We evaluated selected analogs in our in vitro DMPK panel of assays, an assessment of intrinsic clearance (CLINT) and predicted hepatic clearance (CLHEP) in hepatic microsomes and protein binding in plasma (PPB) in multiple species. Interestingly, while the lipophilic HTS hit compound VU714 (cLogP = 5.83) was shown to have high intrinsic clearance (CLHEP (human) = 16.7 mL/min/kg and CLHEP (rat) =64.1 mL/min/kg) and low unbound fraction in plasma (%Fu (mouse) = 0.7, %Fu (rat) = 1.8 and %Fu (human) = 0.5) across species, more potent analogs than VU714 (32, 11 and 19 = ML418) exhibited lower intrinsic clearance in human hepatic microsomes (CLHEP (human) = 10.7, 7.9 and 3.6 mL/min/kg, respectively) and higher unbound fraction in mouse and rat plasma (%Fu (mouse) = 2.0, 5.6 and 11.4 and %Fu (rat) = 2.5, 3.6 and 8.8, respectively) as the cLogPs are reduced. While intrinsic clearance in rat hepatic microsomes of ML418 are slightly reduced (CLHEP (rat) =57.9 mL/min/kg) compared to VU714, subsequent PK study of ML418 by 30 mg/kg dosage of intraperitoneal administration revealed its characteristic PK profile (Fig. 5: Cmax = 0.20 µM and Tmax = 3 hours). It may be attributed to enterohepatic recirculation caused by phenol moiety. In a parallel CNS distribution study in mice, ML418 demonstrated excellent CNS penetration with a mouse brain:plasma ratio (Kp) of 10.9 (average of four mice, brain (323.9 ng/g):plasma (29.5 ng/mL)). Thus, ML418 possesses a DMPK profile suitable for both in vitro and in vivo studies aimed at modulating both peripheral and central Kir7.1 potassium channels.
Figure 5. Time course in vivo PK profile of ML418.

ML418 was dosed at 30 mg/kg in 10% EtOH, 40% PEG 400, 50% saline vehicle. The dosing solution was administered by intraperitoneal injection and whole blood collections via the carotid artery were performed at 0.117, 0.25, 0.5, 1, 2, 4, 7, and 24 hours post dose.
CONCLUSIONS
ML418 is the fifth small-molecule inhibitor of Kir7.1 reported to date and currently represents the state-of-the-art for the field (Table 5). The first Kir7.1 inhibitor, VU590, was identified in a HTS of approximately 225,000 compounds for inhibitors of Kir1.1 (IC50 = 0.24 µM) and subsequently found to have weak off-target activity toward Kir7.1 (IC50 ~ 8 µM)16. Evaluation of eight VU590 analogs revealed a flat SAR against Kir7.117. Another compound, VU573 was identified in the same Kir1.1 HTS, but found to have superior activity toward Kir7.1 (IC50 = 4.9 µM) over Kir1.1 (IC50=19 µM) in Tl+ flux assays. VU573 also inhibits Kir2.3 and Kir3 channels with single-micromolar potency18. Lead optimization efforts failed to generate more potent or selective inhibitors of Kir7.1, but did generate ‘inactive’ analogs that can be useful for determining the specificity of VU573 in pharmacology experiments targeting Kir7.1 (e.g. see ref. 14). ML133 was identified in a HTS of more than 300,000 compounds for modulators of Kir2.1 (IC50 = 0.3 µM at pH 8.0; approximately equal activity toward Kir2.2, Kir2.3, and Kir2.6) and found to possess weak Kir7.1 activity (IC50=33µM)24. MRT00200769 was discovered in an electrophysiology-based screen of 7,087 compounds for Kir7.1 inhibitors, but, despite being one of the most potent Kir7.1 inhibitors identified, was found to exhibit preferential activity toward cardiac hERG K+ channels (IC50=0.3 µM) over Kir7.1 (IC50=1.3µM). MRT00200769 also suffers from flat SAR17. In the present study, a HTS of 5,230 compounds from the VICB library led to the discovery of the novel pore blocker VU714, which was optimized with medicinal chemistry to generate ML418. The salient features of ML418 include: 1) Kir7.1 IC50=0.3 µM; 2) at least 23-fold selectivity for Kir7.1 over Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir4.1 except for Kir6.2/SUR1; 3) clean ancillary pharmacology against 58 GPCRs, ion channels, and transporters, including hERG; 4) PK profile in rat (Cmax = 0.20 µM and Tmax = 3 hours at 30 mg/kg i.p. dosing) and highly CNS penetrant (mouse brain:plasma Kp of 10.9). Further investigations of ML418 in order to probe the detailed physiological functions of Kir7.1 will be reported in due course.
Table 5.
Comparison with known inhibitors of Kir7.1
| Compound | Structure | Kir7.1 IC50 (µM) | ROMK IC50 (µM) | cLogPd | pKad | ||
|---|---|---|---|---|---|---|---|
| Tl+ | EP | Tl+ | EP | ||||
| ML418 | 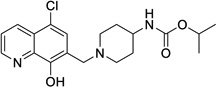 |
1.3 | 0.31 | >30 | Not Tested |
3.25 | 5.8 |
|
VU590 (ref. 16) |
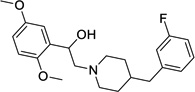
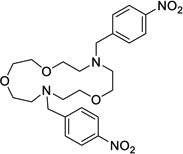 |
Not Tested |
8 | 0.3 | 0.24 | 7.46 | 7.5 |
|
ML133 (ref. 24a) |
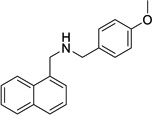 |
Not Tested |
33 | Not Tested |
>300 | 3.76 | 9.1 |
|
VU573 (ref. 18) |
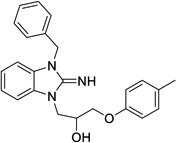 |
4.8 | 0.9 | 19 | Not Tested |
5.09 | 13.3 |
|
MRT00200769 (ref. 17b) |
 |
Not Tested |
1.3 | (38)c | Not Tested |
4.47 | 8.8 |
Kir2.1 IC50 = 0.29 µM
hERG IC50 = 0.3 µM
Calculated by ChemDraw
METHODS
Molecular biology
The open reading frame (ORF) of human Kir7.1 was sub-cloned into pcDNA5/TO (Life Technologies) to enable tetracycline-regulated expression (see below). Mutations were introduced into the Kir7.1 ORF using a QuickChange II mutagenesis kit (Agilent Technologies) and sequenced to verify incorporation of the intended mutation.
Tetracycline-inducible stable cell lines
Stably transfected T-REx-HEK-293 cell lines expressing the following Kir channels were generated and maintained in culture as described previously: Kir1.128, Kir2.122, Kir4.121, Kir6.2/SUR122, Kir7.1-M125R18. Kir channel expression was induced by culturing cells overnight in media containing 1 µg/ml tetracycline.
High-throughput screening (HTS)
HTS for Kir7.1-M125R modulators was performed using a Tl+-flux reporter assay essentially as described previously16, 20, 21. The Kir7.1-M125R mutation increases the channel unitary conductance and enables robust Tl+ flux measurement that cannot be achieved with the WT channel18. T-REx-HEK-293-Kir7.1-M125R cells (20,000/well) were plated in clear-bottomed, black-walled, 384-well plates, and cultured overnight in Delbecco’s Modified Eagle’s Medium (DMEM) containing 10% dialyzed FBS and 1 µg/ml tetracycline to induce Kir7.1-M125R expression. The following day, the cells were incubated with dye-loading assay buffer (Hank’s Balanced Salt Solution, 20 mM HEPES, pH 7.3) containing 0.01% (w/v) Pluronic F-127 (Life Technologies) and 1.2 µM Thallos-AM (TEFlabs, Austin, TX) Tl+ reporter dye. The dye-loading solution was replaced after 1hr with 20 µL/well assay buffer. Test compounds from the VICB Library were dispensed into 384-well plates using an Echo555 liquid handler (Labcyte, Sunnyvale, CA) and diluted to a 2X concentration. Cell plates were transferred to a Hamamatsu Functional Drug Screening System 6000 (FDSS6000; Hamamatsu, Tokyo, Japan) where 20 µL/well test compounds were dispensed into wells to a nominal concentration of 10 µM. After a 20-min incubation period, baseline fluorescence (excitation 470 ± 20 nm, emission 540 ± 30 nm) was recorded at 1Hz for 10 sec before addition of thallium stimulus buffer containing (in mM): 125 NaHCO3, 1.8 CaSO4, 1 MgSO4, 5 glucose, and 1.8 Tl2SO4. Fluorescence data were collected for an additional 4 min at 1Hz. Data were analyzed essentially as described previously28, 29 using a combination of Microsoft Excel (Microsoft Corporation, Redmond, WA) with XLfit add-in (IDBS, Guildford, Surrey, UK) and GraphPad Prism™(GraphPad Software, San Diego, CA, USA). Raw data were opened in Microsoft Excel and each data point in a given trace was divided by the first data point from that trace followed by subtraction of data points from control traces that were generated in the presence of vehicle controls. The slope of the fluorescence increase beginning 5 sec after Tl+ addition and ending 20 sec after Tl+ addition was calculated. The data were then plotted in Prism software to generate CRCs. Potencies were calculated from fits to CRC data using a non-linear regression analysis.
Transient transfections and whole-cell patch clamp electrophysiology
Transfections and whole-cell patch clamp experiments were performed essentially as described previously22. Briefly, HEK-293T cells were transfected with plasmids encoding WT or mutant Kir7.1 and EGFP (transfection marker) using Lipofectamine LTX (Life Technologies). The next day, the cells were dissociated with trypsin, plated on poly-L-lysine-coated round glass coverslips, and allowed to recover for at least 1hr before beginning experiments. Coverslips were transferred to a small-volume recording chamber on the stage of an inverted fluorescence microscope and superfused with a control bath solution containing (in mM), 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, 10 HEPES, pH 7.4. Electrodes were pulled with a Flaming-Brown P-1000 micropipette puller and had resistances between 2–3 MΩ when filled with the following solution (in mM): 135 mM KCl, 2 MgCl2, 1 EGTA, 10 HEPES, pH 7.3. The cells were voltage clamped at a holding potential of −75 mV and then stepped to −120 mV for 500 msec before ramping to 120 mV at a rate of 2.4 mv/msec. The cell potential was returned to −75 msec for 5 sec before initiating the step-ramp protocol again. Dose-response experiments were performed by superfusing cells with increasing doses of Kir7.1 inhibitor (e.g. ML418) followed by 4 mM BaCl2 to fully block Kir7.1. Cells exhibiting less than 90% block by BaCl2 were excluded from analysis. IC50 values were determined by fitting the Hill equation to CRCs using variable-slope, unconstrained, nonlinear regression analyses performed with GraphPad Prism (GraphPad Software, San Diego, CA, USA). All experiments yielded acceptable Hill slope (>0.8) and r2 (0.99) values. IC50 values are expressed as mean of n = 5 values. Mean IC50 values and 95% confidence limits were determined with GraphPad InStatTM (GraphPad Software, San Diego, CA, USA). The mean IC50 values were statistically analyzed using a one-way ANOVA measure with Dunnets multiple comparison test with significance being represented by P<0.05. Statistical analyses were performed using InStatTM (GraphPad Software, San Diego, CA, USA).
Comparative homology modeling and in silico docking
A comparative model of Kir7.1 was generated based on the 3.11 Angstrom resolution crystal structure Kir2.2 (PDB ID 3JYC)30. The sequence identity between Kir7.1 and Kir2.2 is 33.5% among the 343 residues aligned in each subunit of the tetramer. MODELLER version 9.9 [REF 2] was used to construct the model. The inhibitor VU0488714 was docked into the model of Kir7.1 using the Molecular Operating Environment (MOE) software package 31. Docking was performed by allowing flexibility of the ligand molecule and also the side chains of the protein model. A representative docking pose was selected from among the 29 lowest-energy results by choosing the best-scoring pose that features multiple interactions between the inhibitor and the experimentally-determined residues glutamate 149 and alanine 150.
Chemical synthesis
General. All NMR spectra were recorded on a 400 MHz AMX Bruker NMR spectrometer. 1H and 13C chemical shifts are reported in δ values in ppm downfield with the deuterated solvent as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, b = broad, m = multiplet), integration, coupling constant (Hz). Low resolution mass spectra were obtained on an Agilent 6120 or 6150 with ESI source. Method A: MS parameters were as follows: fragmentor: 70, capillary voltage: 3000 V, nebulizer pressure: 30 psig, drying gas flow: 13 L/min, drying gas temperature: 350 °C. Samples were introduced via an Agilent 1290 UHPLC comprised of a G4220A binary pump, G4226A ALS, G1316C TCC, and G4212A DAD with ULD flow cell. UV absorption was generally observed at 215 nm and 254 nm with a 4 nm bandwidth. Column: Waters Acquity BEH C18, 1.0 × 50 mm, 1.7 um. Gradient conditions: 5% to 95% CH3CN in H2O (0.1% TFA) over 1.4 min, hold at 95% CH3CN for 0.1 min, 0.5 mL/min, 55 °C. Method B: MS parameters were as follows: fragmentor: 100, capillary voltage: 3000 V, nebulizer pressure: 40 psig, drying gas flow: 11 L/min, drying gas temperature: 350 °C. Samples were introduced via an Agilent 1200 HPLC comprised of a degasser, G1312A binary pump, G1367B HP-ALS, G1316A TCC, G1315D DAD, and a Varian 380 ELSD (if applicable). UV absorption was generally observed at 215 nm and 254 nm with a 4 nm bandwidth. Column: Thermo Accucore C18, 2.1 × 30 mm, 2.6 um. Gradient conditions: 7% to 95% CH3CN in H2O (0.1% TFA) over 1.6 min, hold at 95% CH3CN for 0.35 min, 1.5 mL/min, 45 °C. High resolution mass spectra were obtained on an Agilent 6540 UHD Q-TOF with ESI source. MS parameters were as follows: fragmentor: 150, capillary voltage: 3500 V, nebulizer pressure: 60 psig, drying gas flow: 13 L/min, drying gas temperature: 275 °C. Samples were introduced via an Agilent 1200 UHPLC comprised of a G4220A binary pump, G4226A ALS, G1316C TCC, and G4212A DAD with ULD flow cell. UV absorption was observed at 215 nm and 254 nm with a 4 nm bandwidth. Column: Agilent Zorbax Extend C18, 1.8 µm, 2.1 × 50 mm. Gradient conditions: 5% to 95% CH3CN in H2O (0.1% formic acid) over 1 min, hold at 95% CH3CN for 0.1 min, 0.5 mL/min, 40 °C. For compounds that were purified on a Gilson preparative reversed-phase HPLC, the system comprised of a 333 aqueous pump with solvent-selection valve, 334 organic pump, GX-271 or GX-281 liquid hander, two column switching valves, and a 155 UV detector. UV wavelength for fraction collection was user-defined, with absorbance at 254 nm always monitored. Method: Phenomenex Axia-packed Luna C18, 30 × 50 mm, 5 µm column. Mobile phase: CH3CN in H2O (0.1% TFA). Gradient conditions: 0.75 min equilibration, followed by user defined gradient (starting organic percentage, ending organic percentage, duration), hold at 95% CH3CN in H2O (0.1% TFA) for 1 min, 50 mL/min, 23 °C. Solvents for extraction, washing and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification.
Synthetic scheme and characterization of ML418
Synthetic scheme of ML418 is shown in Figure S2 as an example of general synthetic scheme. Experimental procedure for ML418 is described below. Specific synthetic schemes for each compound are also shown in supplemental information (Figures S3–6).
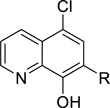
5-Chloro-8-hydroxyquinoline-7-carbaldehyde (49A)
To a solution of 5-chloroquinolin-8-ol 48A (8.08 g, 45.0 mmol) in TFA (75 mL) was added hexamethylenetetramine (12.62 g, 90.0 mmol) at ambient temperature. After a resulting reddish solution was refluxed at 120 −130°C for 5 hours, 1 mol/L HCl-aq (200mL) was added to the reaction mixture at 0°C which was stirred at ambient temperature for 20 min. To the reaction mixture was added ethyl acetate (75 mL) and 5 mol/L NaOH-aq (140mL) at 0°C. The resulting precipitates were collected by filtration and washed with water to give a crude product (6.59 g) which was triturated with mixed solvent of EtOH (25 mL) and Et2O (20 mL) to yield 5-chloro-8-hydroxyquinoline-7-carbaldehyde 49A (3.480 g, 37% yield) as a beige powder.
tert-Butyl (1-((5-chloro-8-hydroxyquinolin-7-yl)methyl)piperidin-4-yl)carbamate (11)
To a suspension of 49A (818 mg, 3.94 mmol) in DCM (25 mL) was added 4-boc-aminopiperidine (1.58 g, 7.89 mmol) at ambient temperature. After a resulting greenish solution was stirred at ambient temperature for 1 hour, sodium triacetoxyborohydride (1.25 g, 5.91 mmol) was added to the reaction mixture which was stirred at ambient temperature for 18 hours. The reaction mixture was poured into NaHCO3-aq (150 mL) and it was extracted with DCM (1st: 125 mL, 2nd: 50 mL). Combined organic extracts were washed with (NaHCO3-aq + water + brine) and dried over MgSO4. The filtrate was evaporated under reduced pressure to give crude product (2.28 g) which was purified on silica gel chromatography (DCM/MeOH) after combined with another crude product (1.41 g) by same reaction conditions from 49A (451 mg, 2.17 mmol) to yield tert-butyl (1-((5-chloro-8-hydroxyquinolin-7-yl)methyl)piperidin-4-yl)carbamate 11 (2.05 g, 86% yield) as a pale yellow powder.
7-((4-Aminopiperidin-1-yl)methyl)-5-chloroquinolin-8-ol trihydrochloride (51)
To a suspension of 11 (1.034 g, 2.64 mmol) in 1,4-dioxane (10 mL) was added HCl/1,4-dioxane (4 mol/L, 20 mL) at ambient temperature. After a reaction mixture was stirred for 24 hours, the precipitate was collected by filtration to yield 7-((4-aminopiperidin-1-yl)methyl)-5-chloroquinolin-8-ol trihydrochloride 51 (1.034 g, 98% yield) as a yellow powder.
iso-Propyl (1-((5-chloro-8-hydroxyquinolin-7-yl)methyl)piperidin-4-yl)carbamate (19 = ML418)
To a solution of 51 (1.015 g, 2.53 mmol) and DIPEA (1.76 mL, 10.1 mmol) in DCM (25 mL) was added a solution of iso-propyl chloroformate in toluene (2 mol/L, 1.30 mL, 2.66 mmol) at 0°C. After a resulting greenish solution was stirred at ambient temperature for 1 hour, it was poured into ice/NaHCO3-aq, which was extracted with DCM (x 2). Combined organic extracts were dried over MgSO4 and the filtrate was evaporated under reduced pressure. The residue was purified by Gilson HPLC separation system using (0.1% TFA in water)/CH3CN as an eluent to give crude product (c.a. 1.1 g) which was recrystallized from Et2O to yield desired product (633 mg 51% yield) as a TFA salt. To a solution of TFA salt of the desired product (1,236 g, 2.51 mmol) in DCM (37 mL) was added saturated NaHCO3-aq (12.5 mL) at ambient temperature. Organic phase was separated and aqueous phase was extracted with DCM (x 2). Combined organic phase was dried over MgSO4. The filtrate was evaporated under reduced pressure to yield iso-propyl (1-((5-chloro-8-hydroxyquinolin-7-yl)methyl)piperidin-4-yl)carbamate 19 (ML418) (915 mg, 96% yield) as a pale yellow powder. 1H NMR (400.1 MHz, DMSO-d6): 8.93 (dd, J = 4.2, 1.3 Hz, 1H), 8.47 (dd, J = 8.5, 1.3 Hz, 1H), 7.69 (dd, J = 8.5, 4.2 Hz, 1H), 7.61 (s, 1H), 7.02 (d, J = 7.7 Hz, 1H), 4.73 (sept, J = 6.2 Hz, 1H), 3.51-3.16 (br, 1H), 2.85-2.82 (m, 2H), 2.15-2.10 (m, 2H), 1.75-1.72 (m, 2H), 1.46-1.39 (m, 2H), 1.16 (d, J = 6.2 Hz, 6H). 13C NMR (100.6 MHz, DMSO-d6): 155.61, 151.63, 149.47, 139.31, 132.80, 128.72, 125.33, 123.09, 121.32, 118.58, 66.86, 56.62, 52.36, 47.98, 32.29, 22.58. LCMS: RT = 0.773 min, m/z = 378 [M + H]+. HRMS calc’d for: C19H24ClN3O3 [M+], 377.1506; found 377.1507.
Table 4.
In vitro DMPK profile for selected analogs
 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R | PPB (%Fu) | CLHEP (mL/min/kg) |
cLogPa | |||
| Mouse | Rat | Human | Rat | Human | |||
| VU714 | 0.7 | 1.8 | 0.5 | 64.1 | 16.7 | 5.83 | |
|
19 (ML418) |
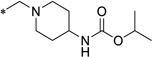 |
11.4 | 8.8 | 1.0 | 57.9 | 3.6 | 3.25 |
| 11 | 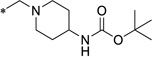 |
5.6 | 3.6 | 1.1 | 56.2 | 7.9 | 3.65 |
| 32 | 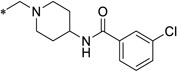 |
2.0 | 2.5 | 1.6 | 60.1 | 10.7 | 4.34 |
Calculated by ChemDraw
Acknowledgments
Funding
This work was funded in part by the Vanderbilt Institute of Clinical and Translational Research pilot grant (D.R.S, J.S.D.), DK082884 (J.S.D.), and MLPCN (C.R.H, C.W.L.).
Footnotes
SUPPORTING INFORMATION
SAR, chemical synthesis of all analogs, in vitro pharmacology procedures, in vitro PK methods, in vivo PK methods.
Author Contributions
Conceived and designed the experiments: D.R.S., H.K., K.S.V, J.S., R.R.R, E.F., J.M., A.L.B., C.W.L., C.R.H., J.S.D. Performed data analysis: D.R.S., H.K., K.S.V., R.R.R., K.R.V., E.F., J.S., J.M., A.L.B., C.W.L., C.R.H., Contributed reagents/materials/analysis tools: D.R.S., H.K., J.S., J.M., C.W.L., Wrote manuscript: D.R.S., H.K., J.S., J.M., A.L.B., C.W.L., C.R.H., J.S.D.
REFERENCES
- 1.Swale DR, Kharade SV, Denton JS. Cardiac and renal inward rectifier potassium channel pharmacology: emerging tools for integrative physiology and therapeutics. Curr Opin Pharmacol. 2014;15:7–15. doi: 10.1016/j.coph.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denton JS, Pao AC, Maduke M. Invited Review - Novel Diuretic Targets. Am J Physiol Renal Physiol. 2013;305(7):F931–F942. doi: 10.1152/ajprenal.00230.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ookata K, Tojo A, Suzuki Y, Nakamura N, Kimura K, Wilcox CS, Hirose S. Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J Am Soc Nephrol. 2000;11(11):1987–1994. doi: 10.1681/ASN.V11111987. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura N, Suzuki Y, Sakuta H, Ookata K, Kawahara K, Hirose S. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+ K+-ATPase. Biochem J. 1999;342(Pt 2):329–336. [PMC free article] [PubMed] [Google Scholar]
- 5.Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron. 1998;20(5):995–1005. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda K, Shimura M, Nakazawa T, Sato H, Tomita H, Sugano E, Tamai M. Expression and functional properties of unique inward rectifier K+ channel Kir7.1 in the porcine iris and retinal pigment epithelium. Curr Eye Res. 2003;27(5):279–287. doi: 10.1076/ceyr.27.5.279.17226. [DOI] [PubMed] [Google Scholar]
- 7.Fujita A, Horio Y, Higashi K, Mouri T, Hata F, Takeguchi N, Kurachi Y. Specific localization of an inwardly rectifying K(+) channel, Kir4.1, at the apical membrane of rat gastric parietal cells; its possible involvement in K+ recycling for the H+-K+-pump. J Physiol. 2002;540(Pt 1):85–92. doi: 10.1113/jphysiol.2001.013439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusaka S, Inanobe A, Fujita A, Makino Y, Tanemoto M, Matsushita K, Tano Y, Kurachi Y. Functional Kir7.1 channels localized at the root of apical processes in rat retinal pigment epithelium. J Physiol. 2001;531(Pt 1):27–36. doi: 10.1111/j.1469-7793.2001.0027j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Derst C, Hirsch JR, Preisig-Muller R, Wischmeyer E, Karschin A, Doring F, Thomzig A, Veh RW, Schlatter E, Kummer W, Daut J. Cellular localization of the potassium channel Kir7.1 in guinea pig and human kidney. Kidney Int. 2001;59(6):2197–2205. doi: 10.1046/j.1523-1755.2001.00735.x. [DOI] [PubMed] [Google Scholar]
- 10.Pattnaik BR, Shahi PK, Marino MJ, Liu X, York N, Brar S, Chiang J, Pillers AM, Traboulsi EI. A Novel KCNJ13 Nonsense Mutation and Loss of Kir7.1 Channel Function Causes Leber Congenital Amaurosis (LCA16) Hum Mutat. 2015 doi: 10.1002/humu.22807. [DOI] [PubMed] [Google Scholar]
- 11.Pattnaik BR, Tokarz S, Asuma MP, Schroeder T, Sharma A, Mitchell JC, Edwards AO, Pillers DA. Snowflake vitreoretinal degeneration (SVD) mutation R162W provides new insights into Kir7.1 ion channel structure and function. PLoS One. 2013;8(8):e71744. doi: 10.1371/journal.pone.0071744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sergouniotis PI, Davidson AE, Mackay DS, Li Z, Yang X, Plagnol V, Moore AT, Webster AR. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum Genet. 2011;89(1):183–190. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hejtmancik JF, Jiao X, Li A, Sergeev YV, Ding X, Sharma AK, Chan CC, Medina I, Edwards AO. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am J Hum Genet. 2008;82(1):174–180. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghamari-Langroudi M, Digby GJ, Sebag JA, Millhauser GL, Palomino R, Matthews R, Gillyard T, Panaro BL, Tough IR, Cox HM, Denton JS, Cone RD. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature. 2015;520(7545):94–98. doi: 10.1038/nature14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCloskey C, Rada C, Bailey E, McCavera S, van den Berg HA, Atia J, Rand DA, Shmygol A, Chan YW, Quenby S, Brosens JJ, Vatish M, Zhang J, Denton JS, Taggart MJ, Kettleborough C, Tickle D, Jerman J, Wright P, Dale T, Kanumilli S, Trezise DJ, Thornton S, Brown P, Catalano R, Lin N, England SK, Blanks AM. The inwardly rectifying K+ channel Kir7.1 controls uterine excitability throughout pregnancy. EMBO Mol Med. 2014;6(9):1161–1174. doi: 10.15252/emmm.201403944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol. 2009;76(5):1094–1103. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright PD, Kanumilli S, Tickle D, Cartland J, Bouloc N, Dale T, Tresize DJ, McCloskey C, McCavera S, Blanks AM, Kettleborough C, Jerman JC. A High-Throughput Electrophysiology Assay Identifies Inhibitors of the Inwardly Rectifying Potassium Channel Kir7.1. J Biomol Screen. 2015 doi: 10.1177/1087057115569156. [DOI] [PubMed] [Google Scholar]
- 18.Raphemot R, Lonergan DF, Nguyen TT, Utley T, Lewis LM, Kadakia R, Weaver CD, Gogliotti R, Hopkins C, Lindsley CW, Denton JS. Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1. Front Pharmacol. 2011;2:75. doi: 10.3389/fphar.2011.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raphemot R, Rouhier MF, Hopkins CR, Gogliotti RD, Lovell KM, Hine RM, Ghosalkar D, Longo A, Beyenbach KW, Denton JS, Piermarini PM. Eliciting renal failure in mosquitoes with a small-molecule inhibitor of inward-rectifying potassium channels. PLoS One. 2013;8(5):e64905. doi: 10.1371/journal.pone.0064905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raphemot R, Rouhier MF, Swale DR, Days E, Weaver CD, Lovell KM, Konkel LC, Engers DW, Bollinger SF, Hopkins C, Piermarini PM, Denton JS. Discovery and Characterization of a Potent and Selective Inhibitor of Aedes aegypti Inward Rectifier Potassium Channels. PLoS One. 2014;9(11):e110772. doi: 10.1371/journal.pone.0110772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raphemot R, Kadakia RJ, Olsen ML, Banerjee S, Days E, Smith SS, Weaver CD, Denton JS. Development and validation of fluorescence-based and automated patch clamp-based functional assays for the inward rectifier potassium channel Kir4.1. Assay Drug Dev Technol. 2013;11(9–10):532–543. doi: 10.1089/adt.2013.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raphemot R, Swale DR, Dadi PK, Jacobson DA, Cooper P, Wojtovich AP, Banerjee S, Nichols C, Denton JS. Direct Activation of beta-cell KATP Channels with a Novel Xanthine Derivative. Mol Pharmacol. 2014 doi: 10.1124/mol.114.091884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swale DR, Sheehan JH, Banerjee S, Husni AS, Nguyen TT, Meiler J, Denton JS. Computational and Functional Analyses of a Small-Molecule Binding Site in ROMK. Biophys J. 2015;108(5):1094–1103. doi: 10.1016/j.bpj.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, Li M, McManus OB. Selective inhibition of the Kir2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol. 2011;6(8):845–856. doi: 10.1021/cb200146a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol. 2009;75(6):1287–1295. doi: 10.1124/mol.108.052936. [DOI] [PubMed] [Google Scholar]
- 26.de Boer TP, Nalos L, Stary A, Kok B, Houtman MJ, Antoons G, van Veen TA, Beekman JD, de Groot BL, Opthof T, Rook MB, Vos MA, van der Heyden MA. The anti-protozoal drug pentamidine blocks Kir2.x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel. Br J Pharmacol. 2010;159(7):1532–1541. doi: 10.1111/j.1476-5381.2010.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T, Rupp J, Sachse FB, Tristani-Firouzi M, Sanchez-Chapula JA. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc Natl Acad Sci U S A. 2008;105(4):1364–1368. doi: 10.1073/pnas.0708153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhave G, Chauder BA, Liu W, Dawson ES, Kadakia R, Nguyen TT, Lewis LM, Meiler J, Weaver CD, Satlin LM, Lindsley CW, Denton JS. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol Pharmacol. 2011;79(1):42–50. doi: 10.1124/mol.110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raphemot R, Weaver CD, Denton JS. High-throughput Screening for Small-molecule Modulators of Inward Rectifier Potassium Channels. J Vis Exp. 2013;(71) doi: 10.3791/4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science. 2009;326(5960):1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chemical Computing Group, I. Molecular Operating Environment (MOE) Canada: Montreal, QC; 2015. [Google Scholar]