

Abstract
Background
It was recently demonstrated that cardiac sodium channels (Nav1.5) localized at the perinexus, an intercalated disc nanodomain associated with gap junctions (GJ), may contribute to electrical coupling between cardiac myocytes via an ephaptic mechanism. Impairment of ephaptic coupling by acute interstitial edema (AIE)-induced swelling of the perinexus was associated with arrhythmogenic, anisotropic conduction slowing. Given that Kir2.1 has also recently been reported to localize at intercalated discs (ID), we hypothesized that Kir2.1 channels may reside within the perinexus and that inhibiting them may mitigate arrhythmogenic conduction slowing observed during AIE.
Methods and Results
Using gSTED and STORM super-resolution microscopy, we indeed find that a significant proportion of Kir2.1 channels reside within the perinexus. Moreover, whereas Nav1.5 inhibition during AIE exacerbated arrhythmogenic conduction slowing, inhibiting Kir2.1 channels during AIE preferentially increased transverse conduction velocity - decreasing anisotropy and ameliorating arrhythmia risk compared to AIE alone. Comparison of our results with a nanodomain computer model identified enrichment of both Nav1.5 and Kir2.1 at intercalated discs as key factors underlying the experimental observations.
Conclusions
We demonstrate that Kir2.1 channels are localized within the perinexus alongside Nav1.5 channels. Further, targeting Kir2.1 modulates intercellular coupling between cardiac myocytes, anisotropy of conduction and arrhythmia propensity in a manner consistent with a role for ephaptic coupling in cardiac conduction.
Introduction
Cardiac conduction has classically been viewed as an electrotonic process occurring by means of direct ionic current flow from cell to cell via gap junctions (GJ).(11) Under this view, direction-dependent changes in action potential propagation in the heart were attributed to intercellular communication - in turn determined by GJ, myocyte geometry and tissue architecture.(11) On the other hand, ionic currents, principally the cardiac sodium current (INa), were thought to be responsible for membrane excitability and thereby, direction-independent changes in cardiac conduction.
The inward-rectifying potassium current (IK1), with its role in setting resting membrane potential, was thought to indirectly affect excitability by modulating the availability of cardiac sodium channels (Nav1.5). However, evidence has emerged to support a more direct role for potassium channels in determining membrane excitability and cardiac conduction (14, 33, 35) Specifically, it has been demonstrated previously that inhibiting the inward-rectifier K+ channels (Kir2.1) in intact ventricles resulted in faster conduction, suggesting that the inward-rectifier K+ current (IK1) may oppose early depolarization driven by the sodium current (INa)(35).
The understanding of cardiac conduction may be undergoing a paradigm shift. Evidence is mounting that ion channels localized at the intercalated disc, actively participate in intercellular coupling.(13, 17, 21) One leading hypothesis envisions ion channels mediating intercellular communication through transient accumulation or depletion of ions within restricted extracellular clefts located within the intercalated disc. In this non-canonical ephaptic model of conduction, intercalated disc-localized ion channels function as part of cell-to-cell junctions. Thus, targeted modulation of these channels should preferentially impact transverse conduction, since the activation wavefront encounters more cell-to-cell junctions per unit distance traveling transverse to the fiber direction than along it. Indeed, in recent work, we provided experimental evidence that Nav1.5 channels, localized at the perinexus adjacent GJ, play a major role in cell-to-cell propagation in the heart via ephaptic mechanisms.(6, 34) We also demonstrated that our observations across multiple experimental conditions were well predicted by a micro-domain computer model incorporating ephaptic coupling as well as preferential localization of sodium channels to the intercalated disc. Given that Kir2.1 channels have been reported to localize at the intercalated disc,(20) we hypothesized that Kir2.1 channels co-reside with Nav1.5 in the perinexus and modulate ephaptic coupling and thereby, anisotropic conduction in the ventricular myocardium. Here we added a further layer of refinement to our computer model in the form of preferential localization of Kir2.1 to the intercalated disc and compared results across six different experimental conditions.
Methods
The investigation was conducted in conformation with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All animal study protocols were approved by Institutional Animal Care and Use Committee (IACUC) at the Virginia Polytechnic University.
Animals
Neonatal myocyte cultures were prepared as previously described using myocytes isolated from ventricles of 2-day-old Sprague-Dawley rats.(24) Additionally, ventricles were isolated as previously described from adult male guinea pigs (800-1000g) and either frozen for cryosectioning or perfused as Langendorff preparations.(34-36)
Neonatal Rat Ventricular Myocyte (NRVM) cultures
Freshly isolated ventricles were placed in cold HBSS, minced and enzymatically dissociated into individual myocytes at 37°C. Myocytes were enriched by centrifugation on a Percoll density gradient 1.08/1.06 and plated in M199/EBSS, 5 % NCS, 10 % HS, and antibiotics onto gelatin-coated coverslips. After attachment at 37°C / 5% CO2, cells were washed in DPBS Ca2+/Mg2+ and maintained in culture for 5 days with maintenance media added every 2 days.
Immunolabeling
Cells and tissue sections were fixed in 2% paraformaldehyde at room temperature for 5 minutes and immunofluorescent staining was performed as previously described.(23, 24) Briefly, Samples were labeled using mouse anti-Cx43 (Millipore MAB3067, 1:100), rabbit anti-Nav1.5 (kindly provided by Dr. Peter Mohler, 1:100) and rabbit anti-Kir2.1 (Alomone Labs, APC-026, 1:100) antibodies. Goat anti-mouse AlexaFluor 546 (1:4000) and goat anti-rabbit AlexaFluor 488 (1:4000) secondary antibodies were used for confocal microscopy while goat anti-rabbit Alexa 647 (1:4000) and donkey anti-mouse Cy3b (1:100) secondary antibodies were used for super-resolution STochastic Optical Reconstruction Microscopy (STORM). Goat anti-rabbit Chromeo 505 (1:100) and anti-mouse biotin (1:200) followed by streptavidin-conjugated Horizon V500 (1:100) secondary antibodies were used for gated STimulated Emission Depletion (gSTED) microscopy.
Confocal, gSTED & STORM Microscopy
Confocal and gSTED Imaging were performed using a TCS SP8 laser scanning confocal microscope equipped with gSTED modules, a Plan Apochromat 63×/1.4 numerical aperture oil immersion objective, Leica HyD hybrid detectors and a 592nm STED depletion laser (Leica, Buffalo Grove, IL). Sequential imaging of individual fluorophores was performed with the excitation wavelength switched between frames. Also, gSTED and confocal imaging were performed sequentially and gSTED images were deconvolved using Huygens STED deconvolution software (Scientific Volume Imaging, Hilversum, The Netherlands). A maximum lateral ‘full width at half maximum’ resolution approaching 22 nm was achieved using gSTED(2, 25).
gSTED images were analyzed using custom Matlab software (Mathworks, Natick, MA) as previously described.(34) Briefly, a nearest-neighbor algorithm was used to identify clusters in each color channel and the geometric properties of the identified clusters were then calculated. The edge-to-edge distance from each cluster of a given species to the closest cluster of the other species was calculated. In addition, the perinexal region (defined as extending 200 nm from the edge of the Cx43 signal(24)) was demarcated for each Cx43 cluster and Nav1.5 and Kir2.1 clusters present within that region were identified.
STORM microscopy was performed using a Vutara 350 microscope equipped with biplane 3D detection and fast-sCMOS imaging achieving 20 nm XY and 50 nm Z resolution. The 3-dimensional location of each identified fluorophore in the STORM dataset was analyzed using the recently published STORM-based Relative Localization Analysis (STORM-RLA) technique.(32) Briefly, clusters of molecules were defined based on their spatial densities,(31) and the interactions between clusters of different types were quantified by the spatial extent of overlap, if any, and distance of closest apposition.
Guinea Pig Langendorff Preparations
Ventricles were extracted from adult male guinea pigs (800-1000g) anaesthetized with 30 mg/kg sodium pentobarbital (Nembutal) IP and perfused (at 40-55 mm Hg) as Langendorff preparations with oxygenated Tyrode's solution (containing, in mM, CaCl2 1.25, NaCl 140, KCl 4.5, dextrose 5.5, MgCl2 0.7, HEPES 10; NaOH 5.5mM pH 7.41) at 37°C as previously described(22, 35). Control Tyrode's solution was perfused for 35 minutes in all optical mapping experiments. Ventricles were paced from the anterior left ventricular (LV) epicardium (midapicobasal) at a basic cycle length of 300 ms with 1 ms pulses at 1.5-times the pacing threshold as described previously.(35)
Acute interstitial edema (AIE) was induced by mannitol (26.1 g/l / 143.2 mOsm) perfusion while the sodium current (INa) and the inward-rectifier potassium current (IK1) were respectively inhibited by flecainide (Flec; 0.5 μM) and barium chloride (BaCl2; 10 μM). Measurements were made 10 minutes following the start of each intervention or combination of interventions.
Electrocardiography
Volume conducted electrocardiograms (ECGs) were collected at 1kHz as previously described. (34-36) Ventricular tachycardia was defined as three or more un-paced consecutive heartbeats with a cycle length shorter than 130 ms.
Optical Mapping
Optical voltage mapping was performed using the voltage sensitive dye di-4-ANEPPS (15 μM) to quantify conduction velocity (CV) and anisotropy (AR; the ratio of longitudinal to transverse CV), as previously described.(22, 35) Briefly, ventricles stained with di-4-ANEPPS by direct coronary perfusion for 10 minutes were excited by LED light sources fitted with 510±5 nm filters (Chroma, Rockingham, VT). A SciMedia MiCam02 HS CCD camera (SciMedia, Irvine CA) in a tandem lens configuration was used to record fluoresced light filtered through a 610 nm LP filter (Newport, Irvine, CA). The system is capable of resolving membrane potential changes as small as 2 mV from 90 × 60 sites (16.5 × 12 mm) simultaneously at 1 kHz temporal resolution.
Motion was reduced with 7.5 mM 2,3-butanedione monoxime (BDM) combined with mechanical stabilization of the anterior epicardium against the front wall of the perfusion chamber. Activation time was defined as the time of the maximum first derivative of the action potential.(7)
Mathematical modeling
Conduction was simulated in a sheet of 3-dimensional myocytes using a previously described nanodomain model.(17, 34) Briefly, cardiac myocytes were represented as rectangular prisms with corner inclusions and were organized into a sheet to simulate anisotropic propagation. Cells were coupled via GJ located at the ends of myocytes. While the extracellular space was finely discretized, the intracellular space was discretized by linearly interpolating triangular elements with a node placed in each corner of each cell. Parameter values for the experimental conditions are summarized in Table 1.
Table 1. Nanodomain model parameters.
| Structural parameters: | ||
| Cell length | 101 μm | |
| Cell width | 24.1 μm | |
| Cellular offset | 50% transverse, 20% longitudinal | |
| Junctional sodium current density | 11 to 90% of total | |
| Junctional potassium current density | 11 to 90% of total | |
| Nominal Conductances: | ||
| GJ coupling | ḡj = 100 mS/cm2 | |
| Extracellular width - junctional | 15 nm | |
| Extracellular width - lateral | 0.1 μm | |
| Lateral extracellular conductivity (σ̄e) | 159.1 mS/cm | |
| Junctional extracellular conductivity (σ̄j) | 17.8 mS/cm | |
| Interventions: | ||
| AIE | σ̄e | 203.5 mS/cm |
| σ̄j | 62.8 mS/cm | |
Both longitudinal and transverse conduction were examined in the model while varying lateral and junctional extracellular conductances (σ̄e and σ̄j), sodium and potassium peak conductances (gNa and gK), and cellular distribution of sodium and potassium channels. For a detailed description of the nanodomain model, the reader is referred to our previous articles.(17, 34)
Statistical analysis
Statistical analysis of data was performed as previously reported.(34, 36) Briefly, parametric data were analyzed using a single factor ANOVA or 2-tailed Student's t-test with Šidák correction for paired and unpaired data. A Fisher's exact test was used to test for significance in nominal data. A p<0.05 was considered statistically significant. All data are reported as mean ± standard error unless otherwise noted.
Results
Inward-rectifier Potassium Channel Distribution
We performed confocal microscopy on sections of guinea pig ventricle in order to assess the cellular distribution of Kir2.1 protein. Immunosignals corresponding to Kir2.1 (red) co-distributed at the intercalated discs along with immunosignals corresponding to Cx43 (green; figure 1) and the confocal co-localization index between the two signals was measured at 44 ± 6%. However, at higher magnifications, the two proteins appeared adjacent to each other rather than overlapping, i.e., the two signals were not directly colocalized (figure 1, insets). This finding closely parallels our recent reports on Nav1.5 and Cx43: Specifically, using super-resolution gSTED(34) and STORM(32) imaging, Nav1.5 was found to be preferentially enriched within the non-junctional perinexal nanodomain surrounding Cx43 GJ in adult guinea pig ventricles.
Figure 1. Kir2.1 at the ID.
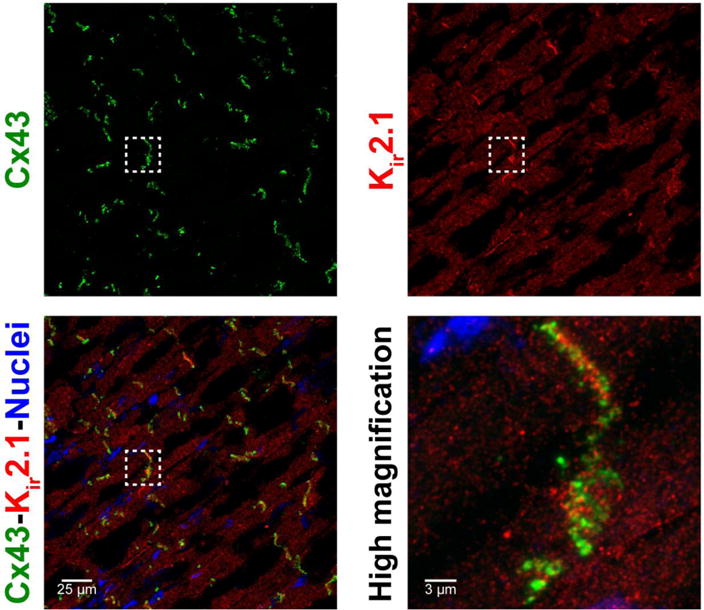
Representative confocal images of A) Cx43, B) Kir2.1 and C) overlay demonstrate co-localization of Cx43 and Kir2.1 in ventricular sections. D) A close up view demonstrates enrichment of Cx43 and Kir2.1 at the ID.
Here, we performed STORM super-resolution imaging to quantitatively assess Kir2.1 localization relative to Cx43 in sections of adult guinea pig ventricles. A representative STORM image in figure 2A shows individual fluorophore molecules corresponding to Cx43 (green) and Kir2.1 (red) depicted as colored spheres, and both proteins can be observed in high density at the intercalated disc. A higher magnification view in figure 2B reveals some areas where Cx43 and Kir2.1 exist in mixed populations; however, most areas of high Cx43 density have dense populations of Kir2.1 located adjacent to them rather than the proteins existing in a mixed cluster. Quantitative assessment by STORM-RLA revealed that only 10.9% of Cx43 clusters (i.e. presumptive GJ) demonstrated direct overlap with Kir2.1 clusters (figure 2C) accounting for 21.1% of intercalated disc-localized Kir2.1 clusters (figure 2D). Importantly, an additional 28.8% of Cx43 clusters had Kir2.1 located less than 200 nm away from the GJ edge (figure 2C) -the previously reported extent of the perinexus - accounting for 31.4% of total Kir2.1 clusters (figure 2D). Overall, the median distance from Cx43 clusters to the nearest Kir2.1 cluster was measured at 341 ± 13 nm. Thus, a sub-population of intercalated disc-localized Kir2.1 exists within juxta-GJ membranes where we previously also identified a high density of Nav1.5.
Figure 2. Cx43 and Kir2.1 relative localization.
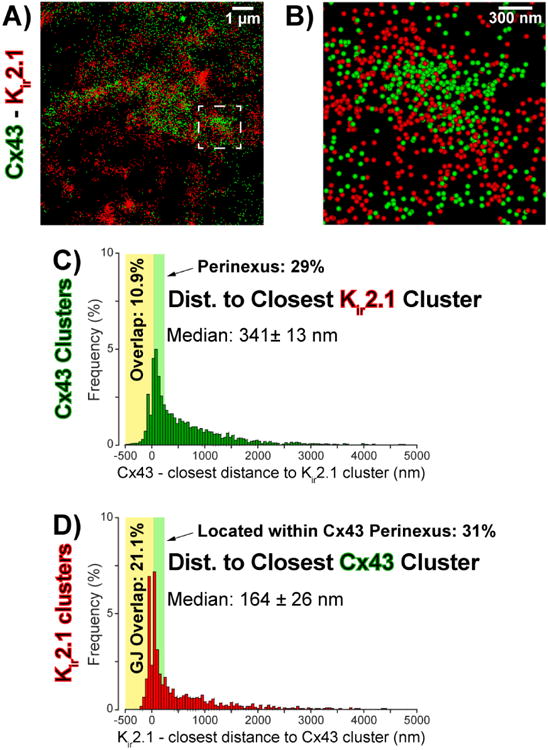
A) Representative STORM micrograph of an ID from guinea pig ventricles showing Cx43 (green) and Kir2.1 (red) immunosignals. B) A high magnification view of the region highlighted by the dashed white box in A. C) Edge-to-edge distance from Cx43 clusters to the closest Kir2.1 cluster. D) Edge-to-edge distance from Kir2.1 clusters to the closest Cx43 cluster.
In order to determine whether juxta-GJ enrichment of Nav1.5 and Kir2.1 varied with age and/or species, we analyzed super-resolution gSTED images of neonatal rat ventricular myocytes (NRVMs). Kir2.1 clusters were located at a median distance of 450 ± 58 nm from the nearest Cx43 clusters (figure 4). Importantly, only 5% of Cx43 GJs had overlapping Kir2.1 signal whereas 40% had Kir2.1 located within 200 nm of the cluster/plaque edge, i.e. within the perinexus. While only 3% of Kir2.1 clusters demonstrated direct overlap with GJ, 12.5% were located within perinexal regions surrounding GJs. These results are consistent with the aforementioned results obtained using STORM in adult guinea pig ventricles.
Figure 4. Cx43 and Kir2.1 relative localization.
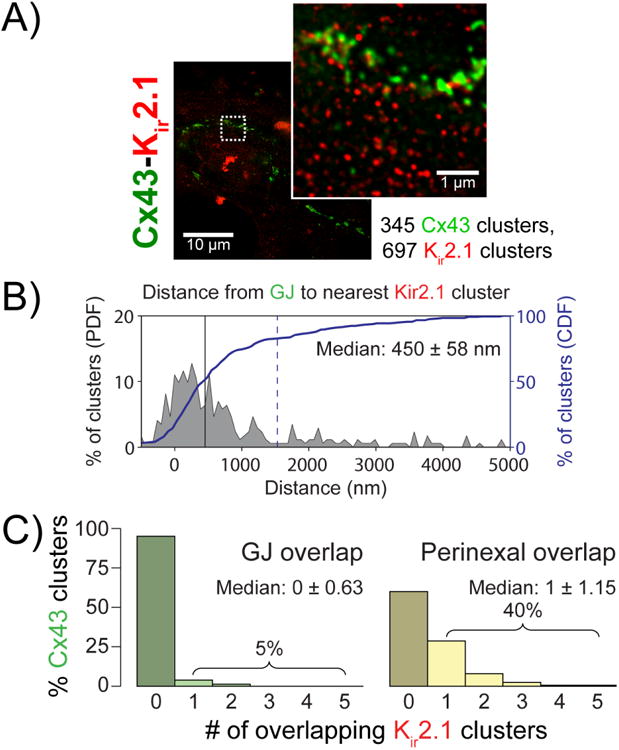
A) Representative gSTED micrograph of NRVMs showing Cx43 (green) and Kir2.1 (red) immunosignals. Inset shows high magnification view of the region highlighted by the dashed white box. B) Edge-to-edge from Cx43 clusters to the closest Kir2.1 cluster. C) Histograms of Cx43 clusters by number of Kir2.1 clusters demonstrating direct overlap (left, green) and located within the perinexus (right, yellow).
Given the close association between Cx43 and NaV1.5 in adult guinea pig ventricles, we next examined these two proteins in NRVMs. The median edge-to-edge distance between Cx43 and Nav1.5 clusters was measured from gSTED images at 129 ± 16 nm (figure 3) in NRVM. While only 9% of Cx43 clusters demonstrated direct overlap with Nav1.5 clusters, 53% had Nav1.5 cluster(s) within 200 nm of their edge, i.e. within the perinexus. Likewise, 9% of all Nav1.5 clusters demonstrated direct GJ overlap, whereas 37% were located within perinexal regions. These results in NRVMs are consistent with our recently published results obtained from adult guinea pig myocardium.(32, 34) Notably, the difference in median distance from Cx43 between NaV1.5 and Kir2.1 suggests a difference in the distribution of these channels between juxta-GJ membranes and the rest of the ID; however, a high proportion of Cx43 GJ had both channels located within their perinexi.
Figure 3. Cx43 and Nav1.5 relative localization.
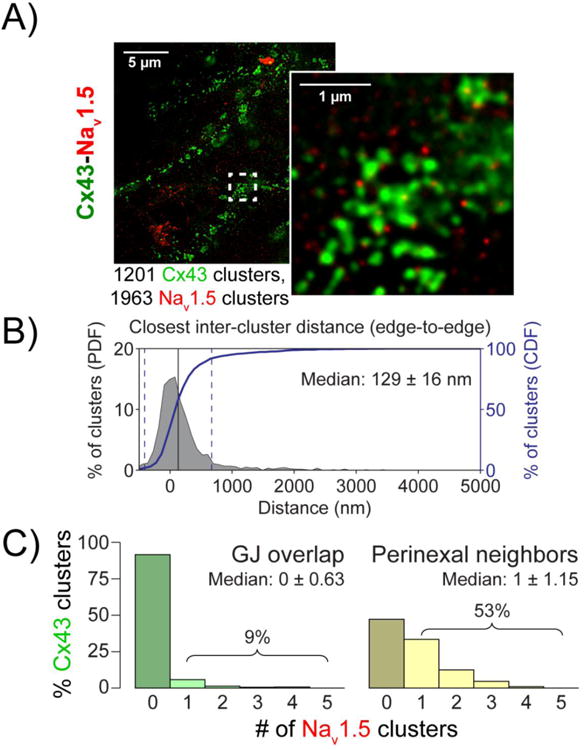
A) Representative gSTED micrograph of NRVMs showing Cx43 (green) and Nav1.5 (red) immunosignals. Inset shows high magnification view of the region highlighted by the dashed white box. B) Edge-to-edge distance from Cx43 clusters to the closest Nav1.5 cluster. C) Histograms of Cx43 clusters by number of Nav1.5 clusters demonstrating direct overlap (left, green) and located within the perinexus (right, yellow).
In summary, we demonstrate in two different preparations that significant sub-populations of intercalated disc-localized NaV1.5 and Kir2.1 exist in close proximity to Cx43 GJs.
Conduction Dependence on AIE, INa and IK1 - Experiments
We previously demonstrated that IK1 inhibition by BaCl2 can increase conduction velocity (CV)(35). Additionally, we demonstrated that acute interstitial edema (AIE) was associated with a selective increase in intermembrane spacing within the perinexus(34), as well as preferential transverse conduction slowing (34, 36). Therefore, we sought to assess whether IK1 inhibition could mitigate conduction slowing during AIE. To this end, we performed optical mapping on Langendorff-perfused guinea pig ventricles and quantified conduction velocity. Under control conditions, longitudinal (CVL) and transverse (CVT) conduction velocities were measured at 55.1 ± 1.4 and 19.6 ± 0.1 cm/s respectively, resulting in an anisotropic ratio (AR) of 2.8 ± 0.1 (figure 5A, F). AIE slowed conduction, particularly in the transverse direction (figure 5B, F), as we previously reported.(34, 36) Importantly, partial IK1 inhibition during AIE preferentially increased CVT more than CVL and decreased AR relative to AIE alone (figure 5C, F). These data suggest that partial IK1 inhibition mitigates anisotropic conduction slowing during AIE. The increase in CV elicited by IK1 inhibition during AIE, taken together with our previously published time control data,(34) also demonstrate that conduction slowing observed in our experiments is not simply a result of run-down of the preparation.
Figure 5. Inhibiting Kir2.1 channels mitigates AIE-induced conduction slowing.
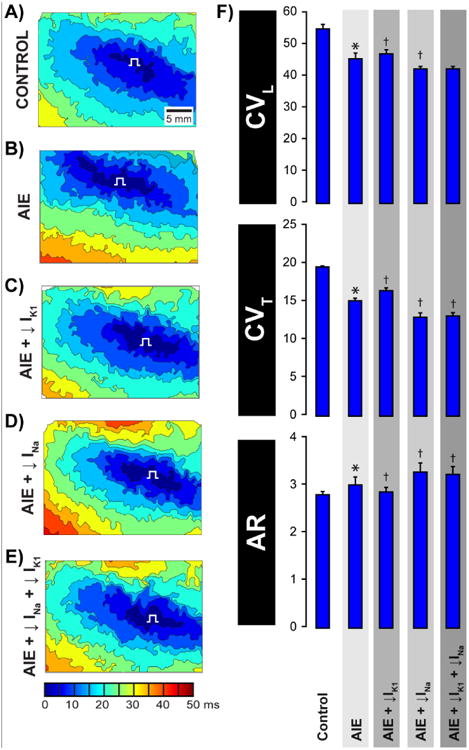
Representative activation isochrones maps during A) Control, B) AIE, C) AIE + IK1 inhibition, D) AIE + INa inhibition and E) AIE + INa inhibition + IK1 inhibition. F) Summary bar graphs of CVL, CVT and AR. n=5 hearts/group, * p<0.05 vs. control, † p<0.05 vs. AIE.
As in our previous study, partial INa inhibition (0.5 μM) during AIE preferentially slowed transverse conduction and increased anisotropy (AR) relative to AIE alone (figure 5D, F). In this setting of AIE and partial INa inhibition, IK1 inhibition did not measurably alter CV (figure 5E, F).
Conduction Dependence on AIE, INa and IK1 - Modeling
Next we compared these experimental observations against two computer models that we previously described(17, 34) - a uniform model where ion channels are uniformly distributed around the myocyte membrane and a polarized model incorporating enrichment of Nav1.5 and Kir2.1 enrichment at the intercalated disc. The polarized model presented here was modified to include subcellular Kir2.1 localization in addition to subcellular Nav1.5 localization, which was included in our previously published model.(34) For all values of potassium channel conductance (gK) tested, the uniform model predicted that increasing extracellular conductivity would increase CVL as evidenced by the upward shift of the CVL curves in Figure 6 (top). Once again, the uniform model predicted that CVT is insensitive to increases in extracellular conductance (σe; figure 6, top). In other words, the uniform model predicted a preferential increase in CVL during AIE and thus, did not match experimental observations.
Figure 6. Modeling results: Conduction dependence on potassium conductance.
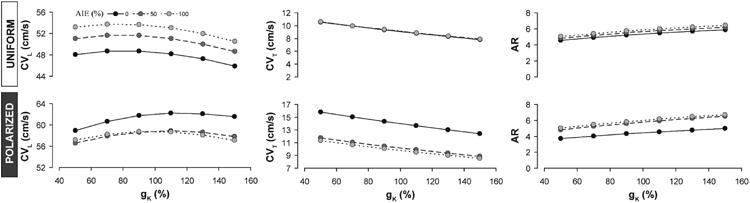
Predictions of CVL, CVT and AR as a function of gK at different levels of σe generated by the uniform model (top) and the polarized model (bottom).
In contrast, increasing sodium and potassium current density within the intercalated disc with the polarized model predicted that AIE should decrease both CVL and CVT, as evidenced by the downward shift in the CVL and CVT curves in Figure 6 (bottom). Additionally, the polarized model correctly anticipated an increase in AR during AIE secondary to the preferential slowing of CVT over CVL. Under these conditions, lowering gK in the polarized model to simulate IK1 inhibition reduced AR relative to simulated AIE alone. Thus, the polarized model was consistent with both experimental AIE and IK1 inhibition during AIE.
We next assessed the relationship between CV, σe and INa, at nominal and reduced gK. The uniform model predicted that with nominal gK (figure 7, top, black lines) increasing gNa should modestly increase AR by preferentially increasing CVL over CVT (figure 7, top panels). Decreasing gK in the uniform model (figure 7, top, red lines) decreased the AR at all gNa and σe tested, once again by preferentially affecting CVL over CVT. Thus, the uniform model's predictions contrasted with experimental results where CVT was more sensitive than CVL to gNa and gK. Further, the uniform model incorrectly predicted a direct relationship between AR and gNa under nominal gK and an inverse relationship between AR and gNa when gK is reduced. In experiments, AR and gNa were inversely related during nominal gK as well as when gK was reduced.
Figure 7. Modeling results: Conduction dependence on sodium conductance.
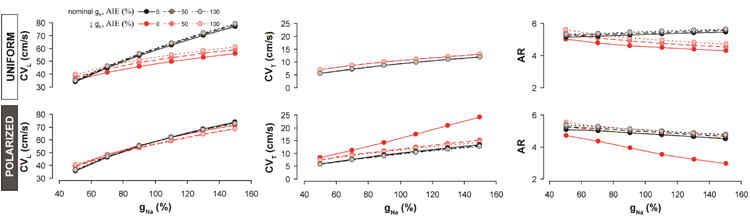
Predictions of CVL, CVT and AR as a function of gNa at different levels of σe under normal (black) and reduced (red) gK generated by the uniform model (top) and the polarized model (bottom).
The polarized model also predicted that, with nominal gK, increasing gNa should preferentially increase CVT over CVL, resulting in decreased AR for all values of σe (figure 7, bottom, black lines), which is consistent with our experimental observations. Additionally, the highest AR was observed in the polarized model when σe was greatest and gNa lowest. These predictions were all consistent with experimental data. Reducing gK in the polarized model (figure 7, bottom, red lines) preferentially increased CVT over CVL, thereby, lowering AR. This was particularly the case when σe was nominal and occurred to a lesser extent when σe was increased. In other words, the polarized model predicts that reducing gK during AIE should significantly reduce AR when gNa is nominal (100%), and this is consistent with experimental results. However, when gNa is reduced, reducing gK has a much lower impact on AR, and in experiments, we found that IK1 inhibition during AIE + INa inhibition did not significantly alter AR. This is because IK1 inhibition during AIE + INa inhibition was incapable of significantly increasing CVL or CVT, just as the polarized model anticipated.
Arrhythmia Incidence
In our previous study, we demonstrated that incidence of spontaneous VTs (polymorphic tachyarrhythmias persisting for at least 1 minute) was elevated during AIE (4/11 preparations; representative traces in figure 8) compared to control (0/12 preparations) and that it was further increased by the combination of AIE and INa inhibition (7/9 preparations).(34) While we determined that partial IK1 inhibition during AIE lowered VT incidence (1/8 preparations, p = ns vs. control), all arrhythmias quantified from the bath ECGs were polymorphic and persisted for more than 1 minute. Therefore, partial IK1 inhibition decreased arrhythmia propensity without observably altering the type of arrhythmias, as we did not observe any change in the arrhythmia morphology or ectopic beat formations for examples. These data are consistent with the relationship between altering cardiac conduction and arrhythmia propensity.
Figure 8. Volume-conducted ECGs: VT's during AIE.
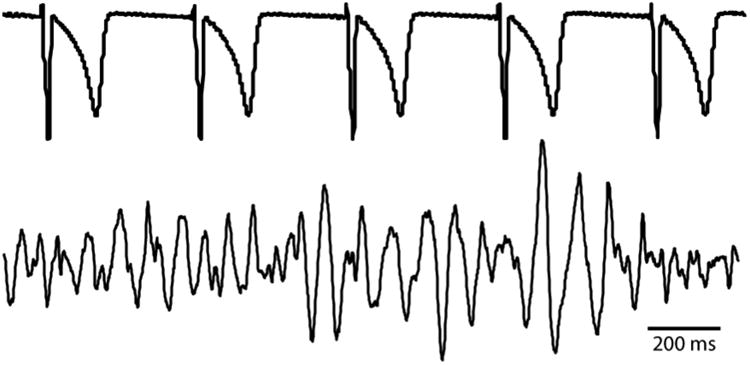
The top trace shows a sample volume-conducted ECG trace of intrinsic rhythm during AIE which had a cycle length of 408 ms. The bottom trace shows a representative ventricular tachyarrhythmia during AIE which had an average cycle length of 70 ms.
Discussion
We recently provided evidence that cardiac sodium channels (Nav1.5) within the perinexus, a specialized nanodomain of non-junctional membrane surrounding Cx43 GJ, play a major role in the propagation of electrical excitation in the heart via ephaptic mechanisms.(34) In this study, we demonstrate that Kir2.1 is enriched along with Nav1.5 in this same nanodomain. Further, we provide evidence that partially inhibiting Kir2.1 channels increases conduction velocity under control conditions,(35) as well as during AIE, provided that INa is not also inhibited. Aside from highlighting IK1 inhibition as a strategy to mitigate arrhythmogenic conduction slowing during AIE, these data suggest that ID-localized potassium channels may be important modulators of ephaptic coupling between myocytes.
Previously, we proposed that close membrane apposition (up to 20 or 30 nm) and enrichment of cardiac sodium channels (Nav1.5) within the perinexus allow the perinexus to function as a cardiac ephapse between ventricular myocytes,(34) which is consistent with theoretical predictions from multiple investigators.(12, 16, 21, 28) In our previous study, using gSTED imaging, we demonstrated that Nav1.5 clusters occurred within the perinexal regions of over half of Cx43 GJs identified in guinea pig ventricles.(34) In a more recent study, we reported a median distance of 71 ± 33 nm from Cx43 clusters to the nearest NaV1.5 clusters in adult guinea pig ventricles as quantified by STORM-RLA, i.e. 50% of GJ had NaV1.5 located less than 71 nm away, well within their perinexi.(32) Here, we demonstrate in neonatal rat ventricular myocytes that a near-identical 53% of GJ had Nav1.5 within their perinexi.
While ventricular myocytes show important differences in the organization of the molecular components of electrical excitation between different species and at different developmental stages, the similarities in perinexal enrichment of Nav1.5 and Kir2.1 adjacent to Cx43 GJ in both the adult guinea pig and the immature rat heart suggest high levels of conservation in the ultrastructure of this nanodomain. Interestingly, Nav1.5 was also reported to be enriched adjacent to desmosomes,(1) however, intermembrane spacing at these locations exceeds 50 nm.(15) Given that computer models stipulate intermembrane distance <=30nm for ephaptic coupling to occur, the perinexal ion channels may represent a specialized sub-population even within ID, capable of modulating ephaptic coupling. Therefore, these data underscore the importance of ultrastructural organization of ion channels within the ID for cardiac conduction.
We previously demonstrated that partial Kir2.1 inhibition alone increases CV during control conditions.(35) Here we evaluated conduction dependence on Kir2.1 when ephaptic coupling is compromised secondary to increased perinexal membrane spacing during mannitol-induced AIE.(34, 36) Interestingly, Kir2.1 inhibition during AIE preferentially increased CVT relative to AIE alone. This is consistent with our previous report that inhibiting another channel found in the perinexus (Nav1.5) can preferentially slow CVT during AIE. To elaborate, if ion channels located within intercalated disc nanodomains, such as the perinexus, are involved in intercellular communication, then it would be anticipated that an activation wavefront traveling transverse to the fiber axis would encounter more disc-localized GJ and associated perinexi per unit distance traveled than a wavefront traveling along the fiber axis. Thus, the observation that CVT was preferentially altered by modulating Nav1.5 and Kir2.1 channels during AIE, supports a role for these channels in intercellular coupling. While Kir2.1 modulation has been previously demonstrated to modulate conduction velocity,(20, 35) to our knowledge, this is the first demonstration that IK1 can anisotropically modulate conduction during AIE.
In addition to Nav1.5 and Kir2.1, previous studies have reported preferential localization of KV1.5,(19) the voltage-gated, shaker-related potassium channel, small conductance calcium-activated potassium (SK) channels(26), and Kir6.2, the ATP-sensitive potassium channel,(9) at the intercalated disc. Also, we have demonstrated that activation of the rapid (HERG)(14) and slow (KvLQT1)(33) delayed rectifier potassium channels as well as Kir6.2 can modulate cardiac conduction. Determining which of these channels, if any, also associate with the perinexus will be a useful avenue of inquiry. If more channels are identified within the perinexus, it would suggest that the narrow region of membrane at the edge of GJs contains multi-component complexes of ion channels associated with cardiac excitation. Thus, these structures may represent molecular machinery central to intercellular propagation in the heart.
We demonstrated in an earlier study that modulating IK1 could not rescue conduction when INa was inhibited.(35) Our observation here that IK1 inhibition did not significantly affect CV during a combination of AIE and INa inhibition is consistent with these earlier results. Overall, the finding that AIE + IK1 inhibition reduced AR and VT incidence to levels not significantly different from control supports the hypothesis that preventing anisotropic conduction slowing is anti-arrhythmic.(3, 11) The inability of IK1 inhibition to increase CV during AIE + INa inhibition further underscores the central roles of Nav1.5 and perinexal width to ephaptic coupling. Importantly, this study suggests that when a single theoretical determinant of ephaptic coupling is reduced (eg. increased perinexal width), then relatively smaller currents like IK1 can significantly modulate conduction. Consistent with double hit hypothesis, i.e. the idea that compromise of multiple determinants of conduction results in a more severe phenotype,(18, 29, 30) we find a diminished role for smaller currents like IK1 in rescuing ephaptic coupling when multiple determinants of ephaptic coupling are simultaneously reduced. Finally, the idea that multiple determinants of cardiac conduction must be simultaneously altered in order to observe conduction slowing has been suggested as the basis of the “Conduction Reserve” hypothesis.(29) We propose that ephaptic coupling may be a principal determinant of conduction reserve.
In earlier studies, we previously proposed the mechanism by which IK1 modulates conduction is by opposing INa depolarization very early in the action potential.(35) Our results herein support a similar mechanism operating locally within the perinexal ephapse whereby efflux of K+ into the perinexal extracellular cleft opposes the local potential change precipitated by the withdrawal of Na+ by Nav1.5 channels. Thus, Kir2.1 channels may constitute an opposing force to ephaptic coupling in much the same way as they oppose membrane depolarization in other parts of the myocyte membrane. However, this hypothesis does not preclude the possibility that Kir2.1 localization within intercalated disc nanodomains may result in a different local resting membrane potentials within these pockets relative to the lateral sarcolemma. It is important to note that Kir6.2 is also located at the intercalated disc and around the sarcolemma.(9) Therefore, the localization-dependent role of potassium channels as direct determinants of cardiac conduction merits further investigation.
While previous mathematical models have suggested that Nav1.5 localization to the intercalated disc can modulate ephaptic coupling by electric fields,(12, 16, 21, 28) the proposal that potassium channel localization to these same nanodomains may modulate ephaptic communication is novel. Importantly, the mathematical modeling results highlight the functional implications of sodium and potassium channel enrichment at the intercalated disc: The polarized model but not the uniform model was able to correctly predict the experimentally observed preferential response of CVT to IK1 and INa modulation during AIE. Furthermore, a purely electrotonic view of cardiac conduction anticipates that modulating an ionic current should affect excitability but not intercellular coupling; therefore, it should affect CV isotropically.(11) Yet, our experimental data show that modulating INa or IK1 during AIE anisotropically alters conduction in a manner consistent with altered intercellular communication. It is also notable that the polarized model incorporating sodium and potassium channel enrichment at cell-cell contacts and ephaptic coupling well predicts experimentally observed behavior across six different experimental conditions. Therefore, our results suggest that K+ channels can modulate anisotropic conduction and that their localization to specific nanodomains of the myocyte membrane may be a key determinant of their role in cardiac conduction.
Limitations
Although our polarized model includes the enrichment of Nav1.5 and Kir2.1 at the intercalated disc, it does not incorporate the full structural complexity of the intercalated disc as observed using electron microscopy.(5, 8, 10) Thus, nanodomain ion accumulation / depletion events, which are key to ephaptic coupling, are only approximated by the model. Additionally, pharmacological agents used in experimental studies have off-target effects: Although flecainide is a potent INa blocker, it has been shown to inhibit the transient outward potassium current (Ito) and a maintained outward potassium current (IK) in rat.(27) Similarly, BaCl2 has some cross-reactivity with the L-type calcium channel,(4) albeit at concentrations far greater than used in this study. The hypothesis forwarded in this manuscript does not preclude a role for protein remodeling in modulating conduction under these conditions. However, the study demonstrates that Kir2.1 enrichment at the intercalated disc is important for computational models of ephaptic coupling.
Conclusion
Anisotropic conduction slowing is strongly correlated to the risk of conduction block leading to ventricular arrhythmias. Previously, the major determinants of anisotropic conduction were theorized to be myocyte size and gap junction coupling. A growing body of evidence, including the data provided herein, supports the hypothesis that ion channels localized to intercalated disc nanodomains such as the perinexus may also play a role in electrically coupling myocytes and thus, emerge as determinants of anisotropic conduction. While these data have yet to be validated in larger vertebrates, and particularly humans, the findings suggest that preserving ephaptic coupling mechanisms may be as viable an anti-arrhythmic target as modulating gap junctions.
Significance.
For over half a century, electrical excitation in the heart has been thought to occur exclusively via gap junction-mediated ionic current flow between cells. Further, excitation was thought to depend almost exclusively on sodium channels with potassium channels being involved mainly in returning the cell to rest. Here, we demonstrate that sodium and potassium channels co-reside within nano-scale domains at cell-to-cell contact sites. Experimental and computer modeling results suggest a role for these channels in electrical coupling between cardiac muscle cells via an ephaptic mechanism working in tandem with gap junctions. This new insight into the mechanism of cardiac electrical excitation could pave the way for novel therapies against cardiac rhythm disturbances.
Acknowledgments
The authors would like to thank Dr. James Smyth for assistance with STORM microscopy. Thanks also to Dr. Gregory Hoeker and Michael Entz for assistance with optical mapping experiments and to Mrs. Jane Jourdan for assistance with NRVM experiments.
Funding Sources: This work was supported by an NIH R01 grant awarded to Dr. Poelzing (R01-HL102298-01A1), an NIH R01 grant awarded to Dr. Gourdie (RO1 HL56728-14A2) and by an American Heart Association post-doctoral fellowship awarded to Dr. Veeraraghavan (2013-15; completed).
Footnotes
Conflicts of Interest: None.
Disclosures: None.
References
- 1.Agullo-Pascual E, Lin X, Leo-Macias A, Zhang M, Liang FX, Li Z, Pfenniger A, Lubkemeier I, Keegan S, Fenyo D, Willecke K, Rothenberg E, Delmar M. Super-resolution imaging reveals that loss of the C-terminus of connexin43 limits microtubule plus-end capture and NaV1.5 localization at the intercalated disc. Cardiovascular research. 2014 doi: 10.1093/cvr/cvu195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clausen Mathias P, Galiani S, de la Serna Jorge B, Fritzsche M, Chojnacki J, Gehmlich K, Lagerholm BC, Eggeling C. Pathways to optical STED microscopy. NanoBioImaging. 2013:1. [Google Scholar]
- 3.Dhein S, Seidel T, Salameh A, Jozwiak J, Hagen A, Kostelka M, Hindricks G, Mohr FW. Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias. Front Physiol. 2014;5:424. doi: 10.3389/fphys.2014.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira G, Yi J, Rios E, Shirokov R. Ion-dependent inactivation of barium current through L-type calcium channels. The Journal of general physiology. 1997;109:449–461. doi: 10.1085/jgp.109.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geisler SB, Green KJ, Isom LL, Meshinchi S, Martens JR, Delmar M, Russell MW. Ordered assembly of the adhesive and electrochemical connections within newly formed intercalated disks in primary cultures of adult rat cardiomyocytes. Journal of biomedicine & biotechnology. 2010;2010:624719. doi: 10.1155/2010/624719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George SA, Sciuto KJ, Lin J, Salama ME, Keener JP, Gourdie RG, Poelzing S. Extracellular sodium and potassium levels modulate cardiac conduction in mice heterozygous null for the Connexin43 gene. Pflugers Arch. 2015 doi: 10.1007/s00424-015-1698-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girouard SD, Laurita KR, Rosenbaum DS. Unique properties of cardiac action potentials recorded with voltage-sensitive dyes. Journal of cardiovascular electrophysiology. 1996;7:1024–1038. doi: 10.1111/j.1540-8167.1996.tb00478.x. [DOI] [PubMed] [Google Scholar]
- 8.Green CR, Severs NJ. Gap junction connexon configuration in rapidly frozen myocardium and isolated intercalated disks. The Journal of cell biology. 1984;99:453–463. doi: 10.1083/jcb.99.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong M, Bao L, Kefaloyianni E, Agullo-Pascual E, Chkourko H, Foster M, Taskin E, Zhandre M, Reid DA, Rothenberg E, Delmar M, Coetzee WA. Heterogeneity of ATP-sensitive K+ channels in cardiac myocytes: enrichment at the intercalated disk. The Journal of biological chemistry. 2012;287:41258–41267. doi: 10.1074/jbc.M112.412122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoyt RH, Cohen ML, Saffitz JE. Distribution and three-dimensional structure of intercellular junctions in canine myocardium. Circulation research. 1989;64:563–574. doi: 10.1161/01.res.64.3.563. [DOI] [PubMed] [Google Scholar]
- 11.Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiological reviews. 2004;84:431–488. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- 12.Kucera JP, Kleber AG, Rohr S. Slow conduction in cardiac tissue, II: effects of branching tissue geometry. Circulation research. 1998;83:795–805. doi: 10.1161/01.res.83.8.795. [DOI] [PubMed] [Google Scholar]
- 13.Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circulation research. 2002;91:1176–1182. doi: 10.1161/01.res.0000046237.54156.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larsen AP, Olesen SP, Grunnet M, Poelzing S. Pharmacological activation of IKr impairs conduction in guinea pig hearts. Journal of cardiovascular electrophysiology. 2010;21:923–929. doi: 10.1111/j.1540-8167.2010.01733.x. [DOI] [PubMed] [Google Scholar]
- 15.Leo-Macias A, Liang FX, Delmar M. Ultrastructure of the intercellular space in adult murine ventricle revealed by quantitative tomographic electron microscopy. Cardiovascular research. 2015;107:442–452. doi: 10.1093/cvr/cvv182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, Keener JP. Ephaptic coupling in cardiac myocytes. IEEE transactions on bio-medical engineering. 2013;60:576–582. doi: 10.1109/TBME.2012.2226720. [DOI] [PubMed] [Google Scholar]
- 17.Lin J, Keener JP. Microdomain effects on transverse cardiac propagation. Biophysical journal. 2014;106:925–931. doi: 10.1016/j.bpj.2013.11.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthes S, Zhang M, Taffet S, Delmar M. Desmosomes and Gap Junctions in Epicardium-Derived Cells. A Possible Role in Arrhythmogenic Right Ventricular Cardiomyopathy? Heart rhythm : the official journal of the Heart Rhythm Society. 2010;7:1715–1716. [Google Scholar]
- 19.Mays DJ, Foose JM, Philipson LH, Tamkun MM. Localization of the Kv1.5 K+ channel protein in explanted cardiac tissue. The Journal of clinical investigation. 1995;96:282–292. doi: 10.1172/JCI118032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2134–2143. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mori Y, Fishman GI, Peskin CS. Ephaptic conduction in a cardiac strand model with 3D electrodiffusion. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6463–6468. doi: 10.1073/pnas.0801089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poelzing S, Veeraraghavan R. Heterogeneous ventricular chamber response to hypokalemia and inward rectifier potassium channel blockade underlies bifurcated T wave in guinea pig. American journal of physiology Heart and circulatory physiology. 2007;292:H3043–3051. doi: 10.1152/ajpheart.01312.2006. [DOI] [PubMed] [Google Scholar]
- 23.Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Molecular biology of the cell. 2011;22:1516–1528. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhett JM, Ongstad EL, Jourdan J, Gourdie RG. Cx43 Associates with Na(v)1.5 in the Cardiomyocyte Perinexus. The Journal of membrane biology. 2012;245:411–422. doi: 10.1007/s00232-012-9465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoonderwoert V, Dijkstra R, Luckinavicius G, Kobler O, van der Voort H. Huygens STED Deconvolution Increases Signal-to-Noise and Image Resolution towards 22 nm. Microscopy Today. 2013;21:38–44. [Google Scholar]
- 26.Skibsbye L, Wang X, Axelsen LN, Bomholtz SH, Nielsen MS, Grunnet M, Bentzen BH, Jespersen T. Antiarrhythmic Mechanisms of SK Channel Inhibition in the Rat Atrium. Journal of cardiovascular pharmacology. 2015;66:165–176. doi: 10.1097/FJC.0000000000000259. [DOI] [PubMed] [Google Scholar]
- 27.Slawsky MT, Castle NA. K+ channel blocking actions of flecainide compared with those of propafenone and quinidine in adult rat ventricular myocytes. The Journal of pharmacology and experimental therapeutics. 1994;269:66–74. [PubMed] [Google Scholar]
- 28.Sperelakis N, McConnell K. Electric field interactions between closely abutting excitable cells. IEEE engineering in medicine and biology magazine : the quarterly magazine of the Engineering in Medicine & Biology Society. 2002;21:77–89. doi: 10.1109/51.993199. [DOI] [PubMed] [Google Scholar]
- 29.Stein M, Boulaksil M, Engelen MA, van Veen TA, Hauer RN, de Bakker JM, van Rijen HV. Conduction reserve and arrhythmias. Neth Heart J. 2006;14:113–116. [PMC free article] [PubMed] [Google Scholar]
- 30.Stein M, van Veen TA, Remme CA, Boulaksil M, Noorman M, van Stuijvenberg L, van der Nagel R, Bezzina CR, Hauer RN, de Bakker JM, van Rijen HV. Combined reduction of intercellular coupling and membrane excitability differentially affects transverse and longitudinal cardiac conduction. Cardiovascular research. 2009;83:52–60. doi: 10.1093/cvr/cvp124. [DOI] [PubMed] [Google Scholar]
- 31.Tran TN, Drab K, Daszykowski M. Revised DBSCAN algorithm to cluster data with dense adjacent clusters. Chemometrics and Intelligent Laboratory Systems. 2013;120:92–96. [Google Scholar]
- 32.Veeraraghavan R, Gourdie R. Stochastic Optical Reconstruction Microscopy-based Relative Localization Analysis (STORM-RLA) for Quantitative Nanoscale Assessment of Spatial Protein Organization. Molecular biology of the cell. 2016 doi: 10.1091/mbc.E16-02-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veeraraghavan R, Larsen AP, Torres NS, Grunnet M, Poelzing S. Potassium channel activators differentially modulate the effect of sodium channel blockade on cardiac conduction. Acta Physiol (Oxf) 2013;207:280–289. doi: 10.1111/j.1748-1716.2012.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch. 2015 doi: 10.1007/s00424-014-1675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veeraraghavan R, Poelzing S. Mechanisms Underlying Increased Right Ventricular Conduction Sensitivity to Flecainide Challenge. Cardiovascular research. 2008;77:749–756. doi: 10.1093/cvr/cvm090. [DOI] [PubMed] [Google Scholar]
- 36.Veeraraghavan R, Salama ME, Poelzing S. Interstitial Volume Modulates the Conduction Velocity- Gap Junction Relationship. American journal of physiology Heart and circulatory physiology. 2012 doi: 10.1152/ajpheart.00868.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]